
Abstract
Choline is an essential nutrient involved in the synthesis of acetylcholine in the cholinergic neurons. The pharmacokinetic properties of choline are well described; however, there is a lack of data about its activity toward the CYP450 superfamily of enzymes. Therefore, the aim of this study was to conduct in silico and in vitro activity assessments of choline against three major CYP450 isoforms—CYP1A2, CYP2D6, and CYP3A4. Preliminary in silico modeling was performed on a specialized DL-CYP Prediction Server to evaluate the affinity of choline toward the enzymes. The in vitro study contained specific cytochrome P450 isoform inhibitors and substrates (for CYP1A2, CYP2D6, and CYP3A4) to determine the inhibition performance of choline at five different concentrations (0.150 − 1 µmol/L). The potential interactions of choline and CYPs were displayed after molecular docking with Glide (Schrödinger). In addition, induced-fit simulations and binding free energy calculations MM/GBSA (Molecular Mechanics-Generalized Born Surface Area) were applied to predict the accessibility in each CYP isoform. The initial in silico simulations revealed that choline lacks inhibition potency against the aforementioned enzymes. The in vitro evaluations confirmed that choline possessed no effect against CYP1A2; however, at 1 µmol/L choline exerted 22% and 27% blocking capacity against CYP2D6 and CYP3A4, respectively. Furthermore, there was good correlation between the in vitro results and the complexes’ free binding energy recalculations. Overall, the assessments indicated that choline is a weak CYP2D6 and CYP3A4 inhibitor. The latter results should be considered as a source of future unwanted drug–drug interactions.
Introduction
Cytochrome P450 is a heme-containing superfamily accountable for the oxidation of various pharmacologically active drugs [Citation1]. The cytochrome P450 enzyme system comprises 57 isoforms of which five (CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) are accountable for approximately 90% of the metabolic reactions. The most abundant isoform, CYP3A4, is located in the liver bile duct and jejunum columnar epithelial cells, where it catalyzes the metabolism of approximately 50% of the registered therapeutic agents [Citation2]. The problems associated with the use of CYP450 inhibitors are of great significance. The inhibition of CYP isoforms could increase the plasma levels of pharmacologically active compounds [Citation3]. An example is the influence of CYP450 inhibitors on the response of antiplatelet therapy in some patients [Citation4]. Such unwanted interactions could be genetically determined or could occur during the concurrent use of CYP inhibitors [Citation5]. Therefore, an initial assessment of compounds for CYP inhibition capacity could avoid potential adverse reactions [Citation6].
Choline is an essential nutrient involved in the synthesis of acetylcholine through acyltransferase in the cholinergic neurons. Subsequently, the formed acetylcholine is released in the synaptic cleft, where it binds to specific receptors [Citation7]. The endogenous synthesis of choline cannot fully meet the physiological requirements [Citation8], which illustrates that it is essential in the diet. Furthermore, dietary reference values for choline were introduced by the European Food Safety Authority [Citation9]. The pharmacokinetic properties of choline are well described [Citation10]; however, there is an insufficient amount of data about its activity toward enzymes from the CYP450 superfamily.
Consequently, the aim of this work was to investigate the inhibition potency of choline toward human CYP1A2, CYP2D6, and CYP3A4 enzymes through in silico and in vitro studies. Initially, the inhibition performance of choline was predicted with the DL-CYP Prediction Server, followed by in vitro studies. Molecular docking simulations demonstrated the possible conformations of choline in the active sites of CYP450 enzymes. Furthermore, Induced-fit docking (IFD) and MM/GBSA free energy recalculations were introduced to acquire reliable in silico results.
Materials and methods
Preliminary in silico simulations
As a preliminary step, the capacity of choline to inhibit human CYP1A2, CYP2D6, and CYP3A4 was predicted with the DL-CYP Prediction Server [Citation11]. It employs a deep autoencoder multi-task neural network and could predict the inhibitory effects toward five major CYP isoforms. Choline, ketoconazole, alpha-naphthoflavone and quinidine were downloaded in .sdf format from PubChem (National Center for Biotechnology Information), and their inhibition capacities were examined against the aforementioned CYP isoforms. The values range from 0 to 1.
