
Abstract
High-quality mRNA extraction is essential for gene expression assays. In this study, we developed a rapid method (20 min) named FACTS for the extraction of intact RNA from Pseudomonas aeruginosa and compared its performance to established rapid techniques like RNAsnap and CiAR. The RNA integrity, yield, purity and presence of residual genomic DNA were adopted as assessment criteria. Multiple assays for RNA integrity were applied, including the Agilent 2200 TapeStation, QIAxcel capillary electrophoresis, and the newly available Qubit RNA Integrity and Quality (IQ) Assay Kit. The RNA purity and DNA/RNA yield were assessed by spectrophotometry and fluorimetry, respectively. Following Dnase treatment, two-step RT-qPCR for the expression of the rpoD reference gene was performed to evaluate the performance of each method. In terms of RNA integrity, FACTS showed the highest RNA integrity, while in terms of purity, CiAR scored best. RNAsnap resulted in a substantial amount of residual DNA. Pilot experiments for RNA extraction with FACTS from other Gram-negative and Gram-positive bacteria revealed promising results. FACTS is a novel RNA extraction method for rapid highly effective extraction of high-quality RNA from P. aeruginosa and can be used as a cost-efficient alternative to other methods in gene expression studies.
Introduction
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) is a widely employed technique for the assessment of gene expression levels within an organism. In order to obtain precise measurements of mRNA levels, the transcript integrity must be preserved. Using low quality RNA for gene expression analysis would have a negative impact on the obtained results [Citation1]. The susceptibility of long mRNA transcripts to degradation [Citation2] is a result of two major factors: the presence of active RNases and storage under suboptimal conditions [Citation3]. Obtaining a representative expression profile by RT-qPCR requires a preliminary quality control of the extracted RNA [Citation4]. The A260/A280 ratio is commonly used to measure RNA purity, but it is unreliable in the presence of contaminants and does not provide information on RNA integrity. An indication of pure RNA is a value greater than 1.8 for both the A260/A230 and A260/A280 ratios. Determining the purity of RNA through agarose gel electrophoresis is a tedious and low-throughput process, and requires a substantial amount of RNA [Citation5].
The introduction of devices such as the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA) and Experion (Bio-Rad Laboratories, Hercules, CA) enabled quality control using a few microliters of RNA sample [Citation6,Citation7]. The operating principle of these devices lies in the microfluidic electrophoretic separation and detection through laser induced fluorescence. Then the 16S/23S rRNA ratio is calculated, and the analysis algorithm determines the so-called RNA Integrity Number (RIN), providing scores between 1 and 10, where 10 and 1 indicate completely intact and highly degraded RNA, respectively. The establishment of the RIN score had a huge impact on the standardization and reproducibility of studies involving RNA [Citation4,Citation6]. Another tool for RNA integrity analysis is the QIAxcel advanced capillary electrophoresis system (Qiagen), which also incorporates the 16S/23S ratio and determines a similar RNA integrity score (RIS). The Ratiometric fluorescence-based method is the newest development that multiplexes several fluorescent dyes with distinct emission channels and can assess RNA integrity in as little as five minutes using a Qubit 4 Fluorometer (Thermo) [Citation8].
Extraction of high-quality RNA depends on both the efficient cell lysis and the rapid inactivation of RNases [Citation9]. The type of tissue or organism from which RNA is isolated must also be taken into consideration [Citation10]. For an RNA extraction method to be robust and efficient, the resulting RNA must be intact, free of contaminants such as genomic DNA, proteins and RNases. RT and PCR inhibitors as well as residual cationic enzyme cofactors must be eliminated during extraction and purification [Citation11].
To the best of our knowledge, there are currently only a few RNA isolation methods that yield high-quality RNA. Furthermore, most methods involve laborious purification procedures that are time, costly and effort consuming. In addition, a disadvantage is the use of toxic agents such as phenol/chloroform, which require additional protective equipment and must be handled in a fume hood. Another problem is the large amount of DNA that is co-isolated with RNA, necessitating, in some cases, several DNАse treatments. Even a moderate amount of DNA leftover may negatively affect the efficiency of the DNAse treatment step and subsequent downstream analysis results.
In the present study, we introduced a robust method for RNA extraction from Pseudomonas aeruginosa that eliminates the need for highly toxic agents and yields high quality RNA with minimal amounts of contaminating DNA. The method was called FACTS (FormAmmide, CDTA, TCEP, Sarcosyl) and was compared to some of the available commercial kits, as well as methods previously described in the literature [Citation4,Citation12–14].
