
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The aim of this study was to evaluate the impact of the most frequent Asn501 polar uncharged amino acid mutations upon important structural properties of SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) Surface Glycoprotein RBD – hACE2 (human angiotensin-converting enzyme 2) heterodimer. Mutations N501Y, N501T and N501S were considered and their impact upon complex solubility, secondary motifs formation and intermolecular hydrogen bonding interface was analyzed. Results and findings are reported based on 50 ns run in Gromacs molecular dynamics simulation software. Special attention is paid on the biomechanical shifts in the receptor-binding domain (RBD) [499-505]: ProThrAsn(Tyr)GlyValGlyTyr, having substituted Asparagine to Tyrosine at position 501. The main findings indicate that the N501S mutation increases SARS-CoV-2 S-protein RBD – hACE2 solubility over N501T, N501 (wild type): ,
. The N501Y mutation shifts
-helix S-protein RBD [366-370]: SerValLeuTyrAsn into π-helix for t > 38.5 ns. An S-protein RBD [503-505]: ValGlyTyr shift from
-helix into a turn is observed due to the N501Y mutation in t > 33 ns. An empirical proof for the presence of a Y501-binding pocket, based on RBD [499-505]: PTYGVGY
’s RMSF peak formation is presented. There is enhanced electrostatic interaction between Tyr505 (RBD) phenolic -OH group and Glu37 (hACE2) side chain oxygen atoms due to the N501Y mutation. The N501Y mutation shifts the
hydrogen bond into permanent polar contact;
;
.
Introduction
Since the coronavirus disease (COVID-19) outbreak in 2019, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome has undergone hundreds of mutations. Virus mutations arise causally during replication, due to the lack of RNA polymerase proofreading activity (Citation1). Although most SARS-CoV-2 mutations have no significant impact upon the general course of COVID-19 (Citation2), specific mutations may increase the infection severity and undermine current vaccination programs. The mutations occurring in SARS-CoV-2 S-protein have attracted special attention, as the virus targets host cells by S-protein–to–hACE2 receptor interaction (Citation3). It has been well-studied that the receptor-binding domain (RBD) of the S-glycoprotein serves as a binding interface to the human Angiotensin-converting enzyme 2 (hACE2) receptor, extracting kinetic and thermodynamic properties of the binding pocket (Citation3). So far, SARS-CoV-2 has evolved into serval VOCs (variants of concern) and VOIs (variants of interest) that may be seen as a direct consequence of the occurrence of specific mutations in the S-protein that resulted in increased infection severity and lucid immune escape, undermining current COVID-19 vaccine development efforts.
The alpha lineage, or B.1.1.7 variant, arose in the United Kingdom in September 2020 (Citation4) and included 7 amino acid substitutions: N501Y, A570D, D614G, P681H, T716I, S982A, D1118H, and two deletions: HV69/70del and Y144del, in the S-protein (Citation5). Substitutions D614G and N501Y were also found in the B.1.351 variant (beta lineage), in addition to: L18F, D80A, D215G, LAL242/244del, R246I, K417N, E484K, N501Y, D614G and A701V S protein mutations (Citation5). The Gamma variant (P1) accumulated totally 12 S-protein substitutions: L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I and V1176F (Citation5). The S-protein of the Delta variant (B.1.617.2) was characterized by 8 non-synonymous substitutions and 1 deletion: T19R, G142D, FR156/157del, R158G, L452R, T478K, D614G, P681R, D950N (Citation6). The recent circulating variant of concern – Omicron (B.1.1.529) – has over 30 mutations in the S-protein (Citation7), some of them: K417N, T478K, N501Y found in common to previous variants (Citation8,Citation9).
Several variants, such as: B.1.1.7, B.1.351 and P.1 share a common mutation N501Y located in the RBD of SARS-CoV-2 spike (S) protein, assumed to be of higher infectivity to humans (Citation10,Citation11). Although a study has discovered that the N501Y mutation diminishes the binding affinity to an animal ACE2 receptor (Citation12), N501Y enhances viral binding to the human ACE2 receptor (hACE2) and confers moderate resistance to certain monoclonal antibodies (Citation12). Liu et al. (Citation10) regards the N501Y mutation as a hot spot for enhanced infection of the upper airway and transmission. Many studies have tried to explain the pathogenic relevance of the N501Y mutation on a biomechanical basis, but there is evident disagreement among the reported findings. Having a Y501 aromatic ring surrounded between residues hACE2K353 and hACE2Y41, which leads to a better-formatted biding pocket (Citation13), the interactions of Y501 to hACE2K353 and π-π stacking of the Y501-hACE2Y41 were provided as main arguments explaining the N501Y increased viral fitness for replication (Citation10–12, Citation14). On the other hand, it has been also found that Y501 increases the unbinding force towards the hACE2 receptor compared to N501 wild type (Citation11). A recent study hypothesizes kinases (the epidermal growth factor receptor, EGFR) as possible factors that involve N501Y mutation binding through Y501 phosphorylation (Citation15).
The aim of this study was to analyze the structural changes caused by the most frequent N501 to polar uncharged amino acids substitutions: N501Y, N501T and N501S in terms of hACE2–S-protein complex stability, solubility, secondary structure organization and intermolecular polar interactions. The change of polar contacts’ affinities of 501-surrounding residues: P499, T500, G502, V503, G504, Y505, due to N501Y mutation, was also analyzed.