In vitro determination of CYPs activity
The inhibitory effects of choline on the catalytic activities of human recombinant CYP1A2, CYP2D6, and CYP3A4 were assayed using fluorometric CYP450 Screening Kits purchased from Abcam. The standard inhibitors for CYP1A2, CYP2D6, and CYP3A4 were α-naphthoflavone, quinidine, and ketoconazole, respectively. Five concentrations were applied: 0.150, 0.250, 0.500, 0.750, and 1 µmol/L.
Molecular docking
Selection and preparation of proteins
The crystallographic structures of CYP1A2 (PDB ID: 2HI4) [Citation12], CYP2D6 (PDB ID: 4WNU) [Citation13], and CYP3A4 (PDB ID: 2V0M) [Citation14] resolved with the co-crystallized ligands alpha-naphthoflavone, quinidine, and ketoconazole, respectively, were retrieved from the Protein Data Bank (PDB). The Protein Preparation Wizard in Maestro (Schrödinger Release 2021-3: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2021) was employed for the protein refinements. Hydrogen bonds and het states at pH 7.0 ± 2.0 were generated followed by the removal of water molecules situated in the active site. Subsequently, the energy of the crystallographic structures was minimized by applying the OPLS2005 force field.
Preparation of ligands
The chemical structure of choline was drawn with the 2D sketcher module in Maestro and converted to its three-dimensional (3D) structure with the Ligprep module (Schrödinger Release 2021-3: LigPrep, Schrödinger, LLC, New York, NY, 2021). Utilizing the latter module, hydrogen bonds, tautomers, and ionization states at pH 7.0 ± 2.0 were generated, and charged groups were neutralized. Furthermore, the energy of the ligand was minimized by applying the OPLS2005 force field.
Docking protocol
The docking module in Maestro–Glide was implemented considering the reported reliable results when CYP enzymes were simulated with this software [Citation15]. Glide utilizes the empirically based GlideScore scoring algorithm, which is presented in three forms: High-throughput screening (HTS), Standard-Precision (SP), and Extra-Precision (XP) modes. The most precise XP docking mode was used. In addition, the induced-fit docking (IFD) in Schrödinger was employed to further validate the examined conformations. The IFD examines the side chains of the protein as fully flexible, which leads to optimization of the active site conformation. MM/GBSA (Molecular Mechanics-Generalized Born Surface Area) recalculations were also introduced to determine the binding free energies of the obtained complexes. The grid box was generated around a co-crystallized ligand with the help of the Receptor Grid Generation module in Maestro.
Statistical analysis
The different CYP450 isoform activities were normalized as percentage of the untreated control set as 100% and the results were expressed as mean values and standard deviation (±SD) (Graph Pad Prizm). Statistical analysis was performed by one-way analysis of variance (ANOVA) with post hoc multiple comparisons procedure (Dunnet’s test) to assess the statistical differences in case of normal distribution. Differences were considered statistically significant at the p < 0.05 and p < 0.001 level.
Results
Preliminary in silico simulations
The potential of choline to inhibit CYP1A2, CYP2D6, and CYP3A4 was initially predicted with the DL-CYP Prediction Server. The positive controls—α-naphthoflavone, ketoconazole, and quinidine, were correctly predicted as inhibitors of CYP1A2, CYP2D6, and CYP3A4. Importantly, the inhibition values of ketoconazole, alpha-naphthoflavone, and quinidine validated the protocol; however, the prediction software showed that choline possesses activity against neither of the isoforms ().
Table 1. Predicted CYP1A2, CYP2D6, and CYP3A4 inhibition potency of choline and the positive controls with the DL-CYP Prediction Server.
