
Abstract
The outbreak of the coronavirus disease 2019, caused by the SARS-CoV-2 virus, has prompted global health concerns. In response, researchers have been conducting investigations on active compounds in plants that may hold the potential to inhibit the proliferation of the virus. The aim of this study was to simulate and predict structural interactions of selected compounds isolated from 28 endemic plants of Bosnia and Herzegovina against the main protease (Mpro), papain-like protease (PLpro), RNA-dependent RNA polymerase (RdRp), spike glycoprotein and uridylate-specific endoribonuclease (NendoU) of SARS-CoV-2. The majority of compounds, especially hesperidin, showed great binding affinity to the target proteins. The highest affinity for Mpro was observed for genistein and hesperidin, while in terms of structural interactions, both compounds achieved interactions of interest. Hesperidin and luteolin were the compounds with the highest binding affinity for PLpro, but no significant interactions were observed. For RdRp, hesperidin and quercetin showed the highest binding affinity, where both compounds formed interactions of interest. Hesperidin and fisetin were the compounds with the highest binding affinity for spike glycoprotein, and both compounds achieved significant interactions. The highest affinity for NendoU was obtained for hesperidin and isorhamnetin, where both compounds formed interactions of interest. Although these findings appear encouraging, further research is needed, which includes in vitro and in vivo assessments, along with clinical trials, to provide evidence for the potential therapeutic uses of these plants.
Introduction
The rising rate of morbidity and mortality caused by viral infections makes them one of the biggest problems for healthcare systems worldwide. Numerous synthetic anti-viral compounds, which have been found by scientific investigation, are frequently restricted for use due to their side effects and high cost of treatment, making them unaffordable for many patients. Currently, one of the key areas of interest for the development of novel medications is plant-derived metabolites and their bioactive potential [Citation10].
The prediction of compounds having inhibitory action against certain proteins is frequently done using the computational approach known as protein-ligand docking. Typically, it is the initial stage in the process of screening potentially active molecules from already-existing chemicals, which is important for future research and the development of new drugs [Citation10]. The development of new drug candidates is a process that involves the design of inhibitors that are characterized by a higher affinity against a particular substrate. One of the most common strategies used in the development of new drug candidates is the design of enzyme inhibitors, where small molecules are used to target a specific enzyme [Citation23].
A novel coronavirus SARS-CoV-2 (Severe acute respiratory syndrome coronavirus 2) caused an unusual viral pneumonia reported in clusters of patients in Wuhan health facilities (Hubei province, China) in December, 2019. The virus caused the coronavirus disease 2019 (COVID-19) and the outbreak of the disease was declared a pandemic in March 2020 by the World Health Organization (WHO) [Citation24]. When the cell is infected, translation of open reading frames ORF1a and ORF1ab is initiated for the production of two polyproteins (pp1a and pp1ab). These polyproteins are then processed for the release of 16 non-structural proteins (NSPs) by 3 C-like protease (3Clpro; also called main protease – Mpro) and papain-like protease (PLpro) [Citation25]. Cleavage of pp1a and pp1ab into functional proteins is a critical step of viral replication [Citation57] and it makes Mpro and PLpro ideal drug targets for the treatment of COVID-19. The conserved dyad with catalytic function (His41-Cys145) serves as the active site of Mpro [Citation20]. This enzyme regulates proteolytic cleavage as well as the processing of the large viral polyprotein orf1ab in conjunction with the PLpro [Citation1]. Papain-like protease is an essential component of the replicase transcriptase complex [Citation34]. It has a role as a de-ubiquitinating and de-ISGylating enzyme [Citation16]. In this way, the activity of de-ubiquitinating affects the regulation of cellular pathways [Citation36] resulting in suppression of inflammation and antiviral signalling [Citation35]. In designing a potential treatment against the spread of the SARS CoV-2 virus, the so-called target sites of PLpro are in focus. Thus, the Zn-binding site within this molecule is important in the analysis because of its influence on the protein folding, or formation of the 3D structure of the nsp3 molecule. Also, there is a catalytic domain consisting of Cys111, His272 and Asp286 [Citation41].
Another potential drug target is the RNA dependent RNA polymerase (RdRp). It is an essential viral enzyme that facilitates viral RNA replication, whose inhibition may prevent the replication of viral RNA, and thus, the development of new viral particles. The 932-amino-acid-long SARS-CoV2 RNA dependent RNA polymerase (Nsp12) protein has two main functional domains: the NIRAN (nidovirus RdRp associated nucleotidyl transferase) domain and the RdRp (RNA dependent RNA polymerase) domain. The proteins Nsp12, Nsp8, and Nsp7 make up the RdRp Complex (Nsp12). The Nsp7-Nsp8 heterodimer stabilizes the closed conformation of Nsp12 and plays a key role in mediating interactions between Nsp12 and RNA [Citation12]. Asp618, Tyr619, Asp760 and Asp761 are amino acid residues that participate in the formation of the active site of RNA-dependent RNA polymerase [Citation9].
Similarly, the spike protein is also a potential drug target, which is present on the surface of mature viruses in the form of homotrimer, while the surface of the trimer is densely decorated with heterogeneous N-glycans [Citation59]. The spike protein is made up of two large subunits, S1 and S2. The primary role of S1 is to bind to host cell surface receptors, whereas S2 enables membrane fusion between host cells and viruses. The S1 subunit includes the N-terminal domain (NTD), C-terminal domain 1 (CTD1) and C-terminal domain 2 (CTD2). In addition to these domains, the S1-subunit also contains three receptor-binding domains (RBDs), which are responsible for recognizing the angiotensin-converting enzyme 2 (ACE2) receptor located on host cell membranes [Citation14]. On the contact surface between ACE2 and the receptor-binding domain of SARS-CoV-2, key amino acids responsible for maintaining their mutual association have been identified. These include Glu35, Tyr83, Asp38, Lys31, Glu37 and His34 of the ACE2 receptor, and amino acid residues Gln493, Gln498, Asn487, Tyr505 and Lys417 of the SARS-CoV-2 receptor-binding domain [Citation58].
In addition to previously mentioned proteins, another potential drug target for SARS-CoV-2 is Nsp15, which is a uridine-specific endoribonuclease with C-terminal catalytic domain. It belongs to EndoU family characteristic for Corona viruses and NendoU is named used for viral subfamily [Citation30]. It plays an important role in viral replication, and is also associated with blocking the function of MDA-dependent pathogen-associated molecular patterns (PAMPs) [Citation30], found as mediator of interferon response [Citation21]. The active site of Nsp15 is structured by a catalytic triad (two histidines and a lysine): His235, His250, Lys290; reminiscent of the RNAse A endoribonuclease which supports transesterification process [Citation21].
As it is mentioned above, metabolites of plants and their bioactive potential appear to be one of the prime points of focus for the discovery of new drugs, including ones for COVID-19 treatment [Citation39]. In this regard, the aim of our study was to investigate the antiviral potential of phenolic compounds isolated from 28 endemic plant species of Bosnia and Herzegovina against uridylate-specific endoribonuclease, main protease, papain-like protease, RNA-dependent RNA polymerase and spike glycoprotein of SARS-CoV-2 virus using molecular docking methods.
