
Abstract
The richness and diversity of bacterial community in the fermented pit mud from the new and old distilleries of Shedian Liquor were analysed via the Illumina Miseq PE300 high-throughput sequencing technology, so as to further understand the differences of bacterial community structure in fermented pit muds with different pit ages. The results show that the bacterial community is mainly distributed into five dominant phyla (> 1.0%): Firmicutes, Synergistes, Bacteroidetes, Proteobacteria and Verrucomicrobia. There are 13 dominant genera (> 1.0%), of which Lactobacillus is dominant in both samples with a higher content in the new pit (61.12%) and lower in the old pit (1.15%). Caproiciproducens accounting for 43.53% and Syntrophomonas accounting for 6.20%, norank_f__Family_XIV (17.68%), Aminobacterium (17.68%), Sedimentibacter (1.89%), Caldicoprobacter (1.52%) and Sporanaerobacter (1.29%) are the dominant bacteria in the old pit mud. Bacillus accounting for 6.02%, Bacteroides (6.27%), Akkermansia (1.51%), Hydrogenispora (2.15%) and norank_f__Bacteroidales_S24-7_group (1.15%) are the dominant bacteria in the new pit mud. The results showed that there is a great difference in the bacterial community structure of in the new and old pit mud. In addition, compared with the bacterial community composition in the pit mud of Luzhou flavour Baijiu in Sichuan, it was found that the microorganisms that formed caproic acid and butyric acid were different.
Introduction
Luzhou-flavour liquor accounts for 70% of the domestic liquor industry in China. There is a saying that ‘A thousand year pit, ten thousand year vinasse’, indicating that pit mud plays an important role in the fermentation process of Luzhou-flavour liquor. Pit mud is the carrier for the growth and propagation of functional bacteria, and also the main place for the production of flavor substances of Luzhou-flavour liquor [Citation1]. The aroma compounds produced by the metabolism of pit mud microorganisms are the basis of the strong flavour of Luzhou-flavour liquor [Citation2], and the microbial community structure of the new and old pit muds has a great impact on the quality of the liquor it produced, therefore, it is of great significance to study the microbial diversity of the new and old pit muds on the production of Luzhou-flavour liquor.
Traditional microbial analysis methods on pit muds rely on the pure culture bacteria separation technique, which is not conducive to strict nutritional requirements or anaerobic microbial separation and is easy to cause inaccurate results due to factors such as medium selectivity, operation errors or bacterial growth conditions [Citation3,Citation4]. In recent years, molecular biological methods such as molecular cloning technology, 16S rRNA gene clone library, fluorescence quantitative polymerase chain reaction (PCR) technology [Citation5], polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE) technology [Citation6] and high-throughput sequencing technology have been used to study the microorganisms of pit muds [Citation7]. High throughput sequencing is also called ‘next generation’ sequencing, and the Illumina sequencing platform can sequence hundreds of thousands to millions of DNA molecules at one time, with outstanding advantages such as high accuracy, high throughput, high sensitivity and low operation cost [Citation8].
Recent years have witnessed research on the analysis of liquor microorganisms by using high-throughput sequencing technology, and attention has been paid to the microbial diversity of liquor pit muds of mixed flavour, Maotai flavour, Qingke health wine, Sichuan style Luzhou-flavour liquor etc., but to a lesser extent on Henan style Luzhou-flavour liquor. For example, Deng et al. [Citation9] used high-throughput sequencing technology to analyse the bacterial community structure of pit muds of 5 and 30 years, and found that the bacteria were mainly distributed into 8 phyla, 13 families and 16 genera. Huang et al. [Citation10] used high-throughput sequencing analysis to find that the bacteria in Baiyubian pit mud were mainly distributed into 4 phyla and 21 genera. Shen et al. [Citation11] used HiSeq 2500 high-throughput sequencing technology to analyse the microbial community structure of Maotai-flavour liquor pit mud, and found four dominant genera.
Different environmental micro-ecologies play a decisive role in the brewing and flavour shaping of liquor, which is why there are differences in liquors produced in different regions and cellar ages. However, the research on the differences in the microbial composition of pit mud of different pit ages is not deep enough, and a systematic explanation cannot be formed for the above problems. Therefore, in this study, high-throughput sequencing technology based on the Illumina Miseq platform was used to analyse the bacterial diversity of the 16S rDNA V3-V4 variable region of Pit mud, and to compare the differences in bacterial community structure between new and old pit mud from Henan Shedian Liquor. A bacterial microbial information resource database was established to provide a theoretical guidance for the effect of pit mud bacterial diversity on liquor quality.
