
Abstract
The bacterial strain LZN01 is a potential biocontrol agent that produces myriocin and inhibits the growth of Fusarium oxysporum f. sp. niveum (Fon). To characterize strain LZN01, the Illumina HiSeq + PacBio sequencing platform was used to sequence its genome. Unicycler software was used to perform genome assembly and antiSMASH to predict gene clusters responsible for secondary metabolite biosynthesis. The genome comprises 3973236 bp and has a 46.52% G + C content. Genome annotation predicts 4031 coding DNA sequences (CDSs), 27 rRNAs and 86 tRNAs. Strain LZN01 was identified as Bacillus velezensis based on phylogenomic analysis. We identified 14 gene clusters associated with the synthesis of antimicrobial secondary metabolites, including surfactin, fengycin, bacillibactin and bacilysin, among others. There are also genes associated with induced systemic resistance and plant growth promotion, such as biofilm formation, swarming motility, root colonisation, phosphate solubilisation, phytohormone production, acetoin and 2,3-butanediol synthesis. Many genes are associated with plant–bacteria interactions, including pectinase and galactosidase genes. We inferred that the genes epsN, capD, dagK, glk and manA are associated with myriocin biosynthesis. Overall, our data provide a valuable reference for further studies of the biocontrol mechanisms of strain LZN01 as a promising agent for application in agricultural practice.
Introduction
Fusarium wilt caused by Fusarium oxysporum f. sp. niveum (Fon) is a soil-borne disease that limits watermelon production [Citation1]. The application of beneficial microorganisms is an effective method for controlling Fusarium wilt in watermelon production [Citation2]. A number of Bacillus species have strong antimicrobial activities and are effective biocontrol agents [Citation3]. It has been reported that Bacillus spp. directly kills plant pathogens by producing antibiotic compounds, inhibits the growth of phytopathogens by competing for nutrients and niches, and induces systemic resistance against different phytopathogens [Citation4, Citation5]. For instance, hentriacontane and 2,4-di-tert-butylphenol, which are produced by B. cereus MH778713, promote tomato seedling growth and show antifungal activity against F. oxysporum [Citation6]. Bacillus spp. inhibits the growth of phytopathogens, and this can be attributed to the production of non-ribosomally synthesised lipopeptides, ribosomally synthesised peptides, siderophore bacillibactin and polyketides [Citation7, Citation8].
Myriocin was first described in 1972 [Citation9]. Myriocin, also known as antibiotic ISP-1 and hermozymocidin, is a sphingosine analogue and an inhibitor of serine palmitoyl transferase (SPT) [Citation10, Citation11]. The structure of SPT inhibitors is similar to that of sphingosine and they have a long hydrophobic tail with a polar head group [Citation12]. Myriocin is a fungal metabolite originally isolated from Myriococcum albomyces, Melanconis flavovirens, Isaria sinclairii and Paecilomyces variotii ATCC 74097 [Citation13]. The strain LZN01, previously identified as Bacillus amyloliquefaciens, produces myriocin [Citation14], which inhibited the growth of Fon via membrane damage and intracellular targets [Citation15, Citation16]. However, the genes or gene clusters encoding myriocin have not been explored in the LZN01 genome. Multiple studies have reported that myriocin is a sphingosine analogue [Citation9, Citation10, Citation17], and the biosynthetic pathway of sphingosine has also been widely reported [Citation12, Citation18–20]. Therefore, it is reasonable to infer that the possible biosynthetic pathway of sphingosine in strain LZN01 is similar to that of myriocin. Understanding the genome of strain LZN01 can provide a valuable reference for the study of the biocontrol potential of antifungal agents active against Fon. Therefore, to provide genomic insights into its antifungal effects, we conducted whole-genome sequencing of strain LZN01 and carried out comparative genome analysis of Bacillus velezensis FZB42, B. velezensis SQR9, B. velezensis LM2303 and B. velezensis LS69 to study the distribution and relatedness of genes associated with secondary metabolite biosynthesis.
Materials and methods
Strain and culture conditions
Bacillus velezensis strain LZN01 was provided by the Laboratory of Microbial Ecology, Department of Life Sciences, Agriculture and Forestry, Qiqihar University in China. The culture of strain LZN01 was performed according to the methods described by Xu et al. [Citation14].
DNA extraction, sequencing and assembly
Genomic DNA of the strain LZN01 was extracted using a Wizard® Genomic DNA Purification Kit (Promega, USA) according to the manufacturer’s instructions. The purified genomic DNA of strain LZN01 was quantified using a TBS-380 fluorometer (Turner BioSystems Inc., Sunnyvale, CA). The DNA extracted was sequenced using the Illumina HiSeq and PacBio RS II Single Molecule Real Time (SMRT) platform by Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China). Sequencing yielded 423052 reads, corresponding to 1453.6 × sequence depth. The raw reads were quality filtered by Fastp v0.23.0 (https://github.com/OpenGene/fastp). The filtered reads were assembled by Unicycler v0.4.8 (https://github.com/rrwick/Unicycler), in which short read sequence alignment and base quality correction were performed by Pilon v1.22 [Citation21].