Materials and methods
Sampling of plant material
The plant material was collected and taxonomic affiliations were determined by a botanist from the University of Sarajevo, who is specialized in the field of systematics of endemic plants, in the period April–June 2021 with a list of species, families and localities. A total of 28 endemic plant species of Bosnia and Herzegovina were collected from 29 populations in the stages of flowering or fruiting: Daphne blagayana, Alyssum moellendorfianum (Konjic region), Crocus vernus (Bjelašnica region), Draba lesiocarpa, Onosma sterullata, Alyssum bosniacum (Visočica region), Iris reichenbachii, Halacsya sendtneri, Euphorbia montenegrina, Potentilla maliana (Kladanj region), Petteria ramentacea, Chrysosplenium cinerariifolium, Onosma echioides, Dictamnus albus, Moltkia petraea, Quercus schneideri (Drežnica region), Iris pumila (Kalinovik region), Narcissus poeticus, Gentiana dinarica, Juniperus sabina, Daphne alpina (Zelengora region), Cerastium grandiflorum, Iris ilirica (Neretva canyon), Sibiraea altaiensis var. croatica (Čabulja region), Menyanthes trifoliate (Treskavica region), Silene sendtneri, Knautia sarajevensis and Dianthus petraeus (Igman region).
Preparation of plant extracts
The plant material was cleaned of impurities, air-dried in a ventilated and darkened room, in a place protected from direct light in order to preserve thermolabile and photosensitive compounds. Rhizome was used as the plant material for Iris reichenbachii, Iris pumila and Iris ilirica, while the whole plants were used for the remaining species. The dried plant material was crushed with an IKA mill, and then larger particles and fibers were removed. Extraction of active components was performed in 50 mL Falcon tubes. To obtain three different extracts from each plant, 1 g of chopped plant material was separately mixed with 20 mL of three different solvents, namely distilled water, 50% ethanol or 96% ethanol. Each mixture was vigorously mixed and incubated for two hours at 60 °C in a water bath with continuous mixing. At the end of the two-hour incubation, the tubes were centrifuged at 4000 rpm for 15 min (Eppendorf centrifuge 5804 R), and then filtered to obtain clear extracts. The solvent was evaporated in an incubator at 40 °C until dry precipitate was isolated.
HPLC analysis
The analysis of the presence of targeted bioactive components in plant extracts was performed via HPLC (High performance liquid chromatography) on an Agilent Infinity II 1260 HPLC device. The mobile phase consisted of a mixture of methanol and 1% formic acid in a 50:50 ratio, which was passed through an HPLC Rastek C18 column. The column dimensions were 150 mm x 4.6 mm, and the particle size was 5 μm. A sample injection volume of 10 μL was used, and the temperature was maintained at a constant 40 °C throughout the analysis. The flow rate of the mobile phase was set at 0.5 mL/min, and the total run time for the analysis was 55 min. The HPLC method was used to analyze the presence and concentration of substances with, according to literature data, proven antiviral activity: fisetin, quercetin, kaempferol, isorhamnetin, luteolin, genistein, pterostilbene, resveratrol, curcumin, hesperidin, scopoletin and hydroxychloroquine.
The results of the HPLC analysis showed the presence of the following active components: Onosma echioides: kaempferol, luteolin, hesperidin; Onosma sterullata: resveratrol; Petteria ramentacea: kaempferol, isorhamentin, luteolin, genistein, scopoletin; Moltkia petraea: genistein, scopoletin; Quercus schneideri: quercetin, kaempferol, luteolin, resveratrol; Alyssum moellendorfianum: quercetin, hesperidin; Draba lesiocarpa: genistein; Halacsya sendtneri: genistein; Alyssum bosniacum: quercetin, kaempferol, resveratrol, scopoletin; Potentilla maliana: fisetin, resveratrol, hesperidin; Euphorbia montenegrina: fisetin, quercetin, isorhamentin resveratrol; Iris reichenbachii: fisetin, quercetin, kaempferol, isorhamentin, hesperidin; Juniperus sabina: fisetin, quercetin, hesperidin, scopoletin; Iris pumila: quercetin, kaempferol, isorhamentin, genistein, hesperidin; Iris ilirica: fisetin, quercetin, isorhamentin; Narcissus poeticus: luteolin; Gentiana dinarica: isorhamentin, pterostilbene, curcumin; Dictamnus albus: luteolin, resveratrol; Chrysosplenium cinerariifolium: fisetin, genistein, hesperidin; Daphne blagayana: fisetin, quercetin, luteolin, hesperidin; Crocus vernus: fisetin, kaempferol, resveratrol; Menyanthes trifoliate: luteolin, resveratrol, curcumin; Cerastium grandiflorum: luteolin; Dianthus petraeus: isorhamentin, luteolin; Knautia sarajevensis: fisetin, quercetin, genistein; Silene sendtneri: genistein, scopoletin; Sibiraea altaiensis: kaempferol, luteolin, resveratrol; Daphne alpina: fisetin, luteolin, hesperidin, scopoletin.
Three-dimensional structures and active sites
The three-dimensional crystal structures of the SARS-CoV-2 target proteins uridylate-specific endoribonuclease (PDB ID: 6WLC), main protease (PDB ID: 7B2J), papain-like protease (PDB ID: 7BF6), RNA-dependent RNA polymerase (PDB ID: 7DFG) and spike glycoprotein (PDB ID: 6YZ5) were obtained from the RCSB Protein Data Bank in PDB format [Citation13] (https://www.rcsb.org/). Each of these proteins was present in a complex with an inhibitor; therefore, the small molecules were removed first and then the structures were used for further analysis. To predict the potential active sites of these proteins, the structures were submitted to PrankWeb (https://prankweb.cz/), a binding site prediction server built on P2Rank, a machine learning-based method [Citation26]. For each of the proteins, PrankWeb displayed multiple pockets, of which, one was selected for each protein with the highest score and probability. The best ranked pocket for uridylate-specific endoribonuclease had a probability score of 0.762 and included 17 amino acids in total: His235, Asp240, His243, Gln245, Leu246, Gly247, Gly248, His250, Lys290, Val292, Ser294, Met331, Trp333, Glu340, Thr341, Tyr343 and Lys345. For papain-like protease the best ranked pocket had a probability score of 0.864 and included the following amino acid residues: Glu104, Pro125, Leu126, Leu127, Ser128, Ala129, Gly130, Ile131, Phe132, Pro136, Ala154, Val155, Phe156, Asp157, Leu160, Ala38, Ala39, Asn40, Val41, Lys44, Gly46, Gly47, Gly48, Val49, Ala50, Ala52, Val96, Gly97 and Asn99. The best ranked pocket for main protease had a probability score of 0.890 and it included the following residues: His41, Met49, Phe140, Leu141, Asn142, Cys145, His163, His164, Met165, Glu166, Asp187, Arg188 and Gln189. For RNA-dependent RNA polymerase the best ranked pocket had a probability score of 0.871 and included the following amino acid residues: Lys545, Lys551, Arg553, Asp618, Tyr619, Pro620, Lys621, Asp623, Thr680, Ser682, Thr687, Asn691, Asp760 and Asp761. A review of the literature [Citation19] determined the amino acids that participate in the binding of the spike protein to ACE2. According to [Citation19], those are: Tyr449, Tyr453, Leu455, Phe456, Phe486, Asn487, Tyr489, Gln493, Gly496, Gln498, Thr500, Asn501, Gly502 and Tyr505.