Materials and methods
Heterodimer – PDB file 6M0J: Crystal structure of SARS-CoV-2 spike receptor-binding domain bound with ACE2 (https://www.rcsb.org/structure/6m0j), (Citation16) was used as the wild type structure. The file was retrieved from Protein Data Bank (https://www.rcsb.org/). Zinc (Zn2+) metal cation bound to hACE2 characterized the receptor environment. Zn2+ ion contacted hACE2 residues His374/His378 and Glu375/Glu402; thus, promoting local stability. A total of five N-acetyl-beta-D-glucosamine (NAG) ligands were attached to the 6M0J hetero dimer. Four NAG glycans were attached to the hACE2 receptor, one NAG glycan bound to S-protein RBD. NAG glycans formed frequent contacts with Asparagine on the opposite side, such as: hACE2 Asn90, hACE2 Asn322 and RBD Asn343. No metal ion, no ligands were attached to the mutagenesis domain of interest in this study, S-protein RBD [499-505]: PTN(Y)GVGY. The wild-type complex with Asparagine at position 501 in RBD was mutated to Tyrosine(Y), Threonine(T) and Serine(S) using PyMol molecular graphics system (https://pymol.org/2/, ver. 2.5.4) mutagenesis object.
All four molecular systems – wild-type Asparagine501(N501) and mutants, Tyrosine501(Y501), Threonine501(T501) and Serine501(S501) – were prepared for molecular dynamics simulation in Gromacs (https://www.gromacs.org/, ver. 2020.1-Ubuntu-2020.1-1), (Citation17). Topology files (.top) were compiled from .pdb files to have each molecule defined as object suitable for MD simulation, including standard properties, such as: atom types, charges, bonds, dihedrals; (command pdb2gmx). Charmm27 (Citation18) all-atom force field was selected for simulation purposes. Long-range energy contributions were calculated by the PME method in Fourier space (particle mesh Ewald), (Citation19,Citation20), with a real-space cutoff for PME calculations of 1 nm. Molecules were placed and centered in a cubic grid box at 1.0 nm minimum distance from the edge of solute-box, providing minimum 2.0 nm between periodic images of the protein.
As SARS-CoV-2 (virus)-to-host cell membrane fusion may occur in blood plasma (Citation21) at physiological pH or near-physiological pH (Citation22), and plasma is predominantly made of water (about 90%), water fits the simulation purpose as a solvent model. An SPC/E (simple point-charge/extended) water model under an SPC216 generic three-point solvent model (Brendsen, 1987) was used as a solvent. In total, 25 SPC/E water molecules were substituted with Na+ ions to neutralize the native total negative charge in the system of −25.000 e. Refined geometry of structure, appropriate water molecules orientation, avoiding steric clashes, was provided through an energy minimization process applying the steepest descent minimization algorithm (Citation23). Tuning maximum force required
energy optimization steps, for potential energy
.
The systems were brought to desired temperature and pressure by NVT, followed by NPT-equilibration, with a coupling time of 2 ps. Then, 150-ps isothermal-isochoric NVT position-restrained equilibration at 310 K controlled by V-rescale thermostat (Citation24) was performed. In this stage, pressure coupling was turned off, while particles initial velocities were assigned from Maxwell distribution at 310 K. To finalize the system equilibration, 150-ps isothermal-isobaric pressure coupling, regulated with Berendsen barostat (Citation25) was performed. Referent coupling pressure of 1 bar and water isothermal compressibility of at NVT equilibrium of 310 K were used. Verlet (Citation26) velocity algorithm was used to integrate Newton’s motion equations. The equilibration process aimed to relax the complex – optimize the water molecules towards the heterodimer. The wild-type system (N501) and mutants (Y501, T501 and S501) were subjected to 50 ns md Gromacs simulation. Output trajectory, index files were used for the analysis.
Backbone dynamics and molecular dynamics convergence analysis
Molecular dynamics equilibrium (convergence) state and systems stability shifts were examined by the means of root mean square deviation (RMSD) analysis upon backbone atoms. By measuring the magnitude of atomic oscillations during the simulation ( relative to the initial conformation at
, one can analyze the impact of induced mutations upon general system dynamics/stability, identify periods of substantial conformational changes and inspect MD simulation convergence. Deviations produced during the course of simulation (Citation27) are taken as stability estimate: flatten RMSD plot for relatively stable dynamics, spike-oscillating RMSD plot for destabilized behavior. The molecular dynamics convergence state was identified based on uninterrupted backbone atoms’ oscillations ranging no more than 0.15 nm (1.5 Å), Appendix RMSD_analysis.xlsx.
Residues fluctuations analysis
Some mutations may disturb the dynamic behavior of a particular protein domains, which is evaluated in terms of the root mean square fluctuations (RMSF). RMSF per residue evaluates the average deviation of each residue relative to the starting, energetically minimized structure (Citation28). Altered RMSFs as a consequence of mutation localized in the spike protein domain found in close proximity to the hACE2 receptor may indicate no-change, lesser or intensified virus-to-receptor interaction. To account for such behavior, molecular RMSF per resides were analyzed. S-protein motifs such that the RMSF per residue equals at least 0.03 nm (0.3 Å) were considered as localized conformational changes in the presence of a particular amino acid substitution. Special attention has focused on the S-protein RBD motif [499-505]: ProThrAsn(Tyr)GlyValGlyTyr, which in the presence of Tyrosine(Y) at position 501 instead of Asparagine(N) and coupled to the hACE2 receptor, exhibits major destabilization relative to the wild type (N501), Appendix RMSF_analysis.xlsx.