Materials and methods
Materials
Pit mud 1 (new distillery, 3 years) and pit mud 2 (old distillery, 40 years) of Henan SheDianLaoJiu Co. Ltd. were taken out by a five-point sampling method, mixed and quickly transported back into an ice box and kept in a refrigerator at −20 °C.
Reagents
TruSeqTM DNA Sample Prep Kit, FastDNA SPIN Kit for Soil Kit, American MP Biomedicals Com.;AxyPrepDNA Gel Extraction Kit, American Axygen Biosciences Com.;QuantiFluor™-ST, American Promega Com.;DNA Polymerase AP221-02, Trans DNA 15KMarker, Beijing Quan shijin Biotechnology Co., Ltd.
Instruments
We used a 9700 GeneAmp® PCR, QuantiFluor™-ST Blue fluorescence quantitative system, American Promega Com.;Illumina Miseq PE 300 high-throughput sequencer, American Illumina Com.;Julabo TW12 thermostat water bath, China Yu Laibo Technology Co., Ltd;Nano Drop 2000 UV-vis spectrophotometer, ST16R high speed freezing centrifuge, PICO17 tabletop centrifuge, China Thermo Fisher Technology Co., Ltd.
Total DNA extraction and PCR amplification
The total DNA extraction was carried out according to the instructions of FastDNA SPIN Kit for Soil. DNA concentration and purity were measured via a NanoDrop 2000 detector, and the quality of DNA extract was measured by 1% agarose gel electrophoresis. Universal primers, 338 F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806 R (5′-GGACTACHVGGGTWTCTAAT-3′) were used for PCR amplification in the V3-V4 variable region [Citation12], and the amplification procedure was: pre denaturation at 95 °C for 3 min; denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 45 s, for 30 cycles; extension at 72 °C for 10 min; preserved at 10 °C. The amplification system was 20 μL, 4 μL 5 * FastpPfu buffer, 2 μL 2.5 mmol/L dNTPs, 0.8 μL primer (5 μmol/L), 0.4 μL FastPfu polymerase, and 10 ng DNA template. The amplification results are shown in .
Figure 1. Bacterial PCR electrophoresis map. The bands in lanes 1 and 2 are bright and clear, there is no smear in the lanes, and the band size is in line with the target.
Purification and quantification of PCR products
The PCR products were recovered by 2% agarose gel, purified by AxyPrep DNA gel extraction kit, eluted by Tris-HCl and detected by 2% agarose gel electrophoresis. Quantifluor™-ST was used for detection and quantification. According to the standard operating procedures of the Illumina MiSeq platform, the purified amplification fragment was used to construct a PE 2*300 bp library.
High-throughput sequencing and data processing
The sequencing was carried out by using the Illumina Miseq PE 300 platform of Shanghai Majorbio Bio-Pharm Technology Co., Ltd. and the original data were uploaded to the NCBI database, accession number: PRJNA859259. To ensure the accuracy of the original data, the data were quality controlled and spliced; then 97% of the remaining high-quality sequences in the sample were divided into operational taxonomic unit (OTU) [Citation13]; finally, the species classification annotation was carried out for each sequence. The analysis of alpha diversity index such as Chao1, Shannon and Simpson index was used to reflect the richness and diversity of the community [Citation14,Citation15]; then through species difference analysis, R language tool mapping and phylogenetic tree, the relative abundance of each microorganism in the sample was analysed.
Results
Rationality analysis of sequencing results
Through OTU clustering, 17 phylum, 30 classes, 58 orders, 107 families, 198 genera and 315 OTUs were identified. The Chao1 index, Sobs index and ACE index represent the total number of species in the sample, and the Shannon index can quantitatively describe the microbial diversity. In order to compare the richness and diversity of microorganisms in the two samples, the sequencing results are analysed by comparison (). The dilution curves were constructed by using the microbial diversity index of the effective sequence of each sample at different depths which reflect the rationality of the sample sequencing quantity directly ().
Figure 2. Sample dilution curve. Taking the randomly selected amount of sequencing data as the abscissa, and the Sobs index of the number of observed species as the ordinate, the red curve represents the new pit mud (sample 1), and the blue curve represents the old pit mud (sample 2).