Materials and methods
P. aeruginosa reference strains (ATCC 27853, PAO1 ATCC 15692 and EARS 5585) and two clinical strains from the collection of the National Reference Laboratory for Control and Monitoring of Antimicrobial Resistance (NRL-CMAR) were included in the study. Cultures were grown overnight at 30 °C in Luria Bertani (LB) broth. Other Gram-negative (Klebsiella pneumoniae, Serratia marcescens and Enterobacter cloacae) and Gram-positive clinical strains (Enterococcus faecalis, Enterococcus gallinarum, Staphylococcus aureus and Bacillus mycoides) from the collection of NRL-CMAR were cultured also at 37 °C overnight in LB. Log phase (OD600 = 0.3–0.5) cultures were then obtained for RNA extraction.
FACTS (Formamide, CDTA, TCEP, Sarcosyl)
Gram-negative bacteria
Bacterial culture corresponding to 0.25 OD600 was mixed with 1/30 volume of 300 mg/mL polyvinyl sulfonic acid (PVSA) (278424, Sigma-Aldrich) and incubated for 5 min at 4 °C for inactivation of RNAses [Citation15]. The suspension was centrifuged at 9000 g for 2 min at 4 °C and the supernatant was discarded. The cell pellet was resuspended in 100 µL lysis buffer composed of 98% deionized formamidе (608082, Beckman Coulter), 2.25 mmol/L trans-1,2-Diaminocyclohexane-N,N,N′,N′-tetraacetic acid (CDTA, 155000250, Thermo Scientific), 1.25 mmol/L Tris(2-carboxyethyl)phosphine (TCEP) (646547, Sigma-Aldrich) and 0.025% N-lauroylsarcosine sodium (SLS) (L9150, Sigma-Aldrich). The suspension was then incubated at 68 °C for 10 min, followed by 2 min on ice and centrifugation at 16,300 g for 6 min at 4 °C. The supernatant (containing the RNA) was then transferred to a new tube and stored at −80 °C.
Gram-positive bacteria
Bacterial culture corresponding to 0.5 OD600 was mixed with 1/30 volume of PVSA (300 mg/mL) and the resulting mixture was incubated for 5 min at 4 °C. The suspension was then centrifuged, and the supernatant was discarded. The cell pellet was resuspended in 200 µL lysis buffer and transferred to a screw cap tube containing ∼50 µL of 0.1 mm and 0.5 mm zirconia beads each and followed by 9 min continuous homogenization on a Vortex Genie-2 equipped with a horizontal 24-tube holder (Scientific Industries, Inc., USA). The rest of the protocol was identical with the one for Gram-negative bacteria.
RNAsnap (Stead et al. 2012) [Citation13]
The method was performed according to the RNAsnap protocol for Gram-negative bacteria described below. Bacterial suspension (0.25 OD600) was centrifuged at 16,000 g for 30 s. The supernatant was discarded and the cell pellet was resuspended in 100 µL RNA extraction solution followed by incubation at 95 °C for 7 min. The mixture was then centrifuged at 16,000 g for 5 min at room temperature (RT). The supernatant was transferred to a new tube and stored at −80 °C.
Citrate-citric acid RNA isolation (CiAR Oñate-Sánchez et al. 2021) [Citation14]
The method was implemented with modifications. Bacterial suspension (0.25 OD600) was centrifuged at 8000 rpm for 1 min. The supernatant was removed and the cell pellet was resuspended in 300 µL lysis solution by pipetting 3 times, followed by incubation for 5 min at 65 °C [Citation14]. Then, 100 µL of modified precipitation solution (4 mol/L potassium acetate/2 mol/L acetic acid), 32 mmol/L citric acid, 16 mmol/L sodium-citrate, Sigma-Aldrich) were added and the suspension was homogenized by gentle inversion six times. After incubation for 10 min at 4 °C tubes are centrifuged for 10 min at 15,000 g 4 °C. The supernatant (∼300 µL) was transferred to a new tube, mixed with 0.46 volumes of isopropanol by gentle inversion and centrifuged for 10 min at RT. Following two washes with 75% ethanol, the sample was allowed to air dry and resuspended in 50 µL DEPC-treated water.
Additional extraction methods
In order to facilitate a more extensive examination, three additional RNA extraction protocols were included. The following two commercial kits were used in accordance with the manufacturer’s instructions: GeneMATRIX Universal RNA/miRNA purification kit (cat. No. E3599, EurX Ltd.) applying the ‘cell culture’ protocol for total RNA and TRIzol™ Reagent (cat. No 15596026) using the ‘cells grown in suspension’ variant. Finally, hot SDS/hot phenol protocol was also performed as described previously [Citation4].