Solubility analysis
Amino acid substitutions at same points may significantly alter the contacts with surrounding solvent molecules, which may be seen as a consequence upon protein stability and protein conformational shifts. Molecular conformational changes were examined by means of solvent accessible surface area (SASA). The most significant conformational alterations are expected to happen due to hydrophilic-to-hydrophobic amino acid mutations and vice versa. Substituting wild-type Asparagine (N501) to also hydrophilic (Citation29) Threonine (T501), Serine (S501) and dual-nature Tyrosine (Y501), primarily classified as a hydrophobic residue because of the aromatic ring in the side chain, being also capable of hydrogen bond formation due to hydroxyl group (-OH) in its side chain, may also imply molecular conformational changes.
As per definition, SASA is the total surface area of a molecule that is accessible to the solvent. Amino acids mutations affect the overall SASA score () in one or another way, which plays an important role in understanding the structure-to-function protein properties. The Double Lattice Method-DCLM (Citation30) is used to compute SASA of the wildtype and mutants, Appendix SASA_analysis.xlsx.
Secondary structure analysis
The impact of induced mutations upon secondary structure changes was monitored by DSSP (dictionary of protein secondary structure) algorithm (Citation31). DSSP predicts secondary structures based on known hydrogen bond patterns found in common secondary motifs, such as: -helix,
-sheet. The algorithm computes electrostatic interaction energy
as a function of interatomic distances between main chain carbonyl and main chain amino group:
, by placing partial charges:
(main chain carbonyl oxygen is considered to be universal acceptor),
(main chain amino group is considered to be universal hydrogen bond donor). The presence of hydrogen bond is confirmed if
is at least 0.5 kcal/mol (Citation32).
Hydrogen bonds dynamics analysis
Hydrogen bonding between molecules plays a crucial role in molecular recognition, as it provides directionality and specificity of the interaction (Citation33). As hydrogen bonding dictates protein folding that is crucial for protein stability, selective macromolecular interactions are governed by specific hydrogen bonding dynamics. In this study, Module gmx hbond was used to analyze the impact of induced mutations upon SARS-CoV-2 S-protein RBD – hACE2 intermolecular hydrogen bonding dynamics and identify major changes in interactions due to induced amino acid substitutions. Several important aspects are reviewed when considering the impact of a particular intermolecular hydrogen bond, such as: the occupancy (continuity) of the bond and h-bond energy (strength). As the strength of a hydrogen bond depends on the electronegativity of engaged atoms (Citation34, pp. 597), (donor-acceptor distance), ∡
(hydrogen-donor-acceptor angle) and environmental factors, such as pressure and temperature (Citation35), hydrogen bonds can be classified as very strong, strong or weak following categorization provided in Andrews et al. (Citation34, , pp. 597).
Table 1. Average RMSD ()/SASA (
) per molecule of N501[Y, T, S] heterodimers.
To get deeper insight into the conformational alterations due to N501Y S-protein mutation and understand the essentials of structural and biomechanical implications, specific aspects of the dynamics of the intermolecular hydrogen bonding interface between SARS-CoV-2 S-protein RBD and hACE2 receptor were analyzed in comparison to N501 wild type, which was used a referent structure. Intermolecular hydrogen bond contacts between S-protein RBD domain [499-505]: ProThrAsn(Tyr)GlyValGlyTyr and hACE2 receptor were analyzed, due to the fact that Asparagine(N) to Tyrosine(Y) transition at position 501 implies major RBD motif destabilization. The electrostatic impact of a specific hydrogen bond was estimated in terms of the occupancy of a h-bond and donor-acceptor distance for bonds formed between atoms of the same electronegativity in the mutant (N501Y) and the wild type (N501), Appendix H_bonds_analysis.xlsx. Since residues typically change their positions during the course of simulation, which leads to dynamic formation/breaking of hydrogen bonds, the criteria for presence of a h-bond at instance
will satisfy (Citation36): 1)
and 2) ∡
. An adjusted version of the equation used in (Citation31) – Eq. 1 was used to calculate the energy (kcal/mol) of the intermolecular main chain hydrogen bond of the highest occupancy
based on charmm27 all atoms force field (Citation37) parameters a) and b), (file: charmm27.ff/aminoacids.rtp), Appendix Gly502_Lys353_H-bond_Energy.xlsx:
mainchain carbonyl oxygen partial charge = -0,51 e;
f = 332 is a dimensional factor;
Results
Of all variants under consideration in the study – N501Y, N501S, N501T and N501(wild type) – the S-protein N501Y mutation implied a major backbone shift, relative to the starting, energetically minimized state at (, ). N501Y and N501T heterodimers exhibited highest backbone deviation relative to the average RMSD score:
,
(). The variants experienced peak backbone deviations at different time stamps during the simulation (). The N501Y variant exhibited peak backbone oscillation of 0.49 nm at t = 6.78 ns, N501S peak backbone oscillation of 0.34 nm at t = 19.68 ns, N501T peak backbone oscillation of 0.42 nm at t = 8.46 ns and N501 peak backbone deviation of 0.35 nm at t = 4.35 ns (). In terms of average backbone deviation,
[0-50] ns, only N501Y scored higher than N501 (). N501S/N501T average RMSD scored less than N501. Regardless of the difference in the magnitude of the backbone shifts, all four structures reached an equilibrium state at t = 42.3 ns (). Throughout the phase [42.3 – 50] ns backbone oscillations ranged no more than 0.15 nm, which indicates that an equilibrium point was reached (). Reaching an equilibrium point confirms that the systems were well-prepared prior to the molecular dynamics simulation phase.