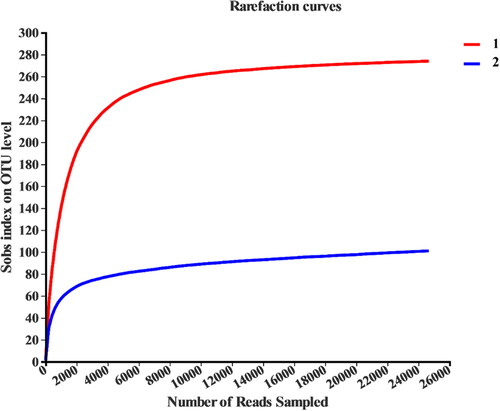
Table 1. High-throughput sequencing diversity index.
As shown in , the dilution curves of the two samples tended to be flatten out to a plateau with the increase of the sampling volume, indicating its rationality. It can be seen from that the Chao1, ACE and Shannon indexes of pit mud in sample 1 are all higher than those in sample 2, indicating that the bacterial microbial richness and diversity in the new pit mud sample are higher than those in the old pit mud. The sequence number obtained in the two samples is more than 30,000, and the coverage index is greater than 99.9%. The results indicated that the sequencing depth is enough to cover all species in the samples, which can reflect the diversity of the bacterial community of the samples.
Analysis of Venn diagram based on OTU level
Venn diagrams can be used to calculate the number of common and unique species in multiple samples (such as OTU), and can be used to show the similarity and overlap of the number of species in different samples. At the 97% similarity level, an OUT-Venn diagram was drawn for the clustering analysis of the two pit mud samples, as shown in .
Figure 3. OTU-Venn diagram of different samples. Sample 1, New pit mud; Sample 2, Old pit mud.
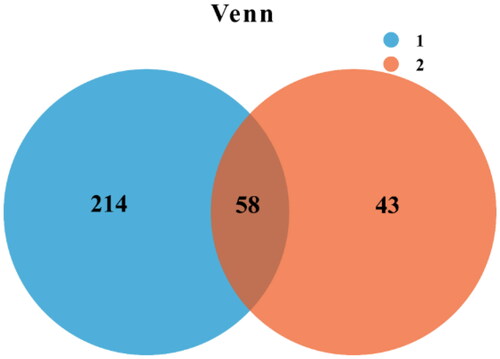
It can be seen from that pit mud samples of 1 and 2 contain 272 and 101 OTUs respectively, of which the number of shared OTUs is 58. The number of unique OTUs to pit mud of samples 1 and 2 is 214 and 43 respectively, indicating that the species number in the pit mud in the new distillery is significantly higher than that in the old one.
Analysis of the community composition of bacterial diversity
We performed taxonomic analysis, to reveal the composition of the community structure of different samples at different taxonomic levels. On the basis of species richness and diversity, this will further describe the microbial community structure of the new and old pit mud at the phylum and genus level.
Analysis of the structure of bacterial community at the phylum level
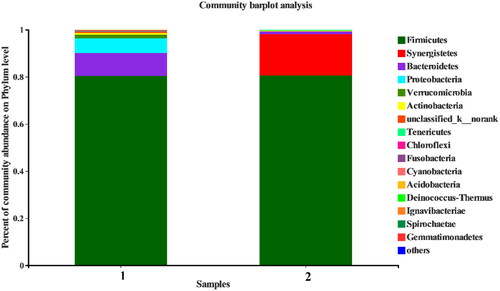
It can be seen from that a total of 16 phyla were obtained from the two samples. With the threshold of abundance >0.5%, the bacteria in the new pit mud were mainly distributed into five phyla: Firmicutes (80.38%), Bacteroidetes (9.70%), Proteobacteria (6.29%), Verrucomicrobia (1.51%) and Actinobacteria (0.75%). The bacteria in the old pit mud were mainly distributed in three phyla: Firmicutes (80.24%), Synergistetes (17.88%) and Bacteroidetes (1.13%).
Figure 4. Phylum level of bacterial community structure. Sample 1, New pit mud; Sample 2, Old pit mud.