Taxonomic classification
To accurately classify strain LZN01, its phylogenetic tree was built via Type Strain Genome Server (https://tygs.dsmz.de/) based on the MASH algorithm (https://github.com/marbl/mash) and the Genome BLAST Distance Phylogeny approach (GBDP) (http://ggdc.dsmz.de).
Genome annotation
The coding DNA sequence of the genome of strain LZN01 was predicted by Glimmer v3.02 (http://ccb.jhu.edu/software/glimmer/index.shtml); repeat sequence was evaluated by Tandem Repeats Finder 4.07b (http://tandem.bu.edu/trf/trf.html). Barrnap v.0.8 (https://github.com/tseemann/barrnap) and tRNAscan-SE v.2.0 (http://trna.ucsc.edu/software/) were used to predict rRNA and tRNA genes, respectively. Functional annotation and assignment of predicted genes were performed using BLAST and HMMER against five databases, including the nonredundant (Nr) (ftp://ftp.ncbi.nlm.nih.gov/blast/db/), Pfam (http://pfam.xfam.org/), Clusters of Orthologous Groups (COGs) (http://www.ncbi.nlm.nih.gov/COG/), Gene Ontology (GO) (http://www.geneontology.org/) and Kyoto Encyclopedia Genes and Genomes (KEGG) databases (http://www.genome.jp/kegg/). The genome map of strain LZN01 was visualized using Circos v.0.64 [Citation22]. Virulence factor analysis was performed using the database VFDB (http://www.mgc.ac.cn/VFs/).
Assessment of secondary metabolic potentials
To assess the potential for secondary metabolite biosynthesis in strain LZN01, the number of putative biosynthetic gene clusters was predicted by antiSMASH 6.6.0rcl (https://dl.secondarymetabolites.org/releases/6.0.0/) and BLAST.
Comparative genomic analysis of B. velezensis
The genomic sequence of B. velezensis LZN01 was compared with genomic sequences of other B. velezensis strains with anti-microbial properties, including B. velezensis FZB42 (accession no. CP000560), B. velezensis SQR9 (accession no. CP006890.1), B. velezensis LS69 (accession no. CP015911) and B. velezensis LM2303 (accession no. CP018152). The genome sequence and annotation information of these four strains were down-loaded from GenBank database (https://www.ncbi.nlm.nih.gov/genbank/). Comparative genomic analyses for the five strains were performed using OrthoMCL v2.0.9 (https://orthomcl.org/common/ downloads/software/v2.0/orthomclSoftware), and core and species-specific gene families were grouped. Pan-genome analysis was performed using PGAP v1.2.1 (https://jaist.dl.sourceforge.net/project/pgap/PGAP).
Data deposition
The complete genome sequence of Bacillus velezensis LZN01 has been deposited at DDJB/ENA/GenBank under accession no. CP081488.
Results
Phylogenetic analysis and genome features of strain LZN01
The phylogenetic analysis indicated that strain LZN01 was most closely related to B. velezensis FZB42 (). Therefore, strain LZN01 was finally identified as B. velezensis. The whole genome of this strain consists of one circular chromosome of 3973236 bp with a GC content of 46.52%. The genome was predicted to contain 4031 coding DNA sequences (CDSs), 27 rRNA genes and 86 tRNA genes. The identified genes were classified into 3013, 2666 and 2163 categories based on COG, GO and KEGG designation, respectively. Fourteen putative secondary metabolite biosynthetic gene clusters and 6 genomic islands were identified in the genome ().
Figure 1. Phylogenetic tree of strain LZN01 based on whole-genome alignments. The genome sequence data were uploaded to type (strain) Genome Server (TYGS) (https://tygs.dsmz.de) for a whole-genome-based taxonomic analysis. The tree was constructed via FastME 2.1.6.1 from GBDP distances calculated from genome sequences. The numbers above branches were inferred pseudo-bootstrap support values > 60% from 100 replications. ‘T’ =typical strains.
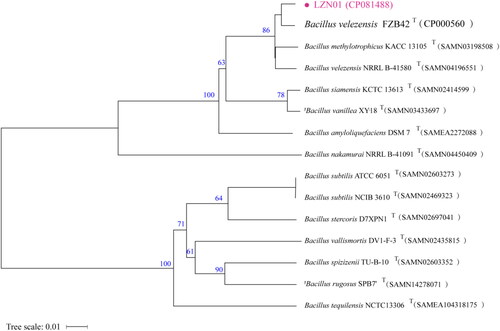
Table 1. General genome features of Bacillus velezensis LZN01 and other Bacillus spp.