Figure 1. Backbone deviation analysis of N501[Y, T, S] hetero dimers (Gromacs gmx rms program).
![Figure 1. Backbone deviation analysis of N501[Y, T, S] hetero dimers (Gromacs gmx rms program).](/cms/asset/28f41463-1897-400e-9acb-a027138b2fcd/tbeq_a_2206492_f0001_c.jpg)
The Asparagine-to-Threonine transition at position 501 did not significantly affect the solvent accessibility surface area, and the scores in terms of complex solubility were very similar: ( and ). Because Serine has a shorter side chain compared to Threonine, Serine exhibits higher polarity, expecting to contribute more favorably to protein solubility than Threonine. The obtained results support this suggestion:
( and ). Average SASA results scored almost the same for N501S and N501Y (). As can be observed in , until 37.5 ns the N501S SASA score was even higher than N501Y, contributing more favorably to the complex solubility than any other hydrophilic amino acid (Citation38).
Figure 2. Solubility analysis of N501[Y, T, S] hetero dimers (Gromacs gmx sasa program).
![Figure 2. Solubility analysis of N501[Y, T, S] hetero dimers (Gromacs gmx sasa program).](/cms/asset/1c97006a-4524-454d-b003-b90c8417bdc6/tbeq_a_2206492_f0002_c.jpg)
shows the S-protein RBD (alpha-carbon) root mean square fluctuation curves per residue for all four variants of interest (N501, N501Y, N501S and N501T), having coupled the SARS-CoV-2 S-protein RBD to the hACE2 receptor. The variants N501, N501S and N501T followed similar trends of RMSF curves, although a few domain exemptions were specific to one or another variant, such as S-protein RBD [367-373] ValLeuTyrAsnSerAlaSer where N501S and N501T variants’ residues exhibited significantly lower
fluctuations than N501 and N501Y. On the other hand, the S-protein RBD domain [499-505] ProThrTyrGlyValGlyTyr in N501Y mutant exhibited unique behavior, as it tended to introduce higher
RMSF scores than any other variant of interest in this study (). This indicates that S-protein RBD motif [499-505] ProThrTyrGlyValGlyTyr deviation can be uniquely associated to the presence of Tyrosine (Y) at position 501, which results in local RBD pocket formation, nearby hACE2 Y41 (). shows the
RMSF scores for each amino acid in the domain of interest, for the N501(wild type)/N501Y mutation.
Figure 3. Mean fluctuation per residue (S-protein RBD) (Gromacs gmx rmsf program).
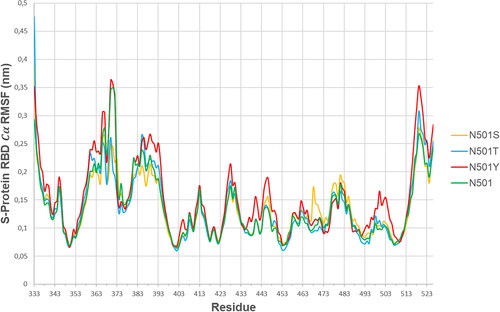
Table 2. S-Protein RBD domain [499-505]: ProThrAsn(Tyr)GlyValGlyTyr RMSF analysis.
shows graphical outputs of do_dssp secondary structure analysis (Citation31) for S-protein RBD in the case of N501 (wild type structure) and N501Y (mutant structure). The actual position of every amino acid in the S-protein can be computed as:
, such as
is the position of the corresponding amino acid on the vertical axis plotted on . For example, the first RBD amino acid (Thr,
is at position 333 in the S-protein (
, thus
is calculated as
, the second RBD amino acid (Asn,
) is at position 334 in the S-protein (
) and is computed as
and so on.
Figure 4. S-protein RBD secondary structure prediction (Gromacs gmx do_dssp module).
Note: Time is plotted on the horizontal axis (0–50 ns), while S-protein RBD residues in the range of 1–194 are shown on the vertical axis.
summarizes major RBD folding differences between N501 and N501Y dimers. S-protein RBD [366-370]: SerValLeuTyrAsn forms stable -helix for
ns in N501, while frequent
-helix to π-helix transitions were observed in N501Y in
ns, . RBD turn [384-387]: ProThrLysLeu was shifted to
-helix during [0-9.25] ns and [25-30] ns in N501, while no turn to
-helix transitions were observed in N501Y, . An interesting conformational change was observed in the domain of the interest S-protein RBD [499-505]: ProThrAsn(Tyr)GlyValGlyTyr. Namely S-protein RBD [503-505]: ValGlyTyr forms
-helix in N501 for
ns that seems to have been destabilized into a turn in N501Y in the same interval, . In general, N501Y mutation has destabilized some of the most-stable secondary S-protein RBD conformations, such as the
-helix, .