Qualitative and quantitative analysis
Quality analysis of the resulting RNA was performed using the QIAxcel advanced capillary electrophoresis system (Qiagen), Agilent 2200 TapeStation (Agilent Technologies), and Qubit RNA Integrity and Quality (IQ) Assay Kit (Thermo Scientific) (for FACTS only) according to the manufacturers’ instructions.
The assessment of RNA purity was performed using a BioDrop spectrophotometer, with blank solutions for FACTS and RNAsnap and DEPC-treated water for CiAR. Prior to purity measurement, samples were diluted 10-fold in DEPC water. DNA and RNA concentrations were measured by Qubit 4 and using appropriate kits.
Sample purification
FACTS and SNAP isolated samples were diluted 10-fold and purified using ethanol and linear polyacrylamide (LPA) as a carrier. Ethanol and Solution A (0.025% LPA, 75 mmol/L ammonium acetate) [Citation16] were mixed in 7:3 ratio, then six volumes of this mixture were added to each sample, followed by vortexing and centrifugation at 16,300 g for 10 min at 4 °C. The supernatant was decanted, pellets were air dried and eluted in 15 µL DEPC water.
One-step DNAse treatment and reverse transcription
A total of 500 ng RNA of each sample from each method were subjected to DNAse treatment in a one-step manner as previously described [Citation17]. The reaction mixture (19 µL) consisted of 1× cDNA buffer (EurX), 20 µmol/L random pentadecamers, 0.5 mmol/L dNTPs, 0.5 U/µL thermostable RNAse inhibitor (EurX), 8 U DNAse I (New England Biolabs). After incubation at 37 °C for 30 min, DNAse inactivation and RNA denaturation were conducted at 70 °C for 5 min. The reactions were chilled on ice for 2 min, followed by the addition of 250 U smART RT enzyme (EurX) and incubation for 5 min at 25 °C for 1 h at 42 °C and 30 min at 48 °C, then the reaction was terminated at 85 °C for 5 min.
The resulting cDNA was used in EVAGreen® (31000, Biotium) qPCR targeting the rpoD reference gene to evaluate the performance of each method [Citation18]. qPCR was performed on an iCycler iQ5 Real Time PCR Thermal Cycler, Bio-Rad Laboratories, Inc. with DNase I untreated RNA samples in order to assess the level of residual DNA. DNАse I treated samples which had not been reverse transcribed were assayed in parallel as controls for the DNase reaction efficiency.
Data analysis
Data represented in figures and tables are mean values from three replicates unless otherwise stated. The standard deviation (±SD) values within each group of replications were used to depict the error bars. Statistical analyses (one-way ANOVA and Pearson’s correlation) were performed with SPSS Statistics software v 27.0 (IBM Corp.) and python package scipy 1.10.0. Graphs were created with the matplotlib v3.7.1 python package.
Results
In this study, several techniques for extraction of RNA from Pseudomonas were evaluated, including the GeneMATRIX Universal RNA Purification Kit (EURx), Trizol (Invitrogen), hot SDS/hot phenol [Citation4,Citation12], RNAsnap [Citation13] and Citrate-Citric Acid RNA Isolation (CiAR) [Citation14]. In preliminary experiments, the GeneMATRIX Universal RNA Purification Kit (EURx) and the Trizol (Invitrogen) kits produced minute amounts of RNA from bacteria (data not shown) and were therefore not considered in further analyses. The hot SDS/hot phenol method provided acceptable results, but it was deemed labor-intensive and time-consuming (>24 h) and was discarded as well. Ultimately, three rapid methods were selected for further analysis, including FACTS (described in this study), RNAsnap and CiAR and compared based on factors such as RNA integrity, yield, and the presence of residual DNA.