Functional annotation of strain LZN01
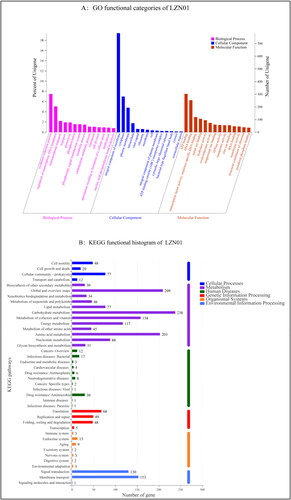
The genome map of strain LZN01 visualized using Circos v.0.64 is presented in . COG analysis revealed that 32.33% of the predicted CDSs are involved in metabolism, 17.42% in information storage and processing, and 26.62% in cellular processes and signaling. The number of genes associated with amino acid transport and metabolism (E), carbohydrate transport and metabolism (G), inorganic ion transport and metabolism (P), energy production and conversion (C), coenzyme transport and metabolism (H), lipid transport and metabolism (I), nucleotide transport and metabolism (F), and secondary metabolites biosynthesis, transport and catabolism (Q) was greater than that of the other function-related genes. GO functional annotation showed that among 2666 genes, approximately 2083 genes are associated with molecular function, 1931 genes in biological process and 1315 ones in cellular component (). In the KEGG pathway annotation, there were more proteins associated with metabolism (1276) than other proteins. Among them, 238 proteins are associated with carbohydrate metabolism, 209 with global and overview maps, 203 with amino acid metabolism, 158 with metabolism of cofactors and vitamins, 170 with genetic information processing, 157 with cellular processes and 117 with energy metabolism ().
Figure 2. Genome map of B. velezensis LZN01. Rings from outer to inner: genome size, forward-strand genes, reverse-strand genes, nomenclature and locations of predicted secondary metabolite gene clusters excluding microcins, GC content and GC skew. Visualisation of genome map of B. velezensis LZN01 via Circos v.0.64 [Citation22].
![Figure 2. Genome map of B. velezensis LZN01. Rings from outer to inner: genome size, forward-strand genes, reverse-strand genes, nomenclature and locations of predicted secondary metabolite gene clusters excluding microcins, GC content and GC skew. Visualisation of genome map of B. velezensis LZN01 via Circos v.0.64 [Citation22].](/cms/asset/97bdb0da-6303-42eb-a760-9a7f262a36ef/tbeq_a_2227731_f0002_c.jpg)
Figure 3. Genome functional annotation of strain LZN01. (A) GO functional annotation of protein-encoding genes in strain LZN01. The X-axis represents function classes, and the Y-axis represents the percent and number of predicted proteins in strain LZN01. (B) KEGG functional annotation of protein-encoding genes in strain LZN01. The X-axis represents the number of predicted proteins, and the Y-axis represents function classes.
Genes associated with plant growth promotion and induction of immunity
Genes involved in sugar utilization and genes associated with cell-wall-degrading enzymes were found in the LZN01 genome (), for example, xynBCD encoding xylanase, yaaH encoding chitinase, csn encoding chitosanase, pel encoding pectinase, lacEFR genes encoding galactosidase, and nagZ, bglA, lacG, abfA, gmuG and pgm encoding glucanase. Additionally, a series of genes was predicted to be involved in root colonization and biofilm formation, including sacB, xerC, xerD, spo0A, sinR, sinI, abrB, resE, lytS and ycbA. The LZN01 genome contained 15 genes of the exo-polysaccharide (EPS) operon epsA-O. Genes coding for factors regulating chemotaxis and motility, i.e. motA and motB; genes responsible for quorum sensing, comA, comP, comQ and comX; and those associated with flagellar hook, i.e. flgK and fliD, were also identified in the LZN01 genome. The LZN01 genome harboured multiple genes encoding enzymes associated with responses to environmental stress, i.e. thiol peroxidase (tpx), alkyl hydroperoxide reductases (ahpC, ahpF) and gamma-glutamyl transpeptidase (ggt), heat-shock proteins (dnaJ, dnaK) and a cold shock protein (cspA). Genes involved in polyamine synthesis, such as speB encoding agmatinase, speA encoding arginine decarboxylase, speD encoding S-adenosyl-methionine decarboxylase and speE encoding spermidine synthase, and metK encoding S-adenosyl-methionine synthase, were detected in the LZN01 genome. Moreover, the potential to produce alkaline phosphatase (phoA) and deferrochelatase (efeB) were excavated in the LZN01 genome. Genes encoding nitrate reductase, nitrite reductase and associated transporters (nasA, nasI, narI, narG, narH, narJ and nirD) were also identified in the genome of LZN01. The trpA-E genes responsible for biosynthesis of L-tryptophan, which acts as a chief precursor for indole-3-acetic acid (IAA), were also found. The gabD and gabT genes are involved in the synthesis of γ-aminobutyric acid (GABA). Genes responsible for the synthesis of acetoin and 2,3-butanediol (pfkA, ilvB, alsD and butB) were also mined in the genome of LZN01.
Table 2. Genes associated with plant-bacterial interactions identified in B. velezensis LZN01.
In contrast, genes associated with pathogenicity in Bacillus spp., including haemolysins (hblA, hblB, hblC and hblD), non-hemolytic enterotoxins (nheA, nheB and nheC), enterotoxin (bceT), cytotoxin (cytK) and cereulide (cesA, cesH, cesP, cesT, cesB, cesC and cesD), were not found in the LZN01 genome. This result suggested that strain LZN01 could be safe for application in agricultural practice.