In vitro evaluations
CYP1A2
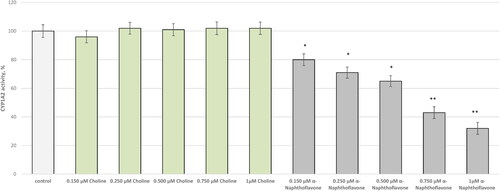
In each CYP isoform choline and a standard inhibitor were applied at five different concentrations: 0.150, 0.250, 0.500, 0.750, and 1 µmol/L. Choline did not demonstrate any inhibition potential against CYP1A2 (). α-Naphthoflavone, which was applied as a selective inhibitor, blocked the isoform as follows: 0.150 µmol/L − 20%; 0.250 µmol/L − 29%; 0.500 µmol/L − 35%; 0.750 µmol/L − 57%, and 1 µmol/L – с 68%.
Figure 1. Effect of choline on CYP1A2 activity in vitro. Five concentrations of choline and α-naphthoflavone (as a selective inhibitor) were assayed: 0.150, 0.250, 0.500, 0.750, and 1 µmol/L. *p < 0.05; **p < 0.01 vs. control (pure CYP1A2).
CYP2D6
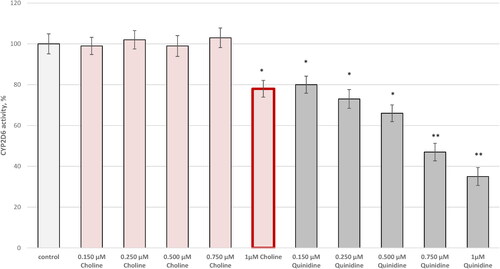
Interestingly, CYP2D6 showed moderate activity when choline was applied at 1 µmol/L concentration. The inhibitory effect was 22% compared to the control (pure CYP2D6). The standard control (quinidine) inhibited the enzyme in a dose-dependent manner ().
Figure 2. Effect of choline on CYP2D6 in vitro. Five concentrations of choline and quinidine (as a selective inhibitor) were assayed: 0.150, 0.250, 0.500, 0.750, and 1 µmol/L. *p < 0.05; **p < 0.01 vs. control (pure CYP2D6).
CYP3A4
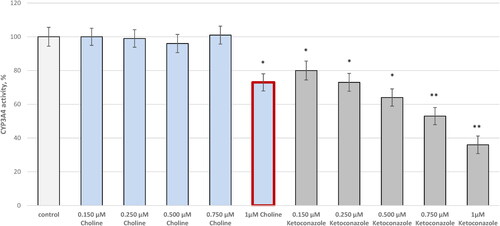
Choline also reduced the CYP3A4 enzyme activity by 27% when applied in 1 µmol/L concentration (). The standard CYP3A4 inhibitor, ketoconazole, exhibited 64% activity at the same concentration.
Figure 3. Effect of choline on CYP3A4 activity in vitro. Five concentrations of choline and ketoconazole (as a selective inhibitor) were assayed: 0.150, 0.250, 0.500, 0.750, and 1 µmol/L. *p < 0.05; **p < 0.01 vs. control (pure CYP3A4).
Re-docking simulations
Initially, we carried out self-docking simulations to assess the reliability of the docking software to correctly place the co-crystallized ligand back into the active sites of CYP1A2 (PDB:2HI4), CYP2D6 (PDB:4WNU), and CYP3A4 (PDB:2V0M). In the most precise Glide docking mode, XP, the RMSD values between the co-crystallized ligands poses and the conformations obtained with Glide, were 0.255, 0.893, and 0.999 Å in 2HI4, 4WNU, and 2V0M, respectively. This indicated that the protocol in Glide is reliable when performing docking simulations in these crystallographic enzymes ().