Table 3. Secondary structure assessment.
shows the number of intermolecular hydrogen bonds formed between S-protein RBD–hACE2 and S-protein RBD [499-505]–hACE2 for N501 and N501Y structures in t = [0-50] ns. On average, during the simulation, the wild-type structure (N501) S-protein RBD formed a higher number of intermolecular polar contacts than N501Y. On the other hand, Tyrosine at position 501 increased the average number of h-bond interactions in domain [499-505] relative to the wild-type structure ( and ).
Figure 5. Hydrogen bonds dynamics in N501 and N501Y hetero dimers (Gromacs gmx hbond program).
Note: red, number of intermolecular hydrogen bonds in N501Y complex during [0–50] ns MD simulation; green, number of intermolecular hydrogen bonds in N501 complex during [0–50] ns MD simulation.
![Figure 5. Hydrogen bonds dynamics in N501 and N501Y hetero dimers (Gromacs gmx hbond program).Note: red, number of intermolecular hydrogen bonds in N501Y complex during [0–50] ns MD simulation; green, number of intermolecular hydrogen bonds in N501 complex during [0–50] ns MD simulation.](/cms/asset/f4867074-e632-4da4-97ec-defb2cd64eb8/tbeq_a_2206492_f0005_c.jpg)
Table 4. Average number of hydrogen bonds formed in [0–50] ns, wild-type or mutant S-protein RBD [499-505]: ProThrAsn(Tyr)GlyValGlyTyr.
The intermolecular hydrogen bonds formed between S-protein RBD [499-505]: ProThrAsn(Tyr)GlyValGlyTyr and hACE2 receptor were subject of special analysis ( and ). Hydrogen bonds were identified as DonorHydrogen…Acceptor atomic structures in N501 and N501Y heterodimers. The impact of a hydrogen bond was measured by means of occupancy – life span of h-bond in % and (donor-acceptor distance, measured in nm). The prime aim was to identify major shifts in intermolecular polar contacts due to the presence of Tyrosine at position 501 instead of Asparagine ( and ).
Table 5. S-protein RBD [499-505]: ProThrAsnGlyValGlyTyr–hACE2 hydrogen bonds dynamics in wild type N501.
Table 6. S-protein RBD [499-505]: ProThrTyrGlyValGlyTyr–hACE2 hydrogen bonds dynamics in mutant N501Y.
The obtained results ( and ) showed that the N501Y mutation shifts the intermolecular hydrogen bond to permanent polar contact, stronger than the one observed in the wild type. In the N501 variant, a Gly502 to Lys353 main chain intermolecular h-bond was observed 79.5% of the time (83.1% occupancy during convergence period [42.3-50] ns) and it was shifted to 98.4% occupancy (97.9% occupancy during [42.3-50] ns) in N501Y. A stronger h-bond
:
in t = [0-50] ns (
in the convergence phase, for t = [42.3-50] ns), compared to
(
) in the wild type, was observed in the presence of the N501Y mutation.
The N501Y mutation also influenced the intermolecular hydrogen bond, resulting in much stronger and more persistent polar contact ( and ). The H-bond occupancy and strength significantly increased during the convergence phase, [42.3-50] ns:
versus
, having increased contact occupancy for 10.6% relative to N501.
In the course of simulation, prior to position 501, Thr500 (S-protein) in N501 formed a highly persistent h-bond with Asp355 (hACE2), 86.8% occupancy in N501 versus 66.5% occupancy in N501Y ( and ). In terms of strength, the h-bond scored almost equally, in [0-50] ns ([42.3-50] ns). During the convergence state ([42.3-50] ns), the N501Y structure intensified the h-bond occupancy (88.8%), but still under 98.4% of occupancy in N501. However, Thr500 (S-protein) also formed periodic contacts to Tyr41 (hACE2), by means of the same Thr500 side chain oxygen atom (acting as a donor), when not engaged in an h-bond to Asp355 (hACE2) (, ). Much of the time when no h-bond existed between Thr500 (S-protein) and Asp355 (hACE2), the N501Y structure favored hydrogen bond formation between the Thr500 (S-protein) side chain hydroxyl group and the Tyr41 (hACE2) phenolic oxygen (), compensating for the less frequent polar contact to Asp355 in N501Y. The h-bond
was highly intensified during the convergence state in N501Y:
against
in N501 ( and ).
Figure 6. Visualization of hydrogen bonds between hACE2 and SARS-CoV-2 spike protein – N501 against N501Y heterodimer drawn in PyMol molecular graphics program.
Note: red, high occupancy of a hydrogen bond; yellow, average occupancy of a hydrogen bond.
Neither Asparagine, nor Tyrosine at position 501, formed highly persistent intermolecular hydrogen bonds. Only temporal polar contacts to Tyr41, Lys 353, Asp 355 in N501 and Asp38, Lys353 in N501Y complex were observed ( and ).
Binding energy shift (kcal/mol) of the most influenced hydrogen bond due to the N501Y mutation was computed applying Eq. 1 (). There was an increased binding affinity for 0.91 kcal/mol during the simulation and 1.06 kcal/mol during convergence state.