Secondary metabolite cluster analysis
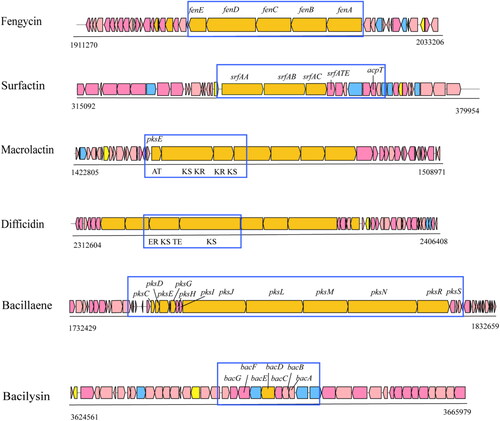
Of these, four gene clusters encode non-ribosomal peptides (bacillibactin, fengycin, surfactin and bacilysin), five enzymes from the polyketide synthase pathway, including four enzymes for type I polyketides of the AT-less type (macrolactin, bacillaene, butirosin and difficidin), one for type III polyketide synthesis, two ones responsible for terpene biosynthesis, and one encoding ribosomal peptides (lantipeptide) ().
Genome analysis showed a five-gene cluster (fenA-E) to be responsible for the synthesis of fengycin in B. velezensis LZN01 (). The gene clusters SrfAA, SrfAB, SrfAC and an external thioesterase-encoding gene SrfATE were found in strain LZN01, which are involved in the biosynthesis of surfactin. In addition, the acpT gene (encoding 4′-phosphopantetheinyl transferase), which is an essential enzyme for non-ribosomal lipopeptide synthesis and polyketide synthesis, was identified in the LZN01 genome. In the LZN01 genome, the gene cluster pdhA organizes trans-ATPKS for the synthesis of macrolactin. It was predicted that the gene cluster for macrolactin encodes two basic domains, acyltransferase (AT) and ketone synthase (KS), by antiSMASH. The gene cluster for difficidin in the LZN01 genome encoded enol reductase (ER) and thioesterase (TE). In LZN01, the biosynthesis of bacillaene was predicted based on the presence of a PKS-NRPS hybrid cluster consisting of 12 genes (pksC-E, pksG-J, pksL-N, pksRS). The LZN01 genome contains a pksE gene encoding malonyl-CoA: ACP transacylase, which is related to the biosynthesis of polyketones. The gene cluster bacABCDEFG is necessary for bacilysin synthesis. Furthermore, two unknown secondary metabolite synthesis gene clusters were predicted in the LZN01 genome, providing a basis for further exploration of the biocontrol potential and potential discovery of new antibacterial substances.
Figure 4. Secondary metabolite gene clusters with antibacterial metabolites in strain LZN01 predicted by antiSMASH 6.0.0rc1 (https://dl.secondarymetabolites.org/releases/6.0.0/). biosynthetic gene clusters are shown above each ORF.
Comparative genomic analysis
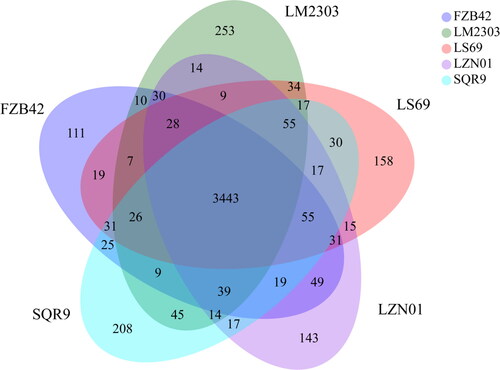
To identify specific genes in B. velezensis LZN01, its genome sequence was compared to four B. velezensis strains. The basic characteristics of the five B. velezensis genomes are shown in . The LZN01 strain had genome size, GC content, protein-encoding genes and number of RNA genes similar to the other B. velezensis strains. However, LZN01 had a larger number of genomic islands than the other B. velezensis strains, indicating that LZN01 possessed unique genetic characteristics. As shown in , the number of clusters of orthologous genes in strains LZN01, FZB42, SQR9, LS69 and LM2303 is 3978, 3932, 4050, 3975 and 4033, respectively. Pan-genome analysis of the five B. velezensis strains revealed that 3443 genes constitute the core genome. In addition, 49 genes were shared by only the LZN01 and FZB42 strains, indicating that LZN01 had higher similarity with FZB42. Furthermore, 143 unique genes were found in the LZN01 genome. These unique genes also conferred unique genetic features to strain LZN01.
Figure 5. Comparison of B. velezensis strain LZN01 genome sequences with the genome sequences of four other Bacillus velezensis strains (FZB42 (CP000560), SQR9 (CP006890.1), LS69 (CP015911) and LM2303 (CP018152)). the Venn diagram shows the number of shared and unique clusters of orthologous genes.