Protein and ligand preparation, molecular docking and visualization
The protein preparation was conducted using AutoDock Tools 1.5.6 [Citation42], where the water molecules were removed, polar hydrogen atoms were added and Kollman charges were assigned. Subsequently, the target protein was converted from PDB to PDBQT format. Also, using AutoDock Tools 1.5.6, a gridbox was defined for each of the proteins with which the best ranked pocket was captured. The gridbox dimensions for uridylate-specific endoribonuclease were 28 × 28 × 28 Å and it was centered at 91.349, −19.493 and −30.304. For papain-like protease the dimensions were set to 22 × 26 × 24 Å and it was centered at 19.073, 17.831 and 1.147. For main protease the gridbox dimensions were 29 × 18 x 22 Å, centered at 12.411, 1.324 and 20.321. The gridbox dimensions for RNA-dependent RNA polymerase were 26 × 24 x 28 Å and it was centered at 133.395, 131.334 and 140.918, while for the spike glycoprotein the dimensions were set to 26 × 30 x 40 and it was centered at 46.809 x −0.448 × 31.859. For each analysis the value of spacing (ångstrom) was set to 1.0. Three-dimensional structures of the isolated components fisetin (CID: 5281614), quercetin (CID: 5280343), kaempferol (CID: 5280863), isorhamentin (CID: 5281654), luteolin (CID: 5280445), genistein (CID: 5280961), pterostilbene (CID: 5280961). 5281727), resveratrol (CID: 445154), curcumin (CID: 969516), hesperidin (CID: 10621), scopoletin (CID: 5280460), hydroxychloroquine (CID: 3652) and the positive control for which selected remdesivir (CID: 121304016), were available in the PubChem database [Citation29] (https://pubchem.ncbi.nlm.nih.gov/). They were downloaded in the appropriate SDF format and then prepared and converted to PDBQT format using OpenBabel 3.1.1 software with default settings. During the preparation, several changes were made to the ligands, such as adding charges, adding hydrogen atoms, assigning atom types, converting bond types, and setting the root. It should be noted that the default settings of OpenBabel generate the molecule’s 3D structure in a neutral state without including any ionization states, which was applied to the PDBQT format of the ligands used in this study [Citation45]. As previously described in [Citation17], all molecular docking simulations were performed using AutoDock Vina 1.1.2, with an energy range value of 4 and exhaustiveness value of 8 [Citation56]. The intermolecular interactions between the receptors and the selected compounds were visualized and analyzed using three different types of software: UCSF Chimera 1.16 [Citation47], PyMOL 2.5.2 [Citation55] and LigPlot+ 2.2.4 [Citation33]. These analyses were conducted on a computer with a Microsoft Windows 10 Pro operating system (Version 10.0.19045 Build 19045), comprising of an 11th Gen Intel(R) Core(TM) i7-11700 CPU @ 2.50 GHz, an Intel(R) UHD Graphics 750 GPU, a 512GB Apacer AS2280P4 M.2 PCIe solid-state drive, and 32GB of Kingston DDR4 3200 MHz RAM (KVR32N22D8/16).
Results
The molecular docking results of phenolic compounds isolated from 28 endemic plant species of Bosnia and Herzegovina against the Mpro, PLpro, RdRp, spike glycoprotein and NendoU of SARS-CoV-2 showed that these compounds can bind with great affinity to the target proteins near their active sites. Supplemental Table S1 displays the best results and corresponding binding affinities of each compound and positive control for the target proteins. Due to the large number of phytochemicals, two compounds were selected for each target protein based on binding affinity to analyze the interactions and types of bonds. In terms of the visualization of interactions between selected compounds and target protein amino acids, UCSF Chimera showed the best results with the largest number of recorded bonds. In order to obtain more reliable results, the same interactions were analyzed using two additional programs, PyMOL and LigPlot+.
Out of 12 isolated phytochemicals, the highest binding affinities for the SARS-CoV-2 main protease were obtained for genistein (–8.4 kcal/mol) and hesperidin (–8.0 kcal/mol), while the binding affinity of the positive control, remdesivir, was −7.6 kcal/mol. The highest binding affinity for the SARS-CoV-2 papain-like protease was obtained for hesperidin (–9.6 kcal/mol) and luteolin (–8.3 kcal/mol), while the binding affinity of the positive control was −7.5 kcal/mol. For the SARS-CoV-2 RNA-dependent RNA polymerase, the highest binding affinities were acquired for hesperidin (–7.5 kcal/mol) and quercetin (–6.7 kcal/mol), while the positive control had an affinity of −6.9 kcal/mol. The highest binding affinity for the SARS-CoV-2 spike glycoprotein was predicted for hesperidin (–7.8 kcal/mol) and fisetin (–6.5 kcal/mol), while the binding affinity of the positive control was −6.7 kcal/mol. For the SARS-CoV-2 uridylate-specific endoribonuclease, the highest binding affinities were acquired for hesperidin (–8.0 kcal/mol) and isorhamnetin (–7.5 kcal/mol), while the positive control had a binding affinity of −7.4 kcal/mol. The highest binding affinities of the selected compounds against SARS-CoV-2 target proteins and their interactions are presented in .
Table 1. Best molecular docking results of selected compounds against the target proteins and their interactions.
Main protease
After analyzing the structural interactions between the selected compounds and SARS-CoV-2 main protease using UCSF Chimera, it was shown that genistein reacted with Cys44 (5.5 Å), Met49 (4.3 Å), Cys145 (3.9 Å), Glu166 (2.3 Å), Arg188 (3.1 Å), Gln189 (2.8 Å), forming six hydrogen bonds (). Taking into consideration the active site of main protease, genistein formed a significant hydrogen interaction with Cys145, which is one of the two amino acids that form the catalytic dyad (His41-Cys145). Utilizing the PyMOL and LigPlot + software, four and two interactions were documented, respectively, none of which were deemed biologically relevant.
Figure 1. Genistein interactions with SARS-CoV-2 main protease, visualized in UCSF Chimera. Genistein formed six hydrogen bonds with amino acids: Cys44, Met49, Cys145, Glu166, Arg188 and Gln189.
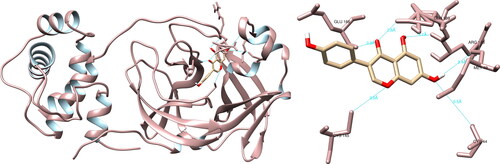
The utilization of UCSF Chimera facilitated the examination of the structural relationships between the specified compounds and the Mpro of SARS-CoV-2. The study revealed that the molecule hesperidin engaged in interactions with residues with Thr26 (1.9 Å), Asn142 (2.2 Å), Cys145 (3.6 Å, 5.2 Å), His164 (2.2 Å), Glu166 (2.3 Å) and Gln189 (2.0 Å), resulting in the formation of a total of seven hydrogen bonds (). Furthermore, the examination of the active site revealed that hesperidin established a substantial double hydrogen interaction with Cys145, which constitutes one half of the crucial catalytic dyad along with His41. Using PyMOL and LigPlot + software, five interactions were recorded in each, none of which were significant.
Figure 2. Hesperidin interactions with SARS-CoV-2 main protease, visualized in UCSF Chimera. Hesperidin formed seven hydrogen bonds with amino acids: Thr26, Asn142, Cys145, His164, Glu166 and Gln189.
Papain-like protease
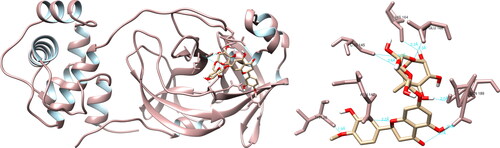
Comprehensive examination of the structural rapport between a curated assemblage of compounds and the SARS-CoV-2 PLpro was performed utilizing UCSF Chimera. It was determined that hesperidin exhibited pronounced interactions with several residues, namely Ile23 (1.999 Å, 3.974 Å), Asn37 (4.386 Å), Ala38 (2.780 Å), Asn40 (2.241 Å), Lys44 (3.902 Å, 4.018 Å), Gly48 (3.812 Å), Ala50 (2.249 Å), Gly51 (2.595 Å) and Ala129 (2.127 Å), via the formation of 11 hydrogen bonds (). However, it is worthy of note that no significant hydrogen bonding was observed between hesperidin and the residues within the active site of the protease. Using PyMOL and LigPlot + software, seven and nine interactions were recorded, respectively, none of which were of significance.
Figure 3. Hesperidin interactions with SARS-CoV-2 papain-like protease, visualized in UCSF Chimera. Hesperidin formed 11 hydrogen bonds with amino acids: Ile23, Asn37, Ala38, Asn40, Lys44, Gly48, Ala50, Gly51 and Ala129.