Molecular docking simulations
After the validation of the crystallographic structures, choline was docked in the active site of CYP1A2 (PDB:2HI4). The co-crystallized ligand alpha-naphthoflavone was utilized as a reference ligand. The docking scores of choline and alpha-naphthoflavone were compared after employing XP, IFD, and MM/GBSA free energy recalculations (). Choline could not obtain a stable complex with CYP1A2: the binding energies were −3.69 and −4.60 in the XP mode and the IFD mode, respectively. The calculated MM/GBSA free binding energy was −38.71, whereas the validated CYP1A2 inhibitor was far more stable (MM/GBSA score of −89.43). In the MM/GBSA free binding energy calculations, an additional amino acid (Asp320) participated in the stabilization of the obtained choline–CYP1A2 complex. Moreover, Asp313 was present in the complex generated with XP, while the active residue Thr385 was involved when IFD was used.
Table 2. Scoring method, docking scores, participating active residues and distance from the heme structure in docking simulations of choline in the active site of CYP1A2, CYP2D6, and CYP3A4 employing XP, IFD, and MM/GBSA recalculations (Schrödinger Inc.).
The major interactions between choline and CYP1A2 are given in . Several amino residues participate in stabilizing polar interactions: Thr124, Thr321, Thr385, and Thr498. The established hydrophobic forces were generated between choline and Phe125, Ala317, Leu382, Ile386, and Leu497. Importantly, most of the major stabilizing interactions were provided by the interaction with the heme group.
Figure 4. Visualized major intermolecular interactions of choline in the active sites of CYP1A2 (A, B), CYP2D6 (C, D), and CYP3A4 (E, F) after employing IFD and MM/GBSA recalculations. The interactions are provided in both 2D and 3 D forms. The enzyme structures are depicted in green, and the heme group is given in red colored sticks.
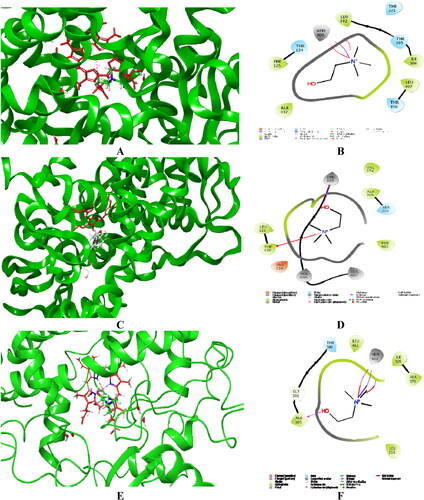
Next, we evaluated the accessibility of choline in CYP2D6 (PDB: 4WNU). The results demonstrated that the induced-fit simulations placed choline closer to the heme group compared to the XP docking algorithm (). In the MM/GBSA free binding energy calculations, an additional amino acid (Asp301) participated in the stabilization of the obtained complex. The acquired score with the molecular mechanics was −58.10, which was lower compared to the MM/GBSA value for quinidine (−101.94). The interactions with the active site are represented in . Various hydrophobic interactions with the active residues Phe120, Leu121, Ala305, Val374, and Phe483 were detected. A pi-cation interaction between choline and Phe120 was also noted.
The next step was docking simulations of choline in the active site of CYP3A4. The XP docking demonstrated that ketoconazole forms a stable complex with a score of −9.11, while the stabilization forces of choline–CYP3A4 were weaker (−4.38) (). However, the distance of the docked choline from the heme group was 3.15 Å. Using IFD, we noticed alterations in the free energy of the complex, the number of participating active residues and the distance from the heme group. After the latter calculations, the binding energy was −5.75, while ketoconazole showed a value of −10.12. Subsequently, the recalculated MM/GBSA value of choline was −63.82, while the complex of CYP3A4–ketoconazole displayed significantly better binding capacity of −115.79.
The interactions between the active site of CYP3A4 and choline are illustrated in . Based on the visualized stabilization forces, we observed that Ala305 and Hem601 were the major drug-binding residues in CYP3A4. The strongest hydrogen bond was formed between the hydroxyl moiety of choline and the active amino residue Ala305. Several weak hydrophobic interactions were formed by Leu216, Ile369, Ala370, and Leu482. Using XP docking, the active pose of choline demonstrated that the N-methyl group is in close proximity to the heme structure. In contrast, IFD, which has greater hardware demands and is more precise, showed different choline orientation in the active site of CYP3A4. In these simulations, the hydroxyl group in the choline molecule was facing the heme moiety.