Table 7. energy (kcal/mol) in N501 and N501Y heterodimers.
Discussion
As of March 3rd 2023, following GISAID database - EpiCoV module (https://gisaid.org/), the N501Y SARS-CoV-2 S-protein mutation was reported in 8 406 386 SARS-CoV-2 isolates worldwide, N501T in 6320 and N501S in 750. The primary aim of this study was to examine the impact of the three most frequent Asn501 polar uncharged amino acid mutations: N501Y, N501T and N501S in contact to the hACE2 receptor.
The findings from this study indicate that the presence of Tyrosine at position 501 in SARS-CoV-2 S-protein RBD implies major destabilization of the complex, which was proved in terms of recorded backbone oscillations during the course of simulation (0.49 nm peek amplitude, ). After 42.3 ns, all variants of interest reached convergence state, which confirmed the appropriate preparation of the structures ().
Highly polar Serine at position 501 favors protein solubility over N501 and N501T complexes (), exhibiting approximately the same solvent accessibility surface area as N501Y: ,
. From the detailed h-bonds analysis of S-protein RBD [499-500] (), the Tyr501 phenolic hydroxyl group serves as a donor complex to Asp38 (hACE2) in 3.5% of the time (0% during convergence state), while the Tyr501 phenolic oxygen atom acts as an acceptor in 2% of the time (2.2% during convergence state). This indicates that most of the time the Tyr501 phenolic -OH group formed polar contacts with surrounding solvent molecules, thus increasing the complex solubility.
Serval S-protein RBD domains were highly destabilized in the presence of Tyr501 ( and ). Of all highly destabilized domains, special attention has been put on the S-protein RBD [499-505]: ProThrAsn(Tyr)GlyValGlyTyr domain, which includes a mutational spot. Following secondary structure assessment analysis ( and ), the N501Y mutation shifts the S-protein RBD [503-505]: ValGlyTyr -helix to turn, increasing the local flexibility after 33 ns. Two other domains were also destabilized in the presence of Tyr501, shifting highly-stable
-helix to
-helix,
-helix to turn at some points ().
Hydrogen bond analysis of the RBD [499-505]: ProThrAsn(Tyr)GlyValGlyTyr region showed that some intermolecular polar contacts were intensified due to Tyr501 ( and ). Tyr501 shifts into a permanent polar contact (), with an increased strength relative to the wild type: 3.909532232 kcal/mol in N501 versus 4.816210332 kcal/mol in N501Y during the course of simulation (3.739474518 kcal/mol versus 4.803593311 kcal/mol during convergence state) (). The computed results [kcal/mol] follow experimentally determined N-H…O = C intermolecular h-bond energy, wich may range within [4 – 6] kcal/mol (Citation39). While the wild-type Tyr505 phenolic -OH group participated in single h-bond formation (), the N501Y Tyr505 -OH group periodically formed polar contacts with two Glu37 side chain oxygen atoms, enhancing the interaction between Tyr505 and Glu37 ( and , ). Although the hydrogen bond formed between Thr500 and Asp355 seems to be of lower occupancy in N501Y relative to the wild type, it was supplemented by frequent polar contacts to Tyr41 that were highly intensified during convergence state ( and , ).
Other studies have also investigated the impact of this crucial N501Y mutation (Citation11, Citation13,Citation14). However, most of them only mention the occurrence of a specific hydrogen bond, without providing further details concerning the dynamics of a hydrogen bond, which is crucial for understanding the biomechanical impactions of the N501Y substitution. Some studies (Citation11, Citation14) explain that N501Y increased the SARS-CoV-2 infectivity due to Tyr501…Tyr41 π-π stacking interaction and Tyr501…Lys353 π-cation interaction. In terms of h-bond analysis, Tian et al. (Citation11) have also confirmed that Thr500 forms two hydrogen bonds with Tyr41, Asp355 in the wild type, but no further details regarding the h-bonds dynamics were provided. Similarly, Thr500 to Asp355 polar connection was also confirmed by Rostami et al. (Citation13).
The present study analyzed the hydrogen bonds dynamics in terms of occupancy and strength (estimated by ) and revealed for the first time intensified Tyr505…Glu37 interaction and Gly502…Lys353 h-bond shift into permanent polar connection due to N501Y mutation. This study points to increased N501Y RBD [499-505] infectivity, nearby mutational spot 501, additionally boosting already confirmed Tyr501…Tyr41 π-π stacking interaction and Tyr501…Lys353 π-cation interaction.
Several Cryo–electron structural studies (Citation40–42), evaluated the role of key spike protein mutations upon the hACE2 binding affinity. Experimental findings (Citation40–42) are in line with in silico findings presented in this study, showing additional insights upon key acting residues.
Cryo–electron experiments (Citation42) identified a pocket-binding structure in Y501- carrying variants, accommodating the Y501 aromatic ring in a sandwich between hACE2 Y41 and K353 residues. Following the findings from the present study, the spike protein RBD [499-505]: PTYGVGY motif nearby hACE2 Y41 is characterized by peak RMSF motif deviation only in the presence of Y501 – structural deviation, which actually leads to RBD pocket formation near the hACE2 Y41 binding interface.