An examination of the structural rapport between a selection of compounds and the SARS-CoV-2 papain-like protease utilizing UCSF Chimera, revealed that luteolin engaged in interactions with His45 (3.809 Å), Gly46 (3.255 Å), Val49 (2.932 Å), Ala50 (4.141 Å) and Ser128 (2.918 Å), effecting the formation of five hydrogen bonds (). In regards to the active site, luteolin did not exhibit any notable hydrogen bonding with the amino acid residues present in the site. Additionally, utilizing PyMOL and LigPlot + software, four and five interactions were documented respectively, none of which were considered significant.
Figure 4. Luteolin interactions with SARS-CoV-2 papain-like protease, visualized in UCSF Chimera. Luteolin formed five hydrogen bonds with amino acids: His45, Gly46, Val49, Ala50 and Ser128.
RNA-dependent RNA polymerase
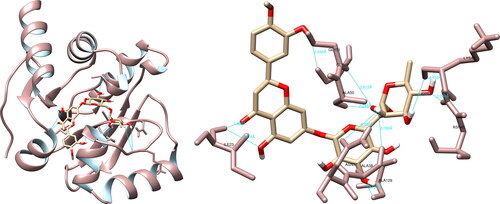
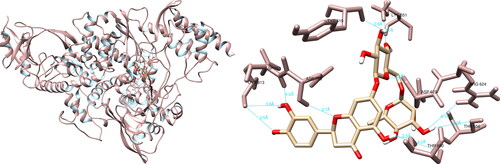
The analysis of the structural interactions between the chosen compounds and the SARS-CoV-2 RNA-dependent RNA polymerase through UCSF Chimera predicted that hesperidin established connections with Lys551 (3.7 Å), Tyr619 (2.8 Å), Arg624 (3.6 Å, 4.1 Å), Thr680 (2.5 Å, 4.2 Å), Asp760 (3.7 Å), Asp761 (4.0 Å) and Cys813 (3.5 Å, 3.8 Å), thereby forming ten hydrogen bonds (). Upon examination of the active site, hesperidin demonstrated three noteworthy interactions with Tyr619, Asp760 and Asp761, which form three of the four amino acids constituting the active site of RdRp. Furthermore, PyMOL and LigPlot + documented five and six interactions, respectively, both of which indicated significant interactions with Tyr619 and Asp760.
Figure 5. Hesperidin interactions with SARS-CoV-2 RNA-dependent RNA polymerase, visualized in UCSF Chimera. Hesperidin formed 10 hydrogen bonds with amino acids: Lys551, Tyr619, Arg624, Thr680, Asp760, Asp761 and Cys813.
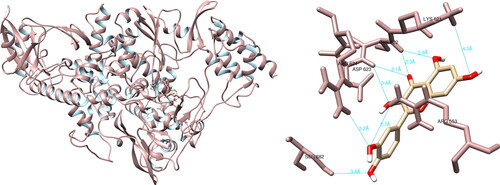
After analyzing the structural interactions between the selected compounds and the SARS-CoV-2 RdRp using the UCSF Chimera software, it was determined that quercetin formed seven hydrogen bonds with Arg553 (3.7 Å), Lys621 (4.3 Å), Cys622 (2.3 Å, 2.9 Å), Asp623 (3.1 Å, 3.4 Å) and Ser682 (3.8 Å) (). Quercetin did not form any hydrogen bonds of significant magnitude with amino acid residues from the active site. The molecular interactions between the selected compounds and the SARS-CoV-2 RNA-dependent RNA polymerase were further analyzed using PyMOL and LigPlot + software. In the case of PyMOL, five interactions were recorded, with two interactions of significance observed with Tyr619 and Asp760. Analysis conducted with LigPlot + revealed five interactions, with one of noteworthy importance with Tyr619.
Figure 6. Quercetin interactions with SARS-CoV-2 RNA-dependent RNA polymerase, visualized in UCSF Chimera. Quercetin formed seven hydrogen bonds with amino acids: Arg553, Lys621, Cys622, Asp623 and Ser682.
Spike glycoprotein
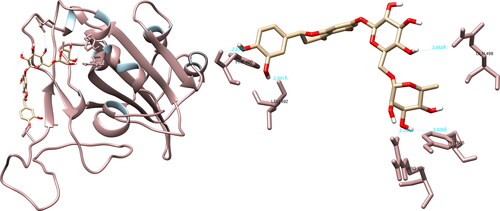
The structural interaction analysis of the selected compounds and SARS-CoV-2 spike glycoprotein using the UCSF Chimera software determined that hesperidin formed hydrogen bonds with Arg408 (2.294 Å), Phe490 (2.275 Å), Leu492 (2.091 Å), Gln498 (3.952 Å) and Tyr505 (3.928 Å) (). Of particular significance, hesperidin exhibited two meaningful hydrogen interactions with Gln498 and Tyr505, both of which play a role in binding the spike glycoprotein to the ACE2 receptor. Further analysis using PyMOL software revealed nine interactions, with Gln493 and Tyr505 being of particular importance. The examination utilizing LigPlot + recorded seven interactions, with one interaction of notable interest involving Tyr505.
Figure 7. Hesperidin interactions with SARS-CoV-2 spike glycoprotein, visualized in UCSF Chimera. Hesperidin formed five hydrogen bonds with amino acids: Arg408, Phe490, Leu492, Gln498 and Tyr505.
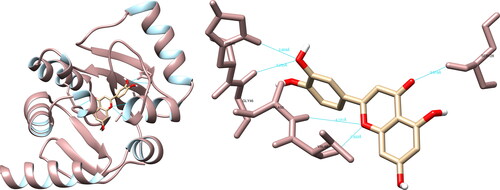
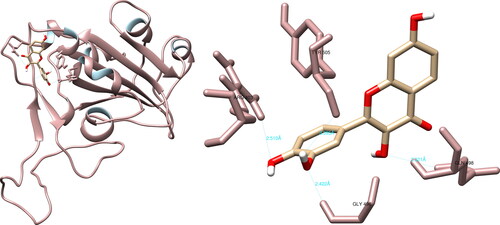
The UCSF Chimera software showed that fisetin could form hydrogen bonds with Arg403 (2.510 Å), Gly496 (2.422 Å), Gln498 (3.321 Å), and Tyr505 (3.395 Å) of the SARS-CoV-2 spike glycoprotein using (). In the active site, fisetin formed two significant hydrogen interactions with the amino acids Gln498 and Tyr505, which play a key role in the binding of the spike glycoprotein to the ACE2 receptor. The PyMOL software was utilized to record five interactions, with one interaction of significance with Gln498. Meanwhile, LigPlot + recorded one non-significant interaction.
Figure 8. Fisetin interactions with SARS-CoV-2 spike glycoprotein, visualized in UCSF Chimera. Fisetin formed four hydrogen bonds with amino acids: Arg403, Gly496, Gln498 and Tyr505.
Uridylate-specific endoribonuclease
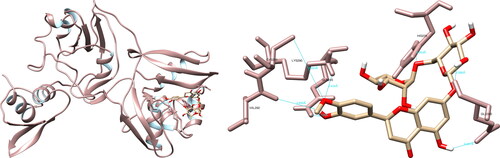
The examination of the structural interactions between the selected compounds and the SARS-CoV-2 NendoU via UCSF Chimera revealed that hesperidin had interactions with His235 (3.954 Å), Gln245 (2.726 Å), Lys290 (2.434 Å), Val292 (3.890 Å), Cys293 (5.624 Å) and Glu340 (2.086 Å, 2.347 Å), resulting in the formation of seven hydrogen bonds (). Of particular significance are the interactions formed by hesperidin with His235 and Lys290, as they are part of the catalytic triad, which also includes His250, and play a crucial role in the active site. The PyMOL and LigPlot + software both recorded four interactions, with PyMOL highlighting interactions of interest with His235 and Lys290, while LigPlot + recorded an interaction with Lys290.