Putative biosynthetic pathway of myriocin
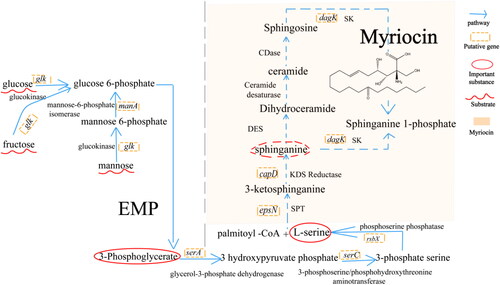
In the sphingosine pathway (), the first step in de novo sphingosine biosynthesis is the condensation of L-serine with palmitoyl-CoA, as catalysed by serine palmitoyltransferases (SPTs), to form 3-ketosphinganine (3-keto-dihydrosphingosine, 3-KDS) [Citation12]. SPTs are ubiquitous in eukaryotes and mammals, whereas all bacterial SPTs are members of the pyridoxal 5′-phosphate (PLP)–dependent α-oxoamine synthase family. The epsN gene, which encodes pyridoxal phosphate-dependent aminotransferase, was found in the LZN01 strain genome. The second enzyme in the committed pathway is 3-ketodihydrosphingosine reductase (3-KDSR), which catalyses the reduction of 3-KDS to sphinganine (2S,3R-dihydrosphingosine (DHS)). The capD gene, which encodes an SDR family oxidoreductase, is similar to 3-ketodihydrosphingosine reductase (KDSR) and was identified in the genome of strain LZN01. Sphingosine kinases (SKs) are ATP-dependent kinases of the diacylglycerol kinase (DAGK) family that phosphorylate the terminal hydroxyl group of sphingoid bases – either sphinganine (from de novo synthesis) or sphingosine (from ceramide breakdown) [Citation12]. The dagK gene, encoding diacylglycerol kinase, was detected in the strain LZN01 genome. Myriocin is similar to sphinganine. Serine palmitoyltransferases, 3-ketodihydrosphingosine reductase and sphingosine kinases are important enzymes for sphingosine biosynthesis. Therefore, we infer that genes epsN, capD and dagK may play important roles in myriocin biosynthesis. L-serine is a substrate for the biosynthesis of sphingosine. In addition, sphingosine synthesis may be involved in L-serine synthesis and the glycolytic pathway [Citation23].
Figure 6. Deduction of the biosynthetic pathway of myriocin in strain LZN01. The putative myriocin synthesis pathway (orange counterstain).
Discussion
In this study, genome sequence analysis of strain LZN01 was performed to discern the molecular cues responsible for the synthesis of potential biocontrol agents. B. subtilis, B. amyloliquefaciens and B. velezensis were difficult to differentiate, leading to several re-classifications; the taxonomic status of B. velezensis was formally established in 2016 [Citation24–26]. The strain LZN01 was previously identified as Bacillus amyloliquefaciens based on 16S rRNA gene sequences [Citation14]. A phylogenetic tree was constructed based on comparison with closely related genomes. The results showed that strain LZN01 was closely related to B. velezensis FZB42. Thus, based on phylogenomic analysis, strain LZN01 was taxonomically redesignated as Bacillus velezensis.
The colonization ability of strains is important for their application as biocontrol agents. Identification of genes related to EPS production (epsA-O), root colonization (sacB, xerC, xerD), biofilm formation (spo0A, sinR, sinI, etc.), chemotaxis and motility (motA, motB), and flagellar hook (flgk, fliD) indicated that strain LZN01 might efficiently colonize inside a plant or on the root surface [Citation27]. The LZN01 genome contains some genes (dnaJ, dnaK and cspA) encoding enzymes associated with response to environmental stress, which suggests the isolate’s potential ability to withstand extreme environments [Citation28].
Acetoin and 2,3-butanediol have been considered as signal molecules in the interrelationship between bacteria and plants, promoting plant growth and diminishing the infective ability of pathogens. The presence of phoA and efeB genes suggests the mineralization of organic phosphorous and utilization of iron by strain LZN01 [Citation29]. The presence of genes responsible for improving nutrient elements (efeB and phoA), secreting hormones (gatA, miaA, nos) and inducing resistance (pfkA, ilvB, alsD and butB) suggests that strain LZN01 possesses the ability to facilitate acquisition of nutrient elements, modulate phytohormones and induce resistance [Citation29, Citation30]. The gabD and gabT genes are involved in the synthesis of γ-aminobutyric acid (GABA), which can reduce the activity of herbivorous insects and the toxicity of bacterial and fungal pathogens [Citation31]. Genes involved in chitin and glucan degradation were presented in LZN01, which suggests that strain LZN01 has the potential to degrade the cell wall of phytopathogens. The chitosanase produced by B. circulas mh-k1 and B. cereus D-11 can inhibit the growth of pathogenic fungi [Citation32, Citation33]. It has been reported that the presence of genes related to spermidine synthesis (speE, speA, SpeB and speD) enhances isolate resistance to abiotic stresses and promotes plant bud differentiation through spermidine biosynthesis [Citation34].
The abundant and diverse secondary metabolites from Bacillus are considered to be important for exerting beneficial effects on plants, such as preventing disease and promoting growth. It has been reported that fengycin and surfactin have antifungal activity against filamentous fungi [Citation7]. Difficidin and macrolactin effectively suppress the growth of plant pathogens [Citation34–36]. Bacillaene, a novel inhibitor of prokaryotic protein synthesis, inhibits both gram-positive and gram-negative bacteria [Citation37].