The presence of a hydrogen bond was identified by Saville et al. (Citation41). The present study also confirm the existence of intermolecular polar contact between Y505 and E37 that has been intensified in terms of occupancy and strength, shifting the weak wild-type
hydrogen bond (
) into a stronger one (
) in the presence of Y501.
As the spike protein RBD [499-505]: PTYGVGY domain remains unchanged in PDB structures: 7SY2, 7MJN, 7SY4, 7SY6, the molecular dynamics findings from the present study are also applicable to the previous Y501-Cryo-EM bearing structures.
Many studies, (Citation11, Citation14, Citation43) try to explain the impact of the N501Y mutation by considering the change of intermolecular contacts at position 501 only. The present study evaluates the change of contacts’ affinities of 501-surrounding residues: P499, T500, G502, V503, G504, Y505, having substituted Asparagine to Tyrosine at position 501, which is a novel finding in the present research.
Despite of the well-documented findings for Y501-K353 π-cation interaction and Y501…Y41 π-π stacking interaction (Citation11, Citation14) that result in increased binding affinity due to the formation of the Y501-binding pocket (Citation43), the present study reveals that the binding affinity is additionally increased by formation of a permanent and stronger G502-K353 hydrogen bond in the presence of Y501. Following the findings from the present study, Y501 also intensifies the Y505-E37 polar interaction. The present study has also found that two stable RBD -helices are destabilized into a
-helix and turn respectively in the presence of Y501. Finally, an empirical proof for the presence of the Y501-binding pocket was presented, estimated by means of
’s RMSF RBD [499-505]: PTYGVGY peak formation, which is adjacent to the hACE2 Y41 binding interface.
Conclusions
This study reveals some important biomechanical aspects of SARS-CoV-2 S-protein RBD–hACE2 dynamics upon Asn501 mutation. While the N501S mutation favors hetero-dimer solubility, the N501Y mutation tends to shift some stable RBD secondary motifs into more flexible ones, such as -helix and turn. The N501Y mutation shifts the
hydrogen bond into permanent intermolecular polar contact, also favoring enhanced Tyr505…Glu37 hydrogen bond interaction. The N501Y variant Thr500 forms periodically polar contacts with two hACE2 amino acids: Tyr41 and Asp355, which may be also seen as a form of increased electrostatic attraction towards the hACE2 receptor due to the N501Y mutation.
Author contributions
Done Stojanov: conceptualization, methodology, simulation, validation, investigation, writing—original draft preparation and revision, visualization.
| Abbreviations | ||
| SARS-CoV-2 | = | Severe acute respiratory syndrome coronavirus 2 |
| S-protein/S-glycoprotein | = | surface/spike glycoprotein |
| RBD | = | receptor-binding domain |
| ACE2 | = | Angiotensin-converting enzyme 2 |
| hACE2 | = | human Angiotensin-converting enzyme 2 |
| VOC | = | variant of concern |
| VOI | = | variant of interest |
| PDB | = | Protein Data Bank |
| PME | = | Particle Mesh Ewald |
| NVT | = | Canonical Ensemble |
| NPT | = | isothermal–isobaric ensemble |
| SPC | = | simple point-charge |
| MD | = | molecular dynamics |
| RMSD | = | root-mean-square deviation |
| RMSF | = | root mean square fluctuations |
| SASA | = | solvent accessible surface area |
| DCLM | = | Double Lattice Method |
| DSSP | = | Dictionary of protein secondary structure |
| E | = | energy |
| h-bond | = | hydrogen bond |
| = | donor-acceptor distance | |
| ∡ | = | hydrogen-donor-acceptor angle |
| GISAID | = | Global Initiative on Sharing Avian Influenza Data. |
Supplemental Material
Download Zip (4.7 MB)Data availability
The data that support the findings reported in this paper are available in the Appendix files: RMSD_analysis.xlsx; RMSF_analysis.xlsx; SASA_analysis.xlsx; H_bonds_analysis.xlsx; Gly502_Lys353_H-bond_Energy.xlsx
Disclosure statement
The author declares that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Funding
References
- Fleischmann WR.Jr. Viral genetics. In Baron S, editor. Medical microbiology. 4th ed. Galveston (TX): University of Texas Medical Branch at Galveston; 1996.
- Chen J, Wang R, Wang M, et al. Mutations strengthened SARS-CoV-2 infectivity. J Mol Biol. 2020;432(19):1–15.
- Yang J, Petitjean SJ, Koehler M, et al. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat Commun. 2020;11(1):1–10.
- Kim Y, Kim EJ, Lee SW, et al. Review of the early reports of the epidemiological characteristics of the B. 1.1. 7 variant of SARS-CoV-2 and its spread worldwide. Osong Public Health Res Perspect. 2021;12(3):139–148.
- Gómez CE, Perdiguero B, Esteban M. Emerging SARS-CoV-2 variants and impact in global vaccination programs against SARS-CoV-2/COVID-19. Vaccines. 2021;9(3):243.
- Harvey WT, Carabelli AM, Jackson B, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19(7):409–424.
- Subramoney K, Mtileni N, Bharuthram A, et al. Identification of SARS-CoV-2 omicron variant using spike gene target failure and genotyping assays, Gauteng, South Africa, 2021. J Med Virol. 2022;94(8):3676–3684.