Analysis of the structure of bacterial community at the genus level
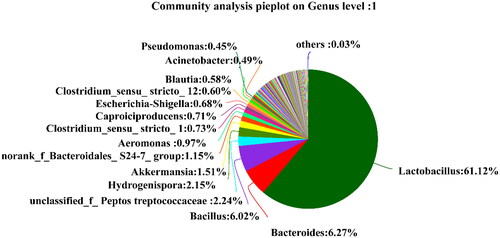
It can be seen from that there are 6 dominant bacterial genera (> 1.0%) in the pit mud of the new distillery: Lactobacillus (61.12%), Bacillus (6.02%), Bacteroides (6.27%), Hydrogenispora (2.15%), norank_f_Bacteroidales_S24-7_group (1.15%) and Akkermansia (1.51%), while other bacterial genera such as Caproiciproproducens, Clostridium_sensu_stricto_12, Clostridium_sensu_stricto_1, Aeromonas, Escherichia-Shigella, Blautia, Acinetobacter and Pseudomonas were less abundant. In addition, unclassified genera accounted for 2.24%, except for the known dominant and non-dominant genera.
Figure 5. Pie chart of genus community distribution of sample 1 (new pit mud).
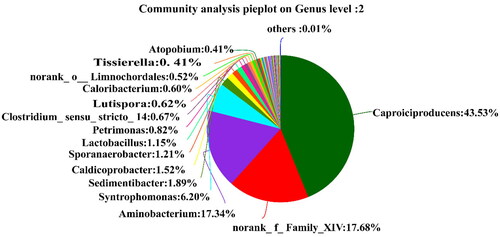
It can be seen from that there are 8 dominant genera (> 1.0%) in the pit mud of the old factory: Caproiciproducens (43.53%), norank_f__Family_XIV (17.68%), Aminobacterium (17.68%), Syntrophomonas (6.20%), Sedimentibacter (1.89%), Caldicoprobacter (1.52%), Sporanaerobacter (1.29%), Lactobacillus (1.29%) and Lactobacillus (1.15%). Other bacterial genera such as Petrimonas, Clostridium_sensu_stricto_14, Lutispora, Caloribacterium, norank_o_Limnochordales, Tissierella and Atopobium were also present but less abundantly.
Figure 6. Pie chart of genus community distribution of sample 2 (old pit mud).
Phylogenetic tree
The species with the top 50 abundance at taxonomic level in the pit mud owned by the new factory and the old factory were selected to construct the phylogenetic tree according to the maximum likelihood method ().
Figure 7. Phylogenetic tree of the top 50 most abundant OTUs in the pit mud of the new factory and the old factory. Each branch in the evolutionary tree represents a class of species, and the branches are coloured according to the advanced taxonomic level to which the species belongs. The length of the branch is the evolutionary distance between two species, that is, the degree of difference between species.
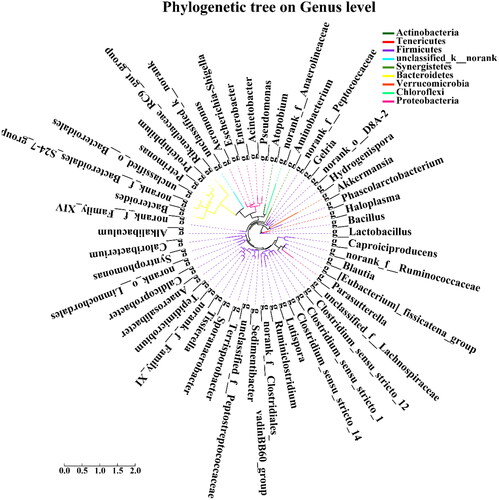
It can be seen from that the phylogenetic relationship between bacterial species in pit mud samples is clearly presented by the evolutionary distance among the species in the ring chart of the top 50 species in the two samples. For example, it can be seen from the figure that Lactobacillus and Bacillus in the pit mud of the new distillery and Caproiciproducens in the pit mud of the old factory both belong to Firmicutes, but the genetic relationship between Lactobacillus and Bacillus bacteria is closer than that between Lactobacillus and Caproiciproducens.