RNA integrity
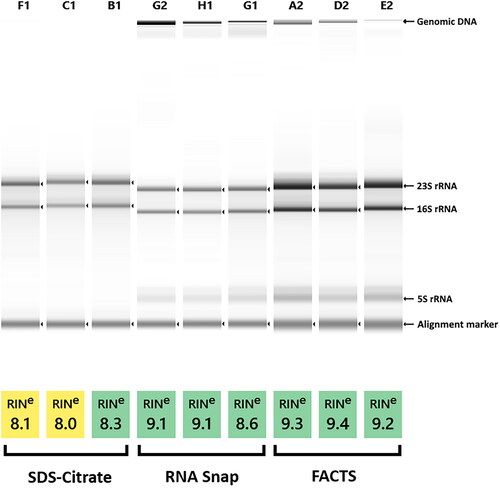
In terms of integrity, with the Agilent TapeStation, the best results were obtained with FACTS. RNAsnap was the fastest and easiest method, but showed the highest genomic DNA contamination. Adequate results were obtained by CiAR, with slightly lower RINs observed for all samples. Notably, no bands corresponding to 5S RNA were present in CiAR, whereas the complete set of rRNA species were evident with the rest of the methods (). The 23S/16S ratios for all methods were ≥1.8 (data not shown).
Figure 1. Agilent TapeStation analysis of RNA integrity of samples isolated by CiAR (SDS-Citrate), RNAsnap and FACTS from three P. aeruginosa reference strains: ATCC 27853 (F1, G2, A2), PAO1 (C1, H1, D2) and EARS 5585 (B1, G1, E2). Yellow coloring is assigned to RNA samples with RIN values ≤ 8.1, indicating moderate to good RNA quality, while green is used to denote the samples with the highest quality.
RNA integrity was also tested using the QIAxcel Advanced system. The observed electrophoretic profiles and 16S/23S rRNA ratios were similar to those from the TapeStation; however, the corresponding RIS values were significantly lower than the RIN ones, with no apparent reason ().
Table 1. Comparison of the RIS (QIAxcel Advanced system) and RIN (TapeStation) integrity values of RNA extracted from P. aeruginosa using three RNA extraction methods.
Finally, for further validation of FACTS, samples were also analyzed using the Qubit RNA Integrity and Quality (IQ) Assay. The results were comparable to those from the TapeStation, indicating high-quality RNA ().
Table 2. Qubit RNA Integrity and Quality (IQ) Assay – P.aeruginosa FACTS samples.
RNA yield versus residual genomic DNA level
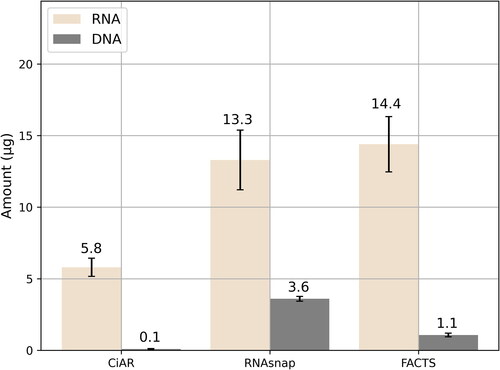
There were no significant differences in the RNA amount isolated with FACTS and RNAsnap methods (p > 0.05), while the CiAR samples resulted in approximately 3-fold lower RNA yield compared to RNAsnap and FACTS (p < 0.05). In contrast, the residual genomic DNA amount varied significantly among methods (p < 0.05). RNAsnap samples were highly contaminated with DNA, FACTS ones were intermediate, whereas CiAR resulted in negligible DNA content ().
Figure 2. Comparison of RNA extraction methods in terms of RNA and DNA amount based on the mean values from three replicates of P. aeruginosa ATCC 27853 for each method.
RNA purity
CiAR showed excellent values in terms of 260/280 (≥2) and 260/230 (2 ≤ 2.2) ratios. The 260/280 values for FACTS were ≥2 and those of 260/230 ratio were slightly higher than 2.2, which is the upper limit of the recommended optimum. Two of the RNAsnap samples showed lower 260/280 (<1.7) and 260/230 values, indicating both protein and carbohydrate contamination. The third sample was within the acceptable range 260/230 = 2.1 ().
Table 3. Comparison of three RNA extraction methods in terms of purity – Absorbance ratios for P.aeruginosa RNA samples.
RNA extraction from other Gram-negative and Gram-positive bacteria with FACTS
Considering the excellent results obtained with P. aeruginosa, the performance of FACTS was further tested with other Gram-negative bacteria (Klebsiella pneumoniae, Serratia marcescens and Enterobacter cloacae) that cause healthcare-associated infections. The results obtained after two replicate RNA extractions from each strain were comparable to those obtained for Pseudomonas with similar RNA concentrations as well as high RIN and lower RIS values (). The 23S/16S ratios were in the range between 1.7 and 2 (data not shown).
Table 4. FACTS RNA extraction from additional Gram-negative and Gram-positive bacteria; results from two replicate extractions.