The whole B. velezensis LS69 genome consists of over 8.5% sequences devoted to antimicrobial gene clusters, including gene clusters for fengycin, surfactin, and iturin A, among others [Citation38]. B. velezensis FZB42, a broad-spectrum antagonist isolated from the beet rhizosphere, carries 13 antimicrobial gene clusters accounting for approximately 10% of the whole-genome length [Citation39]. Remarkably, B. velezensis LZN01 harbours 14 antimicrobial gene clusters, accounting for 19.85% of its complete genome. It is apparent that the difference in isolates regarding biocontrol potential may be attributed to variations in biosynthetic gene clusters. Thirteen gene clusters were related to the synthesis of antimicrobial secondary metabolites in Bacillus velezensis WB [Citation40]. Fourteen gene clusters responsible for secondary metabolites (fengycin, surfactin, difficidin and macrolactin, etc.) in the LZN01 genome accounted for antagonizing phytopathogens, inducing systemic resistance, chelating iron ions and forming biofilms [Citation28, Citation41, Citation42], which indicates that strain LZN01 might be used as a biocontrol agent.
Myriocin is a sphingosine analogue with antifungal activity [Citation43]. In this study, we deduced the biosynthetic pathway of sphingosine, and the genes epsN, capD and dagK encode some crucial enzymes in the biosynthetic pathway. SPTs are ubiquitous in eukaryotes and mammals, whereas all bacterial SPTs are members of the pyridoxal 5′-phosphate (PLP)–dependent α-oxoamine synthase family, which catalyses Claisen-like condensation reactions between acyl-CoA substrates and amino acids [Citation12, Citation18, Citation44]. The short-chain dehydrogenase/reductase (SDR) family NAD(P)-dependent oxidoreductase is similar to 3-ketodihydrosphingosine reductase (KDSR), which catalyses reduction of 3-ketodihydrosphingosine (3-KDS) to dihydrosphingosine (DHS) [Citation45, Citation46]. Therefore, we speculated that these genes (epsN, capD, dagK) play crucial roles in myriocin biosynthesis. Glucose, mannose and fructose are substrates in the glycolytic pathway, which may affect myriocin biosynthesis. We confirmed that addition of D-mannose and D-fructose to LB medium can increase myriocin production from strain LZ01 (data not shown). It was reported that D-fructose, D-mannose, and D-glucose contribute to the chemical synthesis of myriocin [Citation47–49]. It was inferred that the genes glk (encoding glucokinase) and manA (encoding mannose-6-phosphate isomerase) () may affect myriocin biosynthesis, which needs to be confirmed in further experiments.
Conclusions
In this study, the complete genome sequence of strain LZN01 was reported and the microorganism was classified as B. velezensis based on the constructed phylogenetic tree. The B. velezensis LZN01 genome harboured genes associated with induced systemic resistance, plant growth promotion, and plant-bacterial interaction. Fourteen gene clusters associated with secondary metabolite biosynthesis were identified by genome mining. The biosynthetic pathway of myriocin in the strain LZN01 was preliminarily inferred. Our results provide insight into B. velezensis LZN01 as a new promising strain for biological control.
Data availability
All data generated during this study are included in this article.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Arnao MB, Hernandez-Ruiz J. Functions of melatonin in plants: a review. J Pineal Res. 2015;59(2):1–11. doi: 10.1111/jpi.12253.
- Everts KL, Himmelstein JC. Fusarium wilt of watermelon: towards sustainable management of a re-emerging plant disease. Crop Prot. 2015;73:93–99. doi: 10.1016/j.cropro.2015.02.019.
- Bhattacharyya PN, Jha DK. Plant growth-promoting rhizobacteria (PGPR): emergence in agriculture. World J Microbiol Biotechnol. 2012;28(4):1327–1350. doi: 10.1007/s11274-011-0979-9.
- Srivastava S, Bist V, Srivastava S, et al. Unraveling aspects of Bacillus amyloliquefaciens mediated enhanced production of rice under biotic stress of Rhizoctonia solani. Front Plant Sci. 2016;7:587. doi: 10.3389/fpls.2016.00587.
- Wu L, Wu HJ, Qiao J, et al. Novel routes for improving biocontrol activity of Bacillus based bioinoculants. Front Microbiol. 2015;6:1395. doi: 10.3389/fmicb.2015.01395.
- Ramirez V, Martinez J, Bustillos-Cristales MDR, et al. Bacillus cereus MH778713 elicits tomato plant protection against Fusarium oxysporum. J Appl Microbiol. 2022;132(1):470–482. doi: 10.1111/jam.15179.
- Rabbee MF, Ali MS, Choi J, et al. Bacillus velezensis: a valuable member of bioactive molecules within plant microbiomes. Molecules. 2019;24(6):1046. doi: 10.3390/molecules24061046.
- Belbahri L, Chenari Bouket A, Rekik I, et al. Comparative genomics of Bacillus amyloliquefaciens strains reveals a core genome with traits for habitat adaptation and a secondary metabolites rich accessory genome. Front Microbiol. 2017;8:1438. doi: 10.3389/fmicb.2017.01438.
- Kluepfel D, Bagli J, Baker H, et al. Myriocin, a new antifungal antibiotic from Myriococcum albomyces. J Antibiot (Tokyo). 1972;25(2):109–115. doi: 10.7164/antibiotics.25.109.