Discussion
In this study, we used high-throughput sequencing to analyse the microbial community structure in pit muds of Shedian Liquor of different pit age. The results showed that the bacterial community in the two pit mud samples was mainly distributed into 6 phyla (abundance > 0.5%) and 14 genera (abundance > 1.0%). The dominant phyla in the pit mud of the new distillery are Firmicutes (80.38%), Bacteroidetes (9.70%), Proteobacteria (6.29%), Verrucomicrobia (1.51%) and Actinobacteria (0.75%); and those in the pit mud of the old distillery, Firmicutes (80.24%), Synergistetes (17.88%) and Bacteroidetes (1.13%). Among the two pit mud samples, Firmicutes are the dominant bacteria, accounting for more than 80% of the total number of bacteria in each sample. With the increase of pit age, the relative abundance of Bacteroidetes decreased in the pit mud of old pits compared with new pits, which was consistent with previous reports [Citation16,Citation17]. Therefore, it can be inferred that the relative abundance of Bacteroidetes can be used as the basis and index to identify the quality and use time of pit mud. Synergistetes only exist in the pit mud of the old factory, which is conducive to improving the quality of pit mud and liquor [Citation18]. Bacteroidetes, Proteobacteria and Actinobacteria are commonly seen in the new pit mud with a large proportion in the old distillery, which is consistent with the research of Zhang et al. [Citation19] that the ripening of pit mud is a long journey. Mud cellar solid-state fermentation is a unique process for Luzhou-flavour liquor, and it is an important factor that distinguishes it from other flavour liquors. Different geographical locations are also divided into production areas of different styles of Luzhou-flavour liquor such as Sichuan-style and Jianghuai-style (Henan, Anhui, Shandong) [Citation20]. Different environmental micro-ecosystems have a great influence on the quality and flavour of Luzhou-flavour liquor [Citation21]. In particular, the microorganisms in the pit mud tend to be stable during the long-term fermentation and domestication process, and there is a saying that ‘the old cellar produces good wine’ [Citation22]. The scientific connotation of this statement is that the bacterial community composition and species abundance of Laojiao pond mud are fixed, which is significantly correlated with the stability of liquor fermentation quality and style [Citation23]. However, there are certain differences in pit mud microorganisms in different geographical locations. In this study, pit mud samples from Henan Province were sequenced. Compared with Sichuan, Shandong, Anhui and other provinces [Citation24], the phylum level is more consistent, with Firmicutes and Bacteroidetes as the main phyla. However, this study found that the relative abundance of Firmicutes in the pit mud of Henan was 80%, while that of Sichuan was around 60%; the relative abundance of Proteobacteria and Bacteroidetes was not significantly different. However, in Henan, the genus Synergistetes is the dominant phylum, while its relative abundance is low in Sichuan [Citation18, Citation25].
Thirteen major genera (abundance >1.0%) were identified in the two pit mud samples. Caproiciproducens (0.71%, 43.53%), Lactobacillus (61.12%, 1.15%), Clostridium_sensu_stricto (1.33%, 1.06%), Syntrophomonas (0.17%, 6.20%), norank_f__Family_XIV (0.14%, 17.68%), Sedimentibacter (0.44%, 1.89%), Caldicoprobacter (0.12%, 1.52%), Hydrogenispora (2.15%, 0.30%) were detected in the pit muds of the new and old distilleries, respectively. The specific bacteria of the pit mud in the new distillery, Bacillus, Bacteroides, norank_f__Bacteroidales_S24-7_group and Akkermansia account for 6.02%, 6.27%, 1.15% and 1.51%, respectively. The special bacteria of pit mud in the old distillery, Aminobacterium and Sporanaerobacter account for 17.68% and 1.29%, respectively. As the main functional bacteria in liquor, Caproiciproducens has the highest abundance of 43.53% in the pit mud of old distillery, the as-produced caproic acid presents fragrance, helps fragrance and balancs liquor, but also can improve Luzhou-flavour liquor by esterification reaction [Citation21, Citation26]. Clostridium_sensu_stricto is a kind of caproic acid producing bacteria in the pit mud that can also produce small organic acids such as butyric acid and acetic acid [Citation27]. Lactobacillus has a high content in the new pit mud, which is conducive to the inhibition of miscellaneous bacteria and the domestication of beneficial bacteria in the early stage of pit mud [Citation28], also can promote Maillard reaction, promote grains sauce fermentation and maintain the microecology of the pit; however, excess Lactobacillus fermentation will lead to an increase in lactic acid and ethyl lactate content in pit mud and a decrease in liquor quality. At the same time, the large-scale reproduction of Lactobacillus and Pediococcus is not conducive to the ecological equilibrium of pit mud [Citation28,Citation29]. Therefore, the representatives of Bacillus, Clostridium_sensu_stricto, Caldicoprobacter and Sporanaerobacter in old pit mud can adjust the pH value of pit by degrading the lactic acid. The result is consistent with the research of Chen et al. [Citation7] that 12 genera of bacteria including Bacillius, Clostridium, Sporanaerobacter and Soehngenia in the pit mud of Luzhou-flavour liquor help degrade lactic acid. Hydrogenispora is generally found in the anaerobic fermentation system, which can improve the volumetric methane production rate [Citation30], and it is also the dominant bacterial genus in the new pit mud, which can promote the synthesis of butyrate [Citation31,Citation32]. Clostridium_sensu_ stricto, Syntrophomonas, Sedimentibacter, Aminobacterium, Caldicoprobacter, etc. are the dominant bacteria in old pit mud, which also have a great effect on the ripening of pit mud and the quality of liquor.