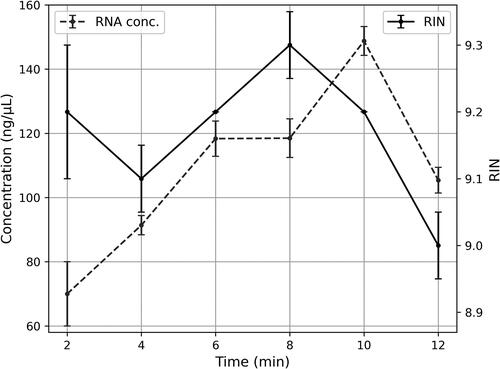
We also attempted FACTS on Gram-positive bacteria that are usually problematic for high-quality RNA extraction. We adopted mechanical lysis (bead-beating) with zirconium beads as previously described for RNAsnap, except that two types of zirconium beads (0.1 mm and 0.5 mm) were used in equal amounts in the FACTS solution. The beating period was determined experimentally, with 9 min being optimal, where the least degradation with the highest RNA concentration was observed (). A positive Pearson’s correlation coefficient of 1.000 was calculated between time and RNA concentration (p < 0.01) up until 9 min threshold of bead-beating. No significant correlation was found between the pairs RIN-RNA concentration and RIN-time (p > 0.05).
Figure 3. Optimization of mechanical lysis (bead-beating time) in FACTS for Gram-positive bacteria (Enterococcus faecalis): RNA concentration and RIN values over beat-beating time - FACTS. This experiment was conducted in two replicates.
All Gram-positive bacteria (S. aureus, E. gallinarum, E. faecalis, B. mycoides) tested showed high RIN values (>8) and 23S/16S ratios between 1.8 and 2 (data not shown). With the exception of B. mycoides, lower RNA concentrations were detected compared to the ones obtained for the Gram-negatives ().
Sample purification
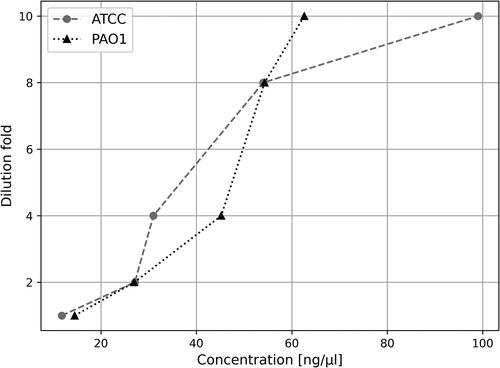
To be RT and qPCR compatible, the RNA isolated with rapid methods must be purified. The impact of formamide (e.g. in RNAsnap and FACTS) on nucleic acids precipitation with ethanol was investigated in accordance with previous research showing that high levels of formamide could be detrimental [Citation19]. We tested varying dilutions (2-, 4-, 8- and 10-fold) of Pseudomonas FACTS RNA samples to determine the formamide concentration that allows the highest RNA recovery after precipitation. Our results indicate that the highest RNA yield was obtained when the formamide concentration was below 10% (10-fold dilution), as illustrated in .
Figure 4. Effect of formamide concentration on recovery rate of precipitated RNA in relation to sample dilution factor (formamide); FACTS RNA samples from P. aeruginosa ATCC 27853 and PAO1 were used in a single repetition for each dilution.
Reverse transcription and qPCR
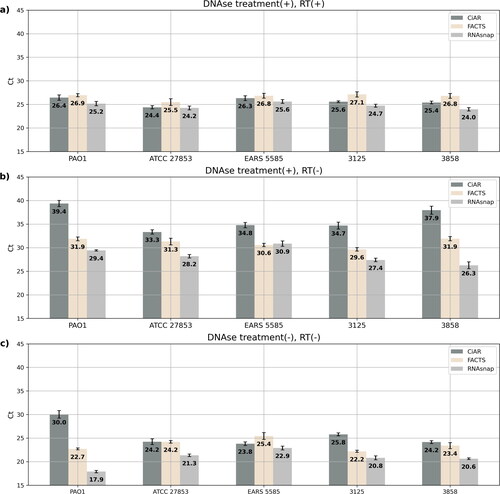
After RNA purification and DNAse treatment, P. aeruginosa RNA samples were subjected to RT-qPCR targeting the rpoD gene. RNAsnap showed the lowest Ct values among the DNAse treated samples (DNAse (+)/RT (+)) (). However, it also had the lowest Ct values with the samples treated with DNAse but not reverse transcribed (DNAse (+); RT(−)), suggesting high levels of undigested residual DNA (). CiAR showed superior results, with the highest Ct values for the DNAse(+)/RT(−) and the untreated DNAse(−)/RT(−) samples () outperforming the other methods, while RNAsnap samples had the lowest Ct values in this group. Finally FACTS resulted in slightly higher Ct values than the other two methods in the (DNAse (+)/RT (+)) group, suggesting lower RNA yield () but at the same time significantly less contamination with DNA compared to RNASnap (p < 0.05), except for strains EARS 5585 () and 3125 ().