Figure 9. Hesperidin interactions with SARS-CoV-2 uridylate-specific endoribonuclease, visualized in UCSF Chimera. Hesperidin formed seven hydrogen bonds with amino acids: His235, Gln245, Lys290, Val292, Cys293 and Glu340.
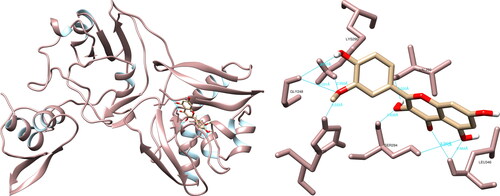
The structural analysis of the selected compounds and the SARS-CoV-2 uridylate-specific endoribonuclease using UCSF Chimera showed that isorhamnetin formed seven hydrogen bonds with Gly248 (3.031 Å, 3.537 Å), His250 (2.222 Å), Lys290 (2.355 Å), Val292 (2.020 Å), Ser294 (2.638 Å, 3.970 Å) and Leu346 (2.464 Å, 2.797 Å). Upon evaluating the active site, isorhamnetin formed two crucial hydrogen bonds with His250 and Lys290, which are integral components of the catalytic triad (His235-His250-Lys290) (). PyMOL allowed for the documentation of seven structural interactions, with His250 and Lys290 being identified as the most significant. LigPlot + resulted in four interactions, one of which was with His250.
Figure 10. Isorhamnetin interactions with SARS-CoV-2 uridylate-specific endoribonuclease, visualized in UCSF Chimera. Isorhamnetin formed seven hydrogen bonds with amino acids: Gly248, His250, Lys290, Val292, Ser294 and Leu346.
The results from the HPLC analysis indicated that the plant species of Chrysosplenium cinerariifolium, Daphne alpina, Daphne blagayana, Iris reichenbachii, Iris pumila, Juniperus sabina and Onosma echioides contained the flavonoid hesperidin. Based on the concentrations of active compounds determined in the plant extracts, the extracts from Chrysosplenium cinerariifolium, Daphne blagayana, Iris reichenbachii and Iris pumila possessed the highest quantity. These findings suggest that the studied natural compounds from endemic plant species could have potential antiviral properties, warranting further investigation through in vitro and in vivo studies to study their potential efficacy in the treatment of COVID-19.
Discussion
Polyphenolic compounds are classified into five classes: flavonoids, stilbenes, hydroxybenzoic acids, hydroxycinnamic acids and lignans. Their bioactive characteristics as antiviral, antioxidant or anti-inflammatory activity, especially towards viruses that affect the respiratory system has been well documented. For example, evidence has shown their antiviral potential against influenza, Epstein-Barr virus, enterovirus, herpes simplex virus and others [Citation53].
Flavonoids (polyphenolic compounds from plants) with bioactive properties [Citation53] have shown PLpro inhibitory effects and antiviral activities against SARS-CoV and MERS-CoV in vitro. In the setting of SARS-CoV-2, several members of the flavonoid family, including coumaroyltyramine, cryptotanshinone, kaempferol, moupinamide, N-cis-feruloyltyramine, quercetin and tanshinone IIa, have been identified as SARS-CoV-2 PLpro ligands using an in silico docking analysis, despite the lack of in vitro evidence for their efficacy [Citation27]. In the group of ligands that we used for molecular docking, there are kaempferol and quercetin.
In this molecular docking study, we used 12 ligands and the best results for SARS-CoV-2 Mpro were obtained with genistein, hesperidin and luteolin, whereas for PLpro, with hesperidin, isorhamnetin, fisetin, quercetin and luteolin. For RdRp the best results were obtained with hesperidin, luteolin and quercetin. For the spike glycoprotein the most promising results were obtained with hesperidin, fisetin, genistein and luteolin. For the nsp15 endoribonuclease of SARS-CoV-2, the best results or binding affinity were found to apply to hesperidin, isorhamnetin, then luteolin and fisetin with similar results. The results we obtained are in agreement with the results we found during the research for the reported studies in the literature.
Flavonoids are the ligands with the highest binding energy for Mpro and RdRp proteins. Hesperidin is a flavonoid, and it is said to be one of the most researched regarding the SARS-CoV-2 virus [Citation28]. Binding energy reached −9.02 kcal/mol in some studies where hesperidin inhibition of Mpro was monitored. In this particular study, hesperidin interacted with amino acids Thr24, Thr25, Thr45, His4, Ser46, Cys145, Thr45, Met49 and Cys145 as part of the protein Mpro (PDB: 6Y84) [Citation18].
In studies that used the same protein molecule with PDB ID: 6LU7, the substance with the highest affinity for the SARS-CoV-2 Mpro active site and the highest binding energy was hesperidin [Citation3,Citation32]. The binding energy of hesperidin with Mpro (PDB: 6LU7), in both studies, resulted in −8.5 kcal/mol. Our results support the previous studies’ findings regarding hesperidin’s affinity for the active site of SARS-CoV-2 Mpro, although we observed a slightly lower binding energy of −8.0 kcal/mol.
There are fewer reports on Mpro inhibition by genistein, but one promising study investigated the inhibition of genistein derivatives [Citation37]. In their study, the compounds with the highest binding energy were derivatives ZINC000096114211 and mucusisoflavone C (ZINC000072110832), with a total of −10 kcal/mol and −9.9 kcal/mol, respectively [Citation38]. In another report, genistein reached a binding energy of −7.6 kcal/mol [Citation47], which is an adequate binding affinity for possible inhibition of Mpro. Our findings align with previous studies, as we observed that genistein demonstrated a binding energy of −8.4 kcal/mol and formed six hydrogen bonds with key residues, including Cys145, within the active site of the SARS-CoV-2 main protease.
In our previous study [Citation17], we examined the potential inhibitory effect of active compounds derived from Knautia sarajevensis on the Mpro of SARS-CoV-2. The results of that investigation indicated a potential inhibitory effect, and when taken together with the results of this study, our findings should generate interest in further research and the potential applications of this plant as an adjuvant.
Quercetin has been documented to bind to the papain-like protease with an affinity of 6.2–4.6 kcal/mol [Citation6]. The evidence strongly suggests that this flavonoid has an anti-inflammatory effect against SARS-CoV-2, especially when combined with vitamin C [Citation6].
Hesperidin has long been used in medical treatment due to its pharmacological action and antioxidant activity. A great source of hesperidin and quercetin are citrus fruits, which are considered fruits rich in flavonoids [Citation4]. According to many authors, hesperidin is classified as a top candidate for active site docking of SARS-CoV-2 [Citation4]. In addition to many useful dietary flavonoids, hesperidin, curcumin, quercetin and kaempferol have been marked as important molecules in the design of potential drugs against SARS-CoV-2, including Mpro, spike protein and PLpro as targets [Citation7].
Some compounds from plants have shown various ability to interact with PLpro, and the following ones have been characterized for global docking [Citation7]: hesperidin (–8.9 kcal/mol), curcumin (–7.4 kcal/mol), quercetin (–6.8 kcal/mol) and kaempferol (–6.3 kcal/mol). For local docking, the following results were obtained: hesperidin (–6.62 kcal/mol), curcumin (–5.0 kcal/mol), quercetin (–5.4 kcal/mol), and kaempferol (–4.92 kcal/mol) when combined with PLpro [Citation7]. In another report [Citation2], the binding affinity of PLpro with kaempferol was −6.8 kcal/mol (PDB ID 6WX4). In comparison to the reported compounds, our findings demonstrate that hesperidin, with a binding affinity of −9.6 kcal/mol, exhibits strong interactions with multiple residues of SARS-CoV-2 PLpro, forming 11 hydrogen bonds. Similarly, luteolin, with a binding affinity of −8.3 kcal/mol shows moderate interactions with specific residues, including the formation of five hydrogen bonds.