In the study of bacterial diversity in the pit mud, Lu et al. [Citation33] used PCR-DGGE technologies and found that the dominant microorganisms in pit mud include 4 species: Bacteroides, Actinomyces, Proteobacteria and Firmicutes. Yang et al. [Citation34] used Miseq technology and reported that the bacteria in pit mud of Gu Xiangyang Wine were mainly distributed into 4 phyla, namely Firmicutes, Proteobacteria, Actinobacteria and Bacteroides, and 4 genera, namely Lactobacillus, Clostridium, Bacillus and Thermoactinomyces. Using 16S rRNA gene sequencing analysis, Chen et al. [Citation7] showed that the pit mud bacteria in Yibin (Sichuan) area belonged to Firmicutes, Bacteroidetes, Synergistetes, Actinobacteria and Proteobacteria, and 10 different genera: Ruminiclostridium (17.9%), Gelria (8.4%), Syntrophomonas (3.8%), Lactobacillus (3.6%), Syntrophaceticus (2.8%), Proteiniphilum (2.7%), Aminobacterium (2.7%), Petrimonas (2.4%), Sedimentibacter (2.1%) and Caldicoprobacter (1.7%). Fu et al. [Citation25] found that the main bacterial genus was Syntrophomonas (7.64%), followed by Petrimonas (5.99%), Clostridium (3.47%), Bacillus (2.92%) and Proteiniphilum (2.80%) in a study of the brewery mud from Yibin, Sichuan Province, China. The above results are consistent with the results of this study, but the diversity of the bacterial community structure of this study is relatively abundant.
In addition, it is interesting that the bacteria related to the synthesis of caproic acid and butyric acid in Luzhou-flavour liquors in Sichuan are Ruminiclostridium and Syntrophomonas [Citation35], while those in Henan are Caproiciproducens and Clostridium. These differences may be one of the factors that lead to the different styles, Sichuan-style and Jianghuai-style. This preliminarily pointed out the direction for further research and verification.
By using high-throughput sequencing technology, 1.51% of Akkermansia, 1.15% of norank_f__Bacteroidales_S24-7_group, 0.58% of Blautia were first detected in the pit mud at the genus level. Akkermansia has been detected in soil [Citation36], animal intestines [Citation37] and human intestines [Citation38], and is the only culturable intestinal microorganism in Verrucomicrobia [Citation39], which is also one of the few genera in human intestines that can be easily detected in Metagenomic analysis and phylogeny [Citation40]. Akkermansia is a genus of beneficial bacteria that is considered a health marker in humans [Citation41]. Blautia, a common intestinal microorganism, exists in both the intestine and the soil [Citation42]. Blautia can synthesize acetate and short-chain fatty acids from galactose and amino acids [Citation43].
Conclusions
During the practical production of two distilleries, the quality of wine in the old distillery is better than that in the new one with characteristics such as rich aroma, taste mellow and lingering aftertaste. Therefore, this study used high-throughput sequencing technology to analyse the bacterial community structure of cellar mud of different pit age in Shedian Liquor. of the analysis identified many beneficial microbial functional bacteria in the old distillery, which is conducive to improving the ecological environment of the pit, while the excessive content of lactic acid bacteria in the pit mud of the new distillery affects the quality of liquor. The bacterial richness and diversity of the pit mud in new distillery are higher than those of the old distillery, with a significant difference in dominant bacteria. By analysing the role of important functional bacteria in old and new distilleries, the relationship between the composition of the bacterial community and the pit age of the mud is revealed, which provides a theoretical basis for the study of the effect of the bacterial community on the pit mud quality.