Figure 5. Remaining DNA with each RNA extraction method in strains P. aeruginosa PAO1 ATCC 15692, ATCC 27853, EARS 5585, 3125, 3858. Values represent the mean cts from rpoD qPCR in triplicate. (a) with cDNA from DNAase-treated samples, lower is better; (b) residual genomic DNA after DNAse treatment, higher is better; (c) total genomic DNA untreated with DNAse or RT, higher is better.
Discussion
In this study, we sought to evaluate the effectiveness of various RNA extraction techniques for P. aeruginosa, including the GeneMATRIX Universal RNA Purification Kit (EURx), Trizol (Invitrogen), hot SDS/hot phenol [Citation4,Citation12], RNAsnap [Citation13] and Citrate-Citric Acid RNA Isolation [Citation14]. Upon evaluating the efficiency, ease of execution and the quality of the resulting RNA, we found that RNAsnap and CiAR were the most favorable methods. These two techniques were then compared to FACTS, a new method developed in-house, based on several crucial parameters such as quality, yield, purity and contamination with residual genomic DNA. FACTS was inspired by the prior rapid formamide-based RNA extraction techniques (e.g. RNASnap [Citation13] and the One step hot-formamide method [Citation9]). Besides formamide, FACTS includes chemicals that function synergistically with heating for the bacterial lysis and rapid inactivation of RNases while leaving most of the high molecular DNA aggregated within the cell pellet. First, washing the cells with PVSA, a non-specific RNAse inhibitor, eliminates exogenous RNases at a fraction of the cost of the commercial RNA preservation products [Citation15]. Second, we have optimized the effective concentration of each reagent systematically for maximum RNA yield with minimum residual DNA (data not shown). CDTA was preferred over the well-established EDTA as a chelating agent of choice as it is reportedly more lipophilic and shows stronger metal binding, both contributing to lysis and RNase inactivation [Citation20]. Next, TCEP being irreversible and resistant to oxidation at various pHs is considered a superior reducing agent compared to dithiotreitol and beta-mercaptoethanol, which are widely adopted in RNA isolation protocols [Citation21]. Finally, the low concentration of sarcosyl (anionic detergent) facilitates the cell suspension and permeabilization.
In terms of RNA integrity, RNAsnap and FACTS displayed superior performance. CiAR samples were slightly degraded but were still of acceptable quality. RIN values for FACTS samples were consistently higher than those of the other two methods. One possible reason for the reduced integrity in CiAR samples was the difference in the eluting agents. Our preliminary tests included several freeze-thaw cycles resulting in a gradual integrity decline of CiAR samples, whereas no significant change was observed for RNAsnap and FACTS samples. Previous studies have shown that formamide is a potent RNA preservation agent, and it has been found that even multiple freeze-thaw cycles had minimal impact on the RNA integrity as opposed to samples dissolved in DEPC-treated water [Citation9,Citation22]. Therefore, for long-term preservation of RNA, we recommend storing samples in the original formamide lysis buffer rather than purified.
The RNA concentrations in our study were consistent across the tested methods, while the residual DNA concentrations varied considerably. The lowest levels of DNA were found in samples processed with CiAR, while the highest levels were found in samples processed with RNAsnap. Additionally, CiAR samples showed very high purity, followed by FACTS samples and lastly RNAsnap samples. It is important to acknowledge that the CiAR samples underwent purification as a mandatory step in the protocol, without which accurate measurements would not have been possible. We also recorded higher DNA concentrations in S. marcescens samples, which may be attributed to factors that require further investigation. In line with our expectation, we found lower RNA concentrations in Gram-positive bacteria, which could be due to their higher resistance to lysis. On the other hand, B. mycoides samples showed higher concentration of RNA and DNA with an elevated RNA/DNA ratio compared to other Gram-positive bacteria. This might be attributed to inaccuracies in measurement of the optical density, as the cells in the culture were aggregated.