Isorhamnetin could act effectively on Mpro SARS-CoV-2 and SARS-CoV, considering its pharmacological effectiveness and wide application in the treatment of various diseases [Citation51]. Another research identified isorhamnetin as a potential candidate for inhibiting Mpro, with computational analysis suggesting its potential anti-inflammatory and antioxidant effects [Citation54].
Isorhamnetin, kaempferol and quercetin belong to the flavonols (flavonoids) [Citation6]. Kaempferol, quercetin and fisetin (as well as their derivatives) have been highlighted as very suitable as treatments due to their antiviral effects on SARS-CoV-2 [Citation43]. Isorhamnetin can bind to S-ACE receptors [Citation49].
Luteolin produced the best results of the ligands used in a previous molecular docking study [Citation5]; then, isorhamnetin bound to PLpro with an affinity of 8.5 kcal/mol. The authors also mention the antiviral effect of quercetin from dietary plants [Citation5].
In addition to polyphenols, it is reported that alkaloids, saponins and terpenoids contain antiviral properties. Based on a literature review, curcumin showed antiviral properties against influenza, chikungunya and zika viruses and showed promising results in the design of antiviral drugs [Citation31].
RNA-dependent RNA polymerase, an essential part of the virus’s replication machinery that allows it to create several copies of its RNA genome, is another possible therapeutic target for SARS-CoV-2. Since human polymerases lack a counterpart with a similar sequence or structural similarity to coronavirus RdRp, developing RdRp inhibitors may offer a therapeutic option without running the danger of interfering with human polymerases [Citation52]. This study looked into the influence of polyphenols, among which is hesperidin, which is said to have therapeutic effects on various diseases and was able to inhibit the catalytic pocket of RdRp. The binding energy of hesperidin resulted in −8.8 kcal/mol and hydrogen bonds with Tyr619, Asp618, Lys798, Ser795, Met794, Pro793, Asp164, Val166, Pro620, Lys621, Asp623, Arg555 and Tyr455 [Citation52].
Hesperidin binds close to the RdRp active site, where Thr556, Lys621, Arg624 and Ser628 form H-bonds, while for Phe793 it displays interactions albeit not interacting with the putative active site. The binding energy of hesperidin was reportedly −9.53 kcal/mol, and its dissociation constant was 0.1 mmol/L [Citation50]. In our study, we found that hesperidin displayed a slightly lower binding energy of −7.5 kcal/mol compared to the previous findings. However, similar to previous research, we observed significant interactions between hesperidin and key amino acids within the catalytic pocket of RdRp, including Tyr619, Asp760 and Asp761, which are important for the enzyme activity.
Experimental validation has been performed on quercetin, a hydrophobic citron-yellow crystal generated from plants, to assess its features and biological properties. One of the most common flavonoid compounds is quercetin. The presence of five hydroxyl groups with electron-donating activity at positions 3, 5, 7, 30 and 40 is what makes quercetin unique. Antioxidant, anti-inflammatory, antiviral, anticarcinogenic, cardioprotective, psychostimulant and neuroprotective characteristics are just a few of the biological benefits that quercetin has [Citation22].
SARS-CoV-2 shows similarity to the SARS-CoV virus, for which quercetin is a good inhibitory agent. Due to its inhibitory effects on many viral life cycle stages, quercetin could be used in SARS-CoV-2 treatment. In particular, quercetin can change the expression of 30% of the genes encoding SARS-CoV-2 protein targets in human cells, potentially affecting the functions of 85% of these proteins [Citation22].
The highest docking score (in kcal/mol) for the QueO- molecule is −7.62 for the BNTP binding pocket. The QueO- molecule interacts with His439, Ser549, Lys551, Arg555, Ser814, His816 and Arg836 residues in the BNTP pocket. In contrast, the QueO- molecule projected higher docking scores at the BRNA binding pocket (7.69 kcal/mol). The molecular docking primarily identified interactions between the ligands and Asn496, Asn497, Lys500, Arg 569, Gln573, Lys577 and Tyr689 as the primary contacts [Citation44]. The coronavirus RdRp complex has the ability to remove mutagenic nucleotides that were mistakenly integrated into the viral RNA, resulting in resistance to nucleotide analog drugs. As a result, non-nucleoside inhibitors may possess more advantageous modes of action that prevent the emergence of resistance [Citation44]. [Citation61] conducted a study on the antiviral properties of 4068 substances, including 2924 compounds from the ZINC Drug Database, 1066 natural substances and 78 previously known antiviral drugs. They evaluated their ability to bind to SARS-CoV-2 proteins and found that hesperidin was the most effective in binding to the spike glycoprotein. Upon superimposing the interaction between the ACE2 receptor and the binding domain of the virus, it was discovered that hesperidin overlapped with the ACE2 interface, suggesting that it may interfere with the interaction between ACE2 and the virus binding domain. Hesperidin has been found to bind with the spike protein with a binding energy of −8.99 kcal/mol [Citation11]. The docked structure was stabilized by two hydrogen bonds, with one between the Phe457 of the spike protein and the O7 atom of hesperidin (bond length of 2.618 Å) and another between the H atom of hesperidin and the O atom of Glu455 of the spike protein fragment (distance of 2.067 Å). Molecular dynamics simulation and analysis of the quantitative structure–activity relationship have revealed that hesperidin demonstrated its potential for antiviral activity through the destabilization of the binding of the spike protein to the human host ACE2 receptor. The modulation of hesperidin on the ACE2 protein has attempted to prevent its interaction with the spike protein [Citation11]. In comparison to the previous studies, our structural analysis revealed that hesperidin formed additional hydrogen bonds with key residues involved in the binding of the spike glycoprotein to the ACE2 receptor, including Gln498 and Tyr505. These findings further support the potential of hesperidin in disrupting the interaction between ACE2 and the spike protein, suggesting its promising antiviral activity.
A study found that fisetin and quercetin had the lowest and identical binding energy at −8.5 kcal/mol and also showed similar binding preferences for the S2 domain of the spike protein [Citation46]. Although they both favored the S2 domain, the additional 5-OH group on quercetin’s chromone ring affected its hydrogen bonding interactions compared to fisetin, as seen in their interactions with different residues of the S2 domain. Fisetin formed hydrogen bonds with the residues of Ser730, Thr778 and His1058 and also showed hydrophobic interactions with Ile870, Pro880 and Thr732 residues of the S2 domain of the spike protein. Molecular dynamics simulations showed that kaempferol, quercetin and fisetin bind to the hACE2-S-protein complex near the hACE2 and S-protein binding interface. These results have been confirmed by other analyses, such as the free energy estimation of binding through MM/PBSA and the free energy landscape [Citation46]. Fisetin has a better molecular docking score and MM/GBSA binding energy compared to nafamostat, a known inhibitor of the host protease TMPRSS2 [Citation40]. The MM/GBSA free energy of fisetin and nafamostat, determined after docking, are −42.78 kcal/mol and −21.11 kcal/mol, respectively [Citation40]. The study observed that fisetin formed strong hydrogen bonds with several key residues in the catalytic domain of TMPRSS2, including Val25, His41 and Ser186. Additionally, the post-simulation results for 1000 snapshots showed that fisetin had a better overall interaction with TMPRSS2 than nafamostat, with an MM/GBSA binding energy of −51.87 ± 4.3 kcal/mol and −48.23 ± 4.39 kcal/mol, respectively. These results suggest that fisetin may have potential as a lead compound in the development of drugs against the host protease TMPRSS2, and therefore could be a promising candidate in managing COVID-19 [Citation40]. In line with the previous findings, our analysis revealed that fisetin exhibited favorable binding energy (–6.5 kcal/mol) and formed significant hydrogen bonds with key residues Gln498 and Tyr505, which supports the potential of fisetin as a promising candidate.