Discussion
The CYP450 superfamily of enzymes consists of 57 isoforms involved in the metabolic pathways of various pharmacologically active compounds. The assessment of CYP450-mediated interactions is important as this superfamily is involved in undesirable drug-drug interactions. Therefore, an initial evaluation toward the CYP450 activity for each active molecule is required in the process of drug development [Citation16]. In recent years, the reliability and robustness of in silico methods for the assessment of CYP450 activities has increased substantially. Studies based on the implementation of free web servers [Citation1,Citation17] and molecular docking simulations [Citation2] have displayed good correlation with the in vitro data. Numerous CYP1A2, CYP2D6, and CYP3A4 crystallographic structures have been deposited in the PDB, and are primarily used in the molecular docking analysis. Therefore, the implementation of in silico methods could provide rapid preliminary evaluation of the inhibition or activation of CYP450.
Choline is an essential nutrient involved in formation of acetylcholine through acyltransferase in the cholinergic neurons. The endogenous synthesis of choline is not adequate to support the bodily requirements, which illustrate the essential character of the compound as a diet supplement [Citation8]. The metabolic pathway and the biological functions of choline were reported by Wiedeman et al. [Citation15]. In this work, the inhibition potency of choline toward CYP1A2, CYP2D6, and CYP3A4 was examined in silico and in vitro.
Initially, the DL-CYP Prediction Server was employed for preliminary overview of the inhibitory potential of choline against three CYP isoforms. This software was utilized in several recent works with good reliability [Citation1,Citation17]. However, we noted that the server was unable to correctly score choline as a weak inhibitor of CYP2D6 and CYP3A4. Similarly, inaccurate results were displayed when fucoxanthin and siphonaxanthin were applied for a preliminary evaluation applying the abovementioned software [Citation1].
The in vitro evaluation showed that choline is a weak inhibitor of CYP2D6 and CYP3A4 when compared to the control standards. Therefore, the next stage of the study was to address whether the atoms are accessible to the oxo-ferryl group of the heme group and to determine the stability of the formed complexes through molecular docking. The docking simulations could be employed as a rapid and inexpensive method for the prediction of sites of metabolism of drug molecules [Citation18]. The obtained conformations were considered to denote a possible catalytically active conformation, if the ligand’s pose was within 6 Å from the heme iron, and the binding forces were robust enough to form a stable complex. The molecular docking simulations were carried out with the docking software Glide (Schrodinger), considering the prominent results of this program in similar studies [Citation15]. However, diverse conformations of the active sites of the aforementioned CYP isoforms have been previously described [Citation19]. Thus, it is required to apply more precise and hardware demanding simulations to obtain reliable virtual results. A recent study has shown that the IFD acquired an excellent success rate of 89% [Citation15]; therefore we applied it in the present work. The initial re-docking protocols validated the crystallographic structures of the isoforms. Thereafter, the docking results displayed the robustness of the IFD when employed in the simulations against CYP450 enzymes. The IFD module of Maestro demonstrated that choline is in close proximity to the heme groups of CYP1A2, CYP2D6, and CYP3A4. However, the docking scores in the active site of CYP1A2 were the lowest, which correlates with the in vitro results. The MM/GBSA recalculations demonstrated that choline forms moderately stable complexes in CYP2D6 and CYP3A4 with scores of −58.10 and −63.82, respectively. Based on the visualized stabilization forces, we observed that the heme group is involved in the stabilizations with CYP1A2 and CYP3A4. Moreover, the majority of the amino residues that formed stable bonds with choline are previously reported to interact with other active CYP450 inhibitors [Citation20].
Conclusions
The in silico and in vitro data indicated that choline is a weak CYP1A2 and CYP3A4 inhibitor. These results should be considered in view of future unwanted drug–drug interactions. The virtual simulations confirmed that the IFD followed by free binding energy recalculation with MM/GBSA provided more reliable results compared to other searching and scoring algorithms.