In this study, we chose to use ethanol precipitation with LPA as a carrier for purification due to its efficient and streamlined nature. Our results indicate that this method is effective in RNA precipitation, despite being initially developed for DNA purification [Citation23].
After cDNA synthesis, we proceeded with measuring rpoD levels by qPCR. RNAsnap had the lowest Ct values among the DNAse (−) RT (−) samples (), suggesting that large amount of residual DNA was present and that qPCR results with cDNA from DNAse (+) RT (+) samples may have been overestimated. This may be attributed to the higher incubation temperature used in the lysis step (95 °C as opposed to 68 °C in FACTS), which may have resulted in more thorough lysis and DNA release. Further studies are needed to confirm this hypothesis. Previously, it has been demonstrated that high levels of residual DNA may necessitate multiple DNase treatments in order to completely eliminate it [Citation4]. Our results were consistent with these findings and also indicate that DNA contamination can be genus or even strain-specific [Citation4]. This phenomenon was observed in our samples where Gram-negative bacteria had lower levels of contaminating DNA, with RNA/DNA ratios being consistent. This pattern was also observed in Gram-positive bacteria. However, a notable exception was observed for Pseudomonas and Serratia, where the highest amounts of genomic DNA contamination were found.
In terms of analytical methods, the most robust results were obtained with the TapeStation, followed by the Qubit RNA Integrity and Quality (IQ) Assay. The manufacturer of the QIAxcel system states that its efficiency in analyzing eukaryotic RNA is comparable to that of the Bioanalyzer [Citation24]. Thus, we decided to investigate its capability to assess prokaryotic RNA, as it provides the same 23S/16S rRNA patterns at considerably lower cost per sample than the TapeStation. Results from the QIAxcel system showed inconsistency in the determination of RIS, but nevertheless the observed 23S/16S rRNA peaks had the characteristic profile of high-quality RNA, suggesting that the computation algorithm might be suboptimal for prokaryotic RNA (data not shown).
One of the major limitations of this study was the narrow scope of bacterial species examined. The three methods were only tested on P. aeruginosa, while other Gram-negative and Gram-positive bacteria were only examined using FACTS. As a result, no comparison with other methods was possible for these species. Additionally, FACTS was also tested on Bacillus licheniformis. Despite utilizing bead-beating lysis, the method was unable to extract RNA from this organism, suggesting that it may be inappropriate for other (e.g. sporulating) bacteria that are difficult to lyse. To address these limitations, further research is needed to optimize the protocol for handling these types of microorganisms.
Conclusions
In this study, we introduced an improved rapid method for RNA extraction from P. aeruginosa and compared it to various techniques such as the GeneMATRIX Universal RNA Purification Kit, Trizol, hot SDS/hot phenol, RNAsnap, and Citrate-Citric Acid RNA Isolation. Our findings revealed that RNAsnap and CiAR were the most favorable in terms of efficiency and ease of execution, but the newly developed FACTS method exhibited superior performance in terms of RNA integrity. FACTS appears as cost-efficient, highly effective and promising technique for rapid RNA extraction from P. aeruginosa, other Gram-negative and some Gram-positive bacteria, providing high-quality RNA amenable to expression studies with minimal presence of contaminating DNA.
Author contributions
Conceptualization, methodology and validation—I.N.I. and I.S. Formal analysis, investigation—I.N.I. and I.S. R.H. E.D; Resources I.N.I. and S.S.; Data analysis –- I.S., D.D., S.S, and R.H.; Writing—original draft preparation—I.S. S.S.; Writing—review and editing—I.N.I.,D.D., E.D. and R.H.; Visualization—I.S., D.D., E.D. and S.S.; Supervision, project administration, funding acquisition—I.N.I. All authors have read and agreed to the published version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the findings of this study are available from the corresponding author, Ivan Ivanov, upon reasonable request.
Additional information
Funding
References
- Imbeaud S, Graudens E, Boulanger V, et al. Towards standardization of RNA quality assessment using user-independent classifiers of microcapillary electrophoresis traces. Nucleic Acids Res. 2005;33(6):1–9. doi: 10.1093/nar/gni054.
- Bustin SA. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol. 2002;29(1):23–39. doi: 10.1677/JME.0.0290023.
- Pérez-Novo CA, Claeys C, Speleman F, et al. Impact of RNA quality on reference gene expression stability. Biotechniques. 2005;39(1):52–56. doi: 10.2144/05391BM05.