The NendoU protein actively participates in viral replication and is found in coronaviruses, arteriviruses and toroviruses. In the report by [Citation31], hydroxychloroquine was used as a positive control in phytochemical testing for binding to nsp15. Luteolin was used in this work, which formed five hydrogen bonds with the nsp15 molecule with an affinity of −7.2 kcal/mol in interaction with Pro344, Leu346, Lys290, Gly248, and His250. Then scopoletin, which interacted with Val292, Cys293, His250, His235 and Thr341, also formed five hydrogen bonds and had an affinity of −6.1 kcal/mol [Citation31].
When compared to our results, in the case of hydroxychloroquine, the binding affinity with NendoU is −5.8 kcal/mol, for scopoletin it is −6.0 kcal/mol, and for luteolin it is −7.3 kcal/mol (interacting with: His235, Gln245, Thr341 and Leu346) [Citation31]. Luteolin as well as quercetin have been mentioned as potential substances for inhibition of PLpro [Citation15].
To consider how the reported substances could be potentially used in the future development or design of drugs, after additional studies and checks for effectiveness, it is interesting that scientists have paid attention to testing bio-essential oils against SARS-CoV-2. A study has reported the effectiveness of bio-essential oils against Mpro as well as against Nsp15/NendoU (PDB: 6VWW, 6W01 and 6W02) [Citation49]. Assessment of absorption, distribution, metabolism and excretion (ADME) are some of the main characteristics relevant in drug design [Citation8]. When compared to Mpro inhibitors, PLpro inhibitors are still in the unfolding phase, whereas the inhibitors for the main protease have been the main focus of research [Citation38]. By using in silico methods, bioinformatics enables detailed analysis without the need for exposing living organisms to the risks associated with infections like COVID-19. This makes it an effective tool for exploring potential drugs or treatments, including vaccines [Citation41].
Conclusions
The results of the in silico analysis of the structural interactions between 12 phytochemicals and target proteins of SARS-CoV-2 indicated that hesperidin had the highest binding affinity for proteins of interest, including the main protease, papain-like protease, RNA-dependent RNA polymerase, spike glycoprotein and uridylate-specific endoribonuclease. By forming hydrogen bonds with significant residues of the target proteins, hesperidin demonstrated its theoretical potential as a therapeutic candidate against SARS-CoV-2 and emphasized the need for further research into its potential as an antiviral agent. The analysis of the concentrations of active compounds in the studied plant extracts revealed that Chrysosplenium cinerariifolium, Daphne blagayana, Iris reichenbachii and Iris pumila exhibit the greatest abundance of hesperidin. More studies are required, including in vitro and in vivo evaluations, as well as clinical trials, to substantiate the possible therapeutic applications of these plants.
Authors’ contributions
Conceptualization and design: KB, TC; supervision: KB, NP; methods: AK, DZO, FC, TC; data collection and/or processing: AK, DZO, FC, TC; analysis and/or interpretation: AK, DZO, FC, TC; manuscript writing: AK, TC; critical review: KB, NP. All authors read and approved the final manuscript.
Supplemental Material
Download PDF (69.9 KB)Acknowledgements
We would like to thank Prof. Dr. Faruk Bogunic for collecting the plant samples, Dr. Anesa Ahatovic Hajro for preparing the extracts and Dr. Mirza Dedic for performing the HPLC analysis.
Disclosure statement
The authors report no conflict of interest.
Data availability
The datasets generated and/or analysed during the current study are available in the RCSB Protein Data Bank [https://www.rcsb.org/] and PubChem database repository [https://pubchem.ncbi.nlm.nih.gov/].
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Abdizadeh R, Hadizadeh F, Abdizadeh T. In silico analysis and identification of antiviral coumarin derivatives against 3-chymotrypsin-like main protease of the novel coronavirus SARS-CoV-2. Mol Divers. 2022;26(2):1–15.
- Adel A, Elnaggar MS, Albohy A, et al. Evaluation of antiviral activity of carica papaya leaves against SARS-CoV-2 assisted by metabolomic profiling. RSC Adv. 2022;12(51):32844–32852.
- Adem Ş, Eyupoglu V, Ibrahim IM, et al. Multidimensional in silico strategy for identification of natural polyphenols-based SARS-CoV-2 main protease (mpro) inhibitors to unveil a hope against COVID-19. Comput Biol Med. 2022;145:105452.
- Agrawal PK, Agrawal C, Blunden G. Pharmacological significance of hesperidin and hesperetin, two citrus flavonoids, as promising antiviral compounds for prophylaxis against and combating COVID-19. Nat Prod Commun. 2021;16(10):1934578X2110425.
- Aini NS, Kharisma VD, Widyananda MH, et al. Bioactive compounds from purslane (portulaca oleracea L.) and star anise (illicium verum hook) as SARS-CoV-2 antiviral agent via dual inhibitor mechanism: in silico approach. Pharmacogn J. 2022;14(4):352–357.
- Al-Kuraishy HM, Al-Gareeb AI, Kaushik A, et al. Ginkgo biloba in the management of the COVID-19 severity. Arch Pharm (Weinheim). 2022;355(10):e2200188.
- Altu NS, Budiman C, Razali R, et al. Technical data of In silico analysis of the interaction of dietary flavonoid compounds against spike-glycoprotein and proteases of SARS-CoV-2. Data. 2022;7(11):144.
- Amin SA, Banerjee S, Ghosh K, et al. Protease targeted COVID-19 drug discovery and its challenges: insight into viral main protease (mpro) and papain-like protease (PLpro) inhibitors. Bioorg Med Chem. 2021;29:115860.
- Aranda J, Wieczór M, Terrazas M, et al. Mechanism of reaction of RNA-dependent RNA polymerase from SARS-CoV-2. Chem Catal. 2022;2(5):1084–1099.
- Bachar SC, Mazumder K, Bachar R, et al. A review of medicinal plants with antiviral activity available in Bangladesh and mechanistic insight into their bioactive metabolites on SARS-CoV-2, HIV and HBV. Front Pharmacol. 2021;12:732891.
- Basu A, Sarkar A, Maulik U. Molecular docking study of potential phytochemicals and their effects on the complex of SARS-CoV2 spike protein and human ACE2. Sci Rep. 2020;10(1):17699.
- Begum F, Srivastava AK, Ray U. Repurposing nonnucleoside antivirals against SARS-CoV2 NSP12 (RNA dependent RNA polymerase): in silico-molecular insight. Biochem Biophys Res Commun. 2021;571:26–31.
- Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242.
- Cai Y, Zhang J, Xiao T, et al. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020;369(6511):1586–1592.
- Chen TH, Tsai MJ, Chang CS, et al. The exploration of phytocompounds theoretically combats SARS-CoV-2 pandemic against virus entry, viral replication and immune evasion. J Infect Public Health. 2023;16(1):42–54.
- Cho CC, Li SG, Lalonde TJ, et al. Drug repurposing for the SARS-CoV-2 papain-Like protease. ChemMedChem. 2022;17(1):e202100455.
- Corbo T, Miralem M, Kalajdzic A, et al. In silico prediction of the inhibitory effect of phytochemical components extracted from knautia sarajevensis on the main protease of SARS-CoV-2 virus. GenApp. 2022;6(2):31–40.