Data availability
All data that support the findings from this study are available from the corresponding author upon reasonable request.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Lu LF, Yang Y, Zheng L, et al. Reclassification of Olsenella gallinarum as Thermophilibacter gallinarum comb. nov. and description of Thermophilibacter immobilis sp. nov., isolated from the mud in a fermentation cellar used for the production of Chinese Luzhou-flavour Baijiu. Int J Syst Evol Microbiol. 2021;71(12):005192.
- Gao J, Liu G, Li A, et al. Domination of pit mud microbes in the formation of diverse flavour compounds during Chinese strong aroma-type baijiu fermentation. LWT-Food Sci Technol. 2021;137:110442.
- Overmann J, Huang S, Nübel U, et al. Relevance of phenotypic information for the taxonomy of not-yet-cultured microorganisms. Syst Appl Microbiol. 2019;42(1):22–29.
- Mao P, Hu Y, Liao T, et al. Microbial diversity during fermentation of sweet paste, a Chinese traditional seasoning, using PCR-denaturing gradient gel electrophoresis. J Microbiol Biotechnol. 2017;27(4):678–684.
- Liang H, Li W, Luo Q, et al. Analysis of the bacterial community in aged and aging pit mud of Chinese Luzhou-flavour liquor by combined PCR-DGGE and quantitative PCR assay. J Sci Food Agric. 2015;95(13):2729–2735.
- Xu Y, Zhao J, Liu X, et al. Flavor mystery of Chinese traditional fermented baijiu: the great contribution of ester compounds. Food Chem. 2022;369:130920.
- Chen L, Li Y, Jin L, et al. Analyzing bacterial community in pit mud of Yibin Baijiu in China using high throughput sequencing. PeerJ. 2020;8:e9122.
- Dong W, Yang Q, Liao Y, et al. Characterisation and comparison of the microflora of traditional and pure culture xiaoqu during the baijiu liquor brewing process. J Inst Brew. 2020;126(2):213–220.
- Deng J, Huang ZG, Wei CH, et al. High-throughput sequencing reveals bacterial structure in the mud pits of heavy-fragrance baijiu. Mod Food Sci Technol. 2015;31(7):50–55.
- Huang YN, Xiong XM, Hu YL, et al. Bacterial community and diversity in pit mud of Baiyunbian liquor analyzed by PCR-DGGE and high-throughput sequencing. Microbiol China. 2017;44(2):375–383.
- Shen Y, Cheng W, Deng XB, et al. Fungal diversity in high-temperature Jiangxiang Daqu, fermented grains and pit mud. Liq-Mak Sci Tech. 2019;3:17.
- He G, Huang J, Zhou R, et al. Effect of fortified daqu on the microbial community and flavor in Chinese strong-flavor liquor brewing process. Front Microbiol. 2019;10(56):56.
- Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–998.
- Cao Q, Wang H, Chen X, et al. Composition and distribution of microbial communities in natural river wetlands and corresponding constructed wetlands. Ecol Eng. 2017;98:40–48.
- Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638.
- Hu X, Du H, Ren C, et al. Illuminating anaerobic microbial community and co-occurrence patterns across a quality gradient in Chinese liquor fermentation pit muds. Appl Environ Microbiol. 2016;82(8):2506–2515.
- Liu MK, Tang YM, Guo XJ, et al. Deep sequencing reveals high bacterial diversity and phylogenetic novelty in pit mud from Luzhou Laojiao cellars for Chinese strong-flavor Baijiu. Food Res Int. 2017;102:68–76.
- Li J, Sun H, Wang Q, et al. Microbial community spatial structures in Luzhou-flavored liquor pit muds with different brewing materials. PeerJ. 2022;10:e12987.
- Zhang H, Meng Y, Wang Y, et al. Prokaryotic communities in multidimensional bottom-pit-mud from old and young pits used for the production of Chinese Strong-Flavor Baijiu. Food Chem. 2020;312:126084.
- Hong J, Wang J, Zhang C, et al. Unraveling variation on the profile aroma compounds of strong aroma type of Baijiu in different regions by molecular matrix analysis and olfactory analysis. RSC Adv. 2021;11(53):33511–33521.
- Pang XN, Han BZ, Huang XN, et al. Effect of the environment microbiota on the flavour of light-flavour Baijiu during spontaneous fermentation. Sci Rep. 2018;8(1):1–12.