- Jahn CE, Charkowski AO, Willis DK. Evaluation of isolation methods and RNA integrity for bacterial RNA quantitation. J Microbiol Methods. 2008;75(2):318–324. doi: 10.1016/j.mimet.2008.07.004.
- Bustin SA. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J Biomol Tech. 2004;15(3):155–166.
- Mueller O, Hahnenberger K, Dittmann M, et al. A microfluidic system for high-speed reproducible DNA sizing and quantitation. Electrophoresis. 2000;21(1):128–134. doi: 10.1002/(SICI)1522-2683(20000101)21:1<128::AID-ELPS128>3.0.CO;2-M.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/METH.2001.1262.
- Applied Biosystems. Qubit RNA IQ assay: a fast and easy fluorometric RNA quality assessment. Thermo Fish. 2017; Accessed: Jan. 27, 2023. [Online]:[1 p.]. Available from: https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0012655_GeneJET_Plasmid_Miniprep_UG.pdf%0Ahttps://www.thermofisher.com/document-connect/document-connect.html?url=https
- Shedlovskiy D, Shcherbik N, Pestov DG. One-step hot formamide extraction of RNA from Saccharomyces cerevisiae. RNA Biol. 2017;14(12):1722–1726. doi: 10.1080/15476286.2017.1345417.
- Pfaffl MW, Tichopad A, Prgomet C, et al. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26(6):509–515. [Online]. Available from: http://www.wzw.tum.de/gene-quantification/bestkeeper.html doi: 10.1023/b:bile.0000019559.84305.47.
- In Bustin SA, editor. A-Z of Quantitative PCR. La Jolla, CA, USA: IUL Press; 2004; pp. 90–91
- Lin-Chao S, Bremer H. Effect of the bacterial growth rate on replication control of plasmid pBR322 in Escherichia coli. Mol Gen Genet. 1986;203(1):143–149. doi: 10.1007/BF00330395.
- Stead MB, Agrawal A, Bowden KE, et al. RNAsnapTM: a rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res. 2012;40(20):e156–e156. doi: 10.1093/nar/gks680.
- Oñate-Sánchez L, Verdonk JC. Citrate-citric acid RNA isolation (CiAR) for fast, low-cost, and reliable RNA extraction from multiple plant species and tissues. Curr Protoc. 2021;1(12). doi: 10.1002/cpz1.298.
- Earl CC, Smith MT, Lease RA, et al. Polyvinylsulfonic acid: a low-cost rnase inhibitor for enhanced RNA preservation and cell-free protein translation. Bioengineered. 2018;9(1):90–97. doi: 10.1080/21655979.2017.1313648.
- Fregel R, González A, Cabrera VM. Improved ethanol precipitation of DNA. Electrophoresis. 2010;31(8):1350–1352. doi: 10.1002/elps.200900721.
- Dilworth DD, McCarrey JR. Single-step elimination of contaminating DNA prior to reverse transcriptase PCR. PCR Methods Appl. 1992;1(4):279–282. doi: 10.1101/gr.1.4.279.
- Quale J, Bratu S, Gupta J, et al. Interplay of efflux system, ampC, and oprD expression in carbapenem resistance of Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother. 2006;50(5):1633–1641. doi: 10.1128/AAC.50.5.1633-1641.2006.
- Nadin-Davis S, Mezl VA. Optimization of the ethanol precipitation of RNA from formamide containing solutions. Prep Biochem. 1982;12(1):49–56. doi: 10.1080/00327488208065549.
- Metal Chelates. https://www.dojindo.eu.com/images/Product Photo/Chelate_Table_of_Stability_Constants.pdf. (accessed Jun. 13, 2023).
- Rhee SS, Burke DH. Tris(2-carboxyethyl)phosphine stabilization of RNA: comparison with dithiothreitol for use with nucleic acid and thiophosphoryl chemistry. Anal Biochem. 2004;325(1):137–143. doi: 10.1016/j.ab.2003.10.019.
- Chomczynski P. Solubilization in formamide protects RNA from degradation. Nucleic Acids Res. 1992;20(14):3791–3792. doi: 10.1093/NAR/20.14.3791.
- Gaillard C, Strauss F. Ethanol precipitation of DNA with linear polyacrylamide as carrier. Nucleic Acids Res. 1990;18(2):378. doi: 10.1093/nar/18.2.378.
- QIAxcel®—Pure Excellence. https://www.qiagen.com/∼/media/nextq/resource library/brochures/qiaxcel-pure-excellence.ashx. (accessed Jun. 13, 2023).