- Craveri R, Manachini PL, Aragozzini F. Thermozymocidin new antifungal antibiotic from a thermophilic eumycete. Experientia. 1972;28(7):867–868. doi: 10.1007/BF01923181.
- Momoi M, Tanoue D, Sun Y, et al. SLI1 (YGR212W) is a major gene conferring resistance to the sphingolipid biosynthesis inhibitor ISP-1, and encodes an ISP-1 N-acetyltransferase in yeast. Biochem J. 2004;381(Pt 1):321–328. doi: 10.1042/BJ20040108.
- Harrison PJ, Dunn TM, Campopiano DJ. Sphingolipid biosynthesis in man and microbes. Nat Prod Rep. 2018;35(9):921–954. doi: 10.1039/c8np00019k.
- Pereira CB, de Oliveira DM, Hughes AF, et al. Endophytic fungal compounds active against Cryptococcus neoformans and C. gattii. J Antibiot (Tokyo). 2015;68(7):436–444. doi: 10.1038/ja.2015.11.
- Xu W, Wang H, Lv Z, et al. Antifungal activity and functional components of cell-free supernatant from Bacillus amyloliquefaciens LZN01 inhibit Fusarium oxysporum f. sp. niveum growth. Biotechnol Biotec Eq. 2019;33(1):1042–1052. doi: 10.1080/13102818.2019.1637279.
- Wang H, Wang Z, Liu Z, et al. Membrane disruption of Fusarium oxysporum f. sp. niveum induced by myriocin from Bacillus amyloliquefaciens LZN01. Microb Biotechnol. 2021;14(2):517–534. doi: 10.1111/1751-7915.13659.
- Wang H, Wang Z, Xu W, et al. Comprehensive transcriptomic and proteomic analyses identify intracellular targets for myriocin to induce Fusarium oxysporum f. sp. niveum cell death. Microb Cell Fact. 2021;20(1):69. doi: 10.1186/s12934-021-01560-z.
- Chen JK, Lane WS, Schreiber SL. The identification of myriocin-binding proteins. Chem Biol. 1999;6(4):221–235. doi: 10.1016/S1074-5521(99)80038-6.
- Rocha FG, Moye ZD, Ottenberg G, et al. Porphyromonas gingivalis sphingolipid synthesis limits the host inflammatory response. J Dent Res. 2020;99(5):568–576. doi: 10.1177/0022034520908784.
- Avila-Garcia R, Valdes J, Jauregui-Wade JM, et al. The metabolic pathway of sphingolipids biosynthesis and signaling in Entamoeba histolytica. Biochem Biophys Res Commun. 2020;522(3):574–579. doi: 10.1016/j.bbrc.2019.11.116.
- Parashuraman S, D'Angelo G. Visualizing sphingolipid biosynthesis in cells. Chem Phys Lipids. 2019;218:103–111. doi: 10.1016/j.chemphyslip.2018.11.003.
- Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. doi: 10.1371/journal.pone.0112963.
- Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109.
- Murtas G, Marcone GL, Sacchi S, et al. L-serine synthesis via the phosphorylated pathway in humans. Cell Mol Life Sci. 2020;77(24):5131–5148. doi: 10.1007/s00018-020-03574-z.
- Wang LT, Lee FL, Tai CJ, et al. Bacillus velezensis is a later heterotypic synonym of Bacillus amyloliquefaciens. Int J Syst Evol Microbiol. 2008;58(Pt 3):671–675. doi: 10.1099/ijs.0.65191-0.
- Dunlap CA, Kim SJ, Kwon SW, et al. Phylogenomic analysis shows that Bacillus amyloliquefaciens subsp. plantarum is a later heterotypic synonym of Bacillus methylotrophicus. Int J Syst Evol Microbiol. 2015;65(7):2104–2109. doi: 10.1099/ijs.0.000226.
- Dunlap CA, Kim SJ, Kwon SW, et al. Bacillus velezensis is not a later heterotypic synonym of Bacillus amyloliquefaciens; Bacillus methylotrophicus, Bacillus amyloliquefaciens subsp. plantarum and ‘bacillus oryzicola’ are later heterotypic synonyms of Bacillus velezensis based on phylogenomics. Int J Syst Evol Microbiol. 2016;66(3):1212–1217. doi: 10.1099/ijsem.0.000858.
- Nanjani S, Soni R, Paul D, et al. Genome analysis uncovers the prolific antagonistic and plant growth-promoting potential of endophyte Bacillus velezensis K1. Gene. 2022;836:146671. doi: 10.1016/j.gene.2022.146671.
- Niazi A, Manzoor S, Asari S, et al. Genome analysis of Bacillus amyloliquefaciens subsp. plantarum UCMB5113: a rhizobacterium that improves plant growth and stress management. PLoS One. 2014;9(8):e104651. doi: 10.1371/journal.pone.0104651.
- Kour D, Rana KL, Kaur T, et al. Biodiversity, current developments and potential biotechnological applications of phosphorus-solubilizing and -mobilizing microbes: a review. Pedosphere. 2021;31(1):43–75. doi: 10.1016/S1002-0160(20)60057-1.