- Nguyen HL, Thai NQ, Nguyen PH, et al. SARS-CoV-2 omicron variant binds to human cells more strongly than the wild type: evidence from molecular dynamics simulation. J Phys Chem B. 2022;126(25):4669–4678.
- Ou J, Lan W, Wu X, et al. Tracking SARS-CoV-2 omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Sig Transduct Target Ther. 2022;7(1):1–9.
- Liu Y, Liu J, Plante KS, et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature. 2022;602(7896):294–299.
- Tian F, Tong B, Sun L, et al. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. Elife. 2021;10:e69091.
- Hou X, Zhang Z, Gao J, et al. SARS-CoV-2 spike protein N501Y mutation causes differential species transmissibility and antibody sensitivity: a molecular dynamics and alchemical free energy study. Mol Syst Des Eng. 2021;6(11):964–974.
- Rostami N, Choupani E, Hernandez Y, et al. SARS-CoV-2 spike evolutionary behaviors; simulation of N501Y mutation outcomes in terms of immunogenicity and structural characteristic. J Cell Biochem. 2022;123(2):417–430.
- Chakraborty S. E484K and N501Y SARS-CoV 2 spike mutants increase ACE2 recognition but reduce affinity for neutralizing antibody. Int Immunopharmacol. 2022;102:108424.
- Kazybay B, Ahmad A, Mu C, et al. Omicron N501Y mutation among SARS-CoV-2 lineages: insilico analysis of potent binding to tyrosine kinase and hypothetical repurposed medicine. Travel Med Infect Dis. 2022;45:102242.
- Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220.
- Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25.
- Brooks BR, Bruccoleri RE, Olafson BD, et al. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4(2):187–217.
- Darden T, Perera L, Li L, et al. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure. 1999;7(3):R55–R60.
- Ewald P. Evaluation of optical and electrostatic lattice potentials. Ann Phys. 1921;64:253–287.
- Zhang Q, Xiang R, Huo S, et al. Molecular mechanism of interaction between SARS-CoV-2 and host cells and interventional therapy. Sig Transduct Target Ther. 2021;6(1):1–19.
- Birtles D, Oh AE, Lee J. Exploring the pH dependence of the SARS-CoV-2 complete fusion domain and the role of its unique structural features. Protein Sci. 2022;31(9):e4390.
- Curry HB. The method of steepest descent for non-linear minimization problems. Quart Appl Math. 1944;2(3):258–261.
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126(1):014101.
- Berendsen HJ, Postma JV, Van Gunsteren WF, et al. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690.
- Verlet L. Computer "experiments" on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967;159(1):98–103.
- Aier I, Varadwaj P, Raj U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci Rep. 2016;6:34984.
- Benson NC, Daggett V. A comparison of multiscale methods for the analysis of molecular dynamics simulations. J Phys Chem B. 2012;116(29):8722–8731.
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132.
- Eisenhaber F, Lijnzaad P, Argos P, et al. The double cubic lattice method: efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J Comput Chem. 1995;16(3):273–284.
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers Original Res Biomolecules. 1983;22(12):2577–2637.
- Meyers RA. Encyclopedia of physical science and technology. 3rd ed. Cambridge (MA): Academic Press, 2001.
- Hubbard RE, Muhammad KH. Hydrogen bonds in proteins: role and strength. In Encyclopedia of life sciences (ELS). Chichester: John Wiley & Sons, Ltd; 2010.
- Andrews D, Scholes G, Wiederrecht G. 2010. Comprehensive nanoscience and technology. Cambridge (MA): Academic Press.
- Park SJ, Seo MK. Interface science and composites. 1st ed., Vol. 18. Cambridge (MA): Academic Press; 2011.
- Baker EN, Hubbard RE. Hydrogen bonding in globular proteins. Prog Biophys Mol Biol. 1984;44(2):97–179.
- Vanommeslaeghe K, Hatcher E, Acharya C, et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31(4):671–690.
- Trevino SR, Scholtz JM, Pace CN. Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favorably than the other hydrophilic amino acids in RNase Sa. J Mol Biol. 2007;366(2):449–460.
- Deshmukh SA, Sankaranarayanan SK, Suthar K, et al. Role of solvation dynamics and local ordering of water in inducing conformational transitions in poly (N-isopropylacrylamide) oligomers through the LCST. J Phys Chem B. 2012;116(9):2651–2663.
- Mannar D, Saville JW, Zhu X, et al. SARS-CoV-2 omicron variant: antibody evasion and cryo-EM structure of spike protein–ACE2 complex. Science. 2022;375(6582):760–764.
- Saville JW, Mannar D, Zhu X, et al. Structural and biochemical rationale for enhanced spike protein fitness in Delta and kappa SARS-CoV-2 variants. Nat Commun. 2022;13(1):742.
- Zhu X, Mannar D, Srivastava SS, et al. Cryo-electron microscopy structures of the N501Y SARS-CoV-2 spike protein in complex with ACE2 and 2 potent neutralizing antibodies. PLoS Biol. 2021;19(4):e3001237.
- Çubuk H, Özbi LM. In silico analysis of SARS-CoV-2 spike protein N501Y and N501T mutation effects on human ACE2 binding. J Mol Graph Model. 2022;116:108260.