- Cai W, Xue Y, Tang F, et al. The depth-depended fungal diversity and non-depth-depended aroma profiles of pit mud for strong-flavor Baijiu. Front Microbiol. 2021;12:789845.
- Zheng Q, Lin B, Wang Y, et al. Proteomic and high-throughput analysis of protein expression and microbial diversity of microbes from 30-and 300-year pit muds of Chinese Luzhou-flavor liquor. Food Res Int. 2015;75:305–314.
- Xu Y, Sun B, Fan G, et al. The brewing process and microbial diversity of strong flavour Chinese spirits: a review. J Inst Brew. 2017;123(1):5–12.
- Fu J, Chen L, Yang S, et al. Metagenome and analysis of metabolic potential of the microbial community in pit mud used for Chinese strong-flavor liquor production. Food Res Int. 2021;143:110294.
- Chai LJ, Qian W, Zhong XZ, et al. Mining the factors driving the evolution of the pit mud microbiome under the impact of long-term production of strong-flavor baijiu. Appl Environ Microb. 2021;87(17):e00885-21.
- Su C, Zheng P, Lin X, et al. Influence of amoxicillin after pre-treatment on the extracellular polymeric substances and microbial community of anaerobic granular sludge. Bioresour Technol. 2019;276:81–90.
- Zhao QS, Yang JG, Zhang KZ, et al. Lactic acid bacteria in the brewing of traditional daqu liquor. J Inst Brew. 2020;126(1):14–23.
- Ren C, Gu Y, Du H, et al. Predicting dominant caproate-producing microbes by comparing the microbiotas between new-and aged-pit muds. Food Ferment Ind. 2018;44(12):8–14.
- Yu JD, Zhao LX, Feng J, et al. Influence factors of batch dry anaerobic digestion for corn stalks-cow dung mixture. Trans Chin Soc Agric Eng. 2018;34(15):215–221.
- Chai LJ, Xu PX, Qian W, et al. Profiling the clostridia with butyrate-producing potential in the mud of Chinese liquor fermentation cellar. Int J Food Microbiol. 2019;297:41–50.
- Qian W, Lu ZM, Chai LJ, et al. Cooperation within the microbial consortia of fermented grains and pit mud drives organic acid synthesis in strong-flavor baijiu production. Food Res Int. 2021;147:110449.
- Lu Z, Meng Z, Zhong QD, et al. Application of PCR-DGGE technique in the analysis of bacterial community structure in pit mud with different characters. China Brew. 2018;37(3):28–32.
- Yang XL, Shang XJ, Yu HZ, et al. Bacterial diversity in pit mud of Guxiangyang Baijiu analyzed by Miseq high-throughput sequencing. China Brew. 2018; 37(7):11.
- Gou WJ, Tian Y, Kong XY, et al. Bacterial composition in pit mud of yanghe liquor and identification of acid producing bacteria. Microbiol China. 2020;47(6):1651–1661.
- Qiu J, Hou Y, Xu L, et al. High throughput sequencing analysis of rhizosphere soil bacteria diversity in different mulberry varieties. J South Agric. 2019;50(3):585–592.
- Han W, Zhuang X. Research progress on the next-generation probiotic Akkermansia muciniphila in the intestine. Food Front. 2021;2(4):443–448.
- Ghaffari S, Abbasi A, Somi MH, et al. Akkermansia muciniphila: from its critical role in human health to strategies for promoting its abundance in human gut microbiome. Crit Rev Food Sci. 2022;3:1–21.
- Han W, Wang C, Li XM, et al. The factors influencing the quantity of Akkermansia muciniphilain the intestines: research progress. Chin J Microecol. 2019;31(3):356-364.
- Derrien M, Belzer C, de Vos WM. Akkermansia muciniphila and its role in regulating host functions. Microb Pathog. 2017;106:171–181.
- Cheng D, Xie MZ. A review of a potential and promising probiotic candidate-Akkermansia muciniphila. J Appl Microbiol. 2021;130(6):1813–1822.
- Liu X, Mao B, Gu J, et al. Blautia – a new functional genus with potential probiotic properties? Gut Microbes. 2021;13(1):1–21.
- Park JE, Kim JS, Choi SH, et al. Draft genome sequence of Blautia sp. KGMB01111 isolated from a healthy Korean human faeces. MSK. 2020;56(1):94–97.