Supplemental Material
Download PDF (830.5 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
The author(s) reported there is no funding associated with the work featured in this article.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies). 2016. Scientific opinion on dietary reference values for choline. EFSA J. 2016;14(8):4484.
- Chand RR, Nimick M, Cridge B, et al. In vitro hepatic assessment of cineole and its derivatives in common brushtail possums (Trichosurus vulpecula) and rodents. Biology. 2021;10(12):1326.
- Sevrioukova IF, Poulos TL. Understanding the mechanism of cytochrome P450 3A4: recent advances and remaining problems. Dalton Trans. 2013;42(9):3116–3126.
- Linn CE, Shah NK, Epstein BJ. Role of CYP450 in antiplatelet therapy: considerations for patients at risk for further cardiovascular or cerebrovascular problems. J PharmacyTechnol. 2013;29(2):72–87.
- Molden E. Variability in cytochrome P450-mediated metabolism of cardiovascular drugs: clinical implications and practical attempts to avoid potential problems. Heart Drug. 2004;4(2):55–79.
- Colburn DE, Giles FJ, Oladovich D, et al. In vitro evaluation of cytochrome P450-mediated drug interactions between cytarabine, idarubicin, itraconazole and caspofungin. Hematology. 2004;9(3):217–221.
- Ekroos M, Sjögren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci U S A. 2006;103(37):13682–13687.
- Gao X, Jiang C, Xu J, et al. Serum pharmacokinetics of choline, trimethylamine, and trimethylamine-N-oxide after oral gavage of phosphatidylcholines with different fatty acid compositions in mice. Biosci Biotechnol Biochem. 2016;80(11):2217–2223.
- Hayes C, Ansbro D, Kontoyianni M. Elucidating substrate promiscuity in the human cytochrome 3A4. J Chem Inf Model. 2014;54(3):857–869.
- Li X, Xu Y, Lai L, et al. Prediction of human cytochrome P450 inhibition using a multitask deep autoencoder neural network. Mol Pharm. 2018;15(10):4336–4345.
- Wang A, Stout CD, Zhang Q, et al. Contributions of ionic interactions and protein dynamics to cytochrome P450 2D6 (CYP2D6) substrate and inhibitor binding. J Biol Chem. 2015;290(8):5092–5104.
- Marechal J-D, Yu J, Brown S, et al. In silico and in vitro screening for inhibition of cytochrome p450 CYP3A4 by comedications commonly used by patients with cancer. Drug Metab Dispos. 2006;34(4):534–538.
- Olsen L, Oostenbrink C, Jorgensen F. Prediction of cytochrome P450 mediated metabolism. Adv Drug Deliv Rev. 2015;86:61–71.
- Pang X, Zhang B, Mu G, et al. Screening of cytochrome P450 3A4 inhibitors via in silico and in vitro approaches. RSC Adv. 2018;8(61):34783–34792.
- Ridhwan MJM, Bakar SIA, Latip NA, et al. A comprehensive analysis of human CYP3A4 crystal structures as a potential tool for molecular docking-based site of metabolism and enzyme inhibition studies. J Comput Biophys Chem. 2022;21(03):259–285.
- Sansen S, Yano JK, Reynald RL, et al. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J Biol Chem. 2007;282(19):14348–14355.
- Sevrioukova IF, Poulos TL. Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4. Proc Natl Acad Sci U S A. 2017;114(3):486–491.
- Wiedeman AM, Barr SI, Green TJ, et al. Dietary choline intake: current state of knowledge across the life cycle. Nutrients. 2018;10(10):1513.
- Yim S-K, Kim K, Chun S, et al. Screening of human CYP1A2 and CYP3A4 inhibitors from seaweed in silico and in vitro. Mar Drugs. 2020;18(12):603.
- Zeisel SH. Choline: critical role during fetal development and dietary requirements in adults. Annu Rev Nutr. 2006;26:229–250.