- Das S, Sarmah S, Lyndem S, et al. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn. 2021;39(9):3347–3357.
- Farouk AE, Baig MH, Khan MI, et al. Screening of inhibitors against SARS-CoV-2 spike protein and their capability to block the viral entry mechanism: a viroinformatics study. Saudi J Biol Sci. 2021;28(6):3262–3269.
- Ferreira JC, Fadl S, Villanueva AJ, et al. Catalytic dyad residues His41 and Cys145 impact the catalytic activity and overall conformational fold of the main SARS-CoV-2 protease 3-chymotrypsin-like protease. Front Chem. 2021;9:692168.
- Frazier MN, Dillard LB, Krahn JM, et al. Characterization of SARS2 Nsp15 nuclease activity reveals it’s mad about U. Nucleic Acids Res. 2021;49(17):10136–10149.
- Gasmi A, Mujawdiya PK, Lysiuk R, et al. Quercetin in the prevention and treatment of coronavirus infections: a focus on SARS-CoV-2. Pharmaceuticals (Basel, Switzerland). 2022;15(9):1049.
- Hognon C, Marazzi M, García-Iriepa C. Atomistic-level description of the covalent inhibition of SARS-CoV-2 papain-like protease. IJMS. 2022;23(10):5855.
- Hu B, Guo H, Zhou P, et al. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021;19(3):141–154.
- Ibrahim TM, Ismail MI, Bauer MR, et al. Supporting SARS-CoV-2 papain-like protease drug discovery: in silico methods and benchmarking. Front Chem. 2020;8:592289.
- Jendele L, Krivak R, Skoda P, et al. PrankWeb: a web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019;47(W1):W345–W349.
- Jiang H, Yang P, Zhang J. Potential inhibitors targeting papain-Like protease of SARS-CoV-2: two birds with one stone. Front Chem. 2022;10:822785.
- Kaul R, Paul P, Kumar S, et al. Promising antiviral activities of natural flavonoids against SARS-CoV-2 targets: systematic review. IJMS. 2021;22(20):11069.
- Kim S, Chen J, Cheng T, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49(D1):D1388–D1395.
- Kim Y, Wower J, Maltseva N, et al. Tipiracil binds to uridine site and inhibits Nsp15 endoribonuclease NendoU from SARS-CoV-2. Commun Biol. 2021;4(1):193.
- Kumar S, Kashyap P, Chowdhury S, et al. Identification of phytochemicals as potential therapeutic agents that binds to Nsp15 protein target of coronavirus (SARS-CoV-2) that are capable of inhibiting virus replication. Phytomed. 2021;85:153317.
- Kumar S, Paul P, Yadav P, et al. A multi-targeted approach to identify potential flavonoids against three targets in the SARS-CoV-2 life cycle. Comput Biol Med. 2022;142:105231.
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786.
- Lei J, Kusov Y, Hilgenfeld R. Nsp3 of coronaviruses: structures and functions of a large multi-domain protein. Antiviral Res. 2018;149:58–74.
- Li D, Luan J, Zhang L. Molecular docking of potential SARS-CoV-2 papain-like protease inhibitors. Biochem Biophys Res Commun. 2021;538:72–79.
- Liang JJ, Pitsillou E, Ververis K, et al. Investigation of small molecule inhibitors of the SARS-CoV-2 papain-like protease by all-atom microsecond modelling, PELE monte carlo simulations, and in vitro activity inhibition. Chem Phys Lett. 2022;788:139294.
- Liu J, Zhang L, Gao J, et al. Discovery of genistein derivatives as potential SARS-CoV-2 main protease inhibitors by virtual screening, molecular dynamics simulations and ADMET analysis. Front Pharmacol. 2022;13:961154.
- Ma C, Wang J. Validation and invalidation of SARS-CoV-2 papain-like protease inhibitors. ACS Pharmacol Transl Sci. 2022;5(2):102–109.
- Matondo A, Kilembe JT, Ngoyi EM, et al. Oleanolic acid, ursolic acid and apigenin from ocimum basilicum as potential inhibitors of the SARS-CoV-2 main protease: a molecular docking study. IJPR. 2021;6(2):1–16.
- Mishra A, Kaur U, Singh A. Fisetin 8-C-glucoside as entry inhibitor in SARS CoV-2 infection: molecular modelling study. J Biomol Struct Dyn. 2022;40(11):5128–5137.
- Moradi M, Golmohammadi R, Najafi A, et al. In silico analysis of inhibiting papain-like protease from SARS-CoV-2 by using plant-derived peptides. Int J Pept Res Ther. 2022;28(1):24.
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791.
- Mouffouk C, Mouffouk S, Mouffouk S, et al. Flavonols as potential antiviral drugs targeting SARS-CoV-2 proteases (3CLpro and PLpro), spike protein, RNA-dependent RNA polymerase (RdRp) and angiotensin-converting enzyme II receptor (ACE2). Eur J Pharmacol. 2021;891:173759.
- Munafò F, Donati E, Brindani N, et al. Quercetin and luteolin are single-digit micromolar inhibitors of the SARS-CoV-2 RNA-dependent RNA polymerase. Sci Rep. 2022;12(1):10571.
- O’Boyle NM, Banck M, James CA, et al. Open babel: an open chemical toolbox. J Cheminform. 2011;3(1):33.
- Pandey P, Rane JS, Chatterjee A, et al. Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: an in silico study for drug development. J Biomol Struct Dyn. 2021;39(16):6306–6316.
- Pendyala B, Patras A. In silico screening of cyanobacterial and food bioactive compounds to predict potential inhibitors of COVID-19 main protease (mpro), papain-like protease (PLpro) and RNA-dependent RNA polymerase (RdRp). ChemRxiv. 2022. This content is a preprint and has not been peer-reviewed.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612.
- Ramesh V, Kulkarni SA, Velusamy P, et al. Current update of phytotherapeutic agents in the treatment of COVID-19: in-silico based virtual screening approach for the development of antiviral drug. Front Biosci (Landmark Ed). 2022;27(4):123.
- Rehman MFU, Akhter S, Batool AI, et al. Effectiveness of natural antioxidants against SARS-CoV-2? Insights from the in-silico world. Antibiotics (Basel, Switzerland). 2021;10(8):1011.
- Shahhamzehei N, Abdelfatah S, Efferth T. In silico and in vitro identification of pan-coronaviral main protease inhibitors from a large natural product library. Pharmaceuticals (Basel, Switzerland). 2022;15(3):308.
- Singh S, Sk MF, Sonawane A, et al. Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2 via RNA-dependent RNA polymerase (RdRp) inhibition: an in-silico analysis. J Biomol Struct Dyn. 2021;39(16):6249–6264.
- Srinivasan V, Brognaro H, Prabhu PR, et al. Antiviral activity of natural phenolic compounds in complex at an allosteric site of SARS-CoV-2 papain-like protease. Commun Biol. 2022;5(1):805.
- Tejera E, Pérez-Castillo Y, Toscano G, et al. Computational modeling predicts potential effects of the herbal infusion "horchata" against COVID-19. Food Chem. 2022;366:130589.
- The PyMOL Molecular Graphics System. Version 2.5.4. Schrödinger, LLC; 2022.
- Trott O, Olson AJ. AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461.
- Ullrich S, Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett. 2020;30(17):127377.
- Veeramachaneni GK, Thunuguntla V, Bobbillapati J, et al. Structural and simulation analysis of hotspot residues interactions of SARS-CoV 2 with human ACE2 receptor. J Biomol Struct Dyn. 2021;39(11):4015–4025.
- Walls AC, Park YJ, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292.e6.
- Wang G, Zhu W. Molecular docking for drug discovery and development: a widely used approach but far from perfect. Future Med Chem. 2016;8(14):1707–1710.
- Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10(5):766–788.