- Zhou L, Zhang T, Tang S, et al. Pan-genome analysis of Paenibacillus polymyxa strains reveals the mechanism of plant growth promotion and biocontrol. Anton Van Leeuw. 2020;113(11):1539–1558. doi: 10.1007/s10482-020-01461-y.
- Loper JE, Hassan KA, Mavrodi DV, et al. Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet. 2012;8(7):e1002784. doi: 10.1371/journal.pgen.1002784.
- Tomita M, Kikuchi A, Kobayashi M, et al. Characterization of antifungal activity of the GH-46 subclass III chitosanase from Bacillus circulans MH-K1. Anton Van Leeuw. 2013;104(5):737–748. doi: 10.1007/s10482-013-9982-5.
- Gao X-A, Ju W-T, Jung W-J, et al. Purification and characterization of chitosanase from Bacillus cereus D-11. Carbohydr Polym. 2008;72(3):513–520. doi: 10.1016/j.carbpol.2007.09.025.
- Yuan J, Li B, Zhang N, et al. Production of bacillomycin- and macrolactin-type antibiotics by Bacillus amyloliquefaciens NJN-6 for suppressing soilborne plant pathogens. J Agric Food Chem. 2012;60(12):2976–2981. doi: 10.1021/jf204868z.
- Wu L, Wu H, Chen L, et al. Difficidin and bacilysin from Bacillus amyloliquefaciens FZB42 have antibacterial activity against Xanthomonas oryzae rice pathogens. Sci Rep. 2015;5(1):12975. doi: 10.1038/srep12975.
- Crits-Christoph A, Diamond S, Butterfield CN, et al. Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature. 2018;558(7710):440–444. doi: 10.1038/s41586-018-0207-y.
- Patel PS, Huang S, Fisher S, et al. Bacillaene, a novel inhibitor of procaryotic protein synthesis produced by Bacillus subtilis: production, taxonomy, isolation, physico-chemical characterization and biological activity. J Antibiot (Tokyo). 1995;48(9):997–1003. doi: 10.7164/antibiotics.48.997.
- Liu G, Kong Y, Fan Y, et al. Whole-genome sequencing of Bacillus velezensis LS69, a strain with a broad inhibitory spectrum against pathogenic bacteria. J Biotechnol. 2017;249:20–24. doi: 10.1016/j.jbiotec.2017.03.018.
- Fan B, Wang C, Song X, et al. Bacillus velezensis FZB42 in 2018: the gram-positive model strain for plant growth promotion and biocontrol. Front Microbiol. 2018;9:2491. doi: 10.3389/fmicb.2018.02491.
- Wang KX, Xu WH, Chen ZN, et al. Complete genome sequence of Bacillus velezensis WB, an isolate from the watermelon rhizosphere: genomic insights into its antifungal effects. J Glob Antimicrob Resist. 2022;30:442–444. doi: 10.1016/j.jgar.2022.05.010.
- Yi HS, Ahn YR, Song GC, et al. Impact of a bacterial volatile 2,3-butanediol on Bacillus subtilis rhizosphere robustness. Front Microbiol. 2016;7:993. doi: 10.3389/fmicb.2016.00993.
- Liu Y, Feng H, Chen L, et al. Root-secreted spermine binds to Bacillus amyloliquefaciens SQR9 histidine kinase kind and modulates biofilm formation. Mol Plant Microbe Interact. 2020;33(3):423–432. doi: 10.1094/MPMI-07-19-0201-R.
- de Melo NR, Abdrahman A, Greig C, et al. Myriocin significantly increases the mortality of a non-mammalian model host during candida pathogenesis. PLoS One. 2013;8(11):e78905. doi: 10.1371/journal.pone.0078905.
- Olsen I, Jantzen E. Sphingolipids in bacteria and fungi. Anaerobe. 2001;7(2):103–112. doi: 10.1006/anae.2001.0376.
- Kihara A, Igarashi Y. FVT-1 is a mammalian 3-ketodihydrosphingosine reductase with an active site that faces the cytosolic side of the endoplasmic reticulum membrane. J Biol Chem. 2004;279(47):49243–49250. doi: 10.1074/jbc.M405915200.
- Persson B, Kallberg Y, Bray JE, et al. The SDR (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative. Chem Biol Interact. 2009;178(1–3):94–98. doi: 10.1016/j.cbi.2008.10.040.
- Yoshikawa M, Yokokawa Y, Okuno Y, et al. Total synthesis of a novel immunosuppressant, myriocin (thermozymocidin, ISP-I), and Z-myriocin. Chem Pharm Bull (Tokyo). 1994;42(4):994–996. doi: 10.1248/cpb.42.994.
- Banfi L, Beretta M, Colombo L, et al. Total synthesis of (+)-thermozymocidin (myriocin) from D-fructose. J Chem Soc Chem Commun. 1982;(9):488–490. doi: 10.1039/c39820000488.
- Gajdosíková E, Martinková M, Gonda J, et al. Microwave accelerated aza-Claisen rearrangement. Molecules. 2008;13(11):2837–2847. doi: 10.3390/molecules131102837.