
Abstract
Disease-associated induced pluripotent stem cells (iPSCs) established from patients are now widely used for disease modeling. They can provide an unlimited source of hematopoietic cells that carry the patients’ genetic background, making them advantageous for modeling immunological disorders. To obtain functional immune cells from human iPSCs, we have developed a differentiation system that generates immortalized myeloid cells including neutrophils and monocytic cells. By using this strategy, we have established in vitro models of many immunological disorders. In this review, we focus on autoinflammatory disorders. These models have proven useful for genetic diagnosis and elucidation of the disease mechanism.
1. Introduction
Because of their pluripotency and capacity for self-renewal, human pluripotent stem cells (PSCs), such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), can be potential cellular sources for regenerative medicine, drug screening, toxicology testing and the investigation of disease mechanisms [Citation1,Citation2]. iPSCs are PSCs reprogrammed from somatic cells by introducing sets of transcription factors and possess ESC-like characteristics [Citation3,Citation4]. Disease-associated iPSCs established from patients are now widely used for disease modeling, because these cells share an almost identical genetic background with the patients.
Immunological disorders are abnormalities of the immune system, which consists mainly of hematopoietic cells in the blood, lymphoid system and tissues. To understand their cellular pathophysiology, studies using patient-derived primary hematopoietic cells or animal models are usually conducted. However, these approaches have limitations. For example, obtaining a large number of patient-derived cells from the blood can be invasive, and the in vitro functions of the cells are often affected by the patients’ conditions, such as the presence of therapeutic agents. On the other hand, although genetically modified animals provide an indispensable opportunity to study in vivo pathophysiology, differences in immunological development between humans and other animals can cause discrepancies in the resulting phenotypes. Modeling immunological disorders with patient-derived iPSCs can circumvent these issues, because they can be an unlimited source of hematopoietic cells that inherit the patients’ genetic background [Citation5].
Autoinflammatory syndrome is defined as disorders associated with dysregulation of the innate immune systems [Citation6]. In typical cases, patients with autoinflammatory syndrome show various inflammatory symptoms, including periodic fever, skin rash and sterile serositis [Citation7,Citation8]. PSC models of autoinflammatory syndrome are attractive, because (1) most typical autoinflammatory disorders are monogenic, (2) the responsible cells are usually innate immune cells, which can be robustly differentiated from PSCs, and (3) there is a high demand for better therapeutics and diagnostics. In this review, we introduce our strategy for modeling immunological disorders using human PSCs with special attention on autoinflammatory disorders and application of these models to the identification of the genetic and biological backgrounds of the patients’ pathophysiology.
2. Hematopoietic differentiation and establishment of myeloid cell lines
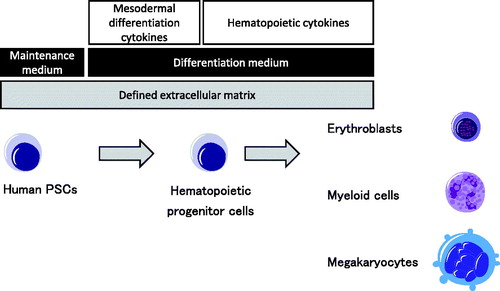
To obtain functional hematopoietic cells from human PSCs, we have developed an original monolayer differentiation system that generates hematopoietic lineages () [Citation9]. In this system, human PSCs can be differentiated into mesodermal lineage cells that give rise to both hematapoietic cells and vascular endothelial cells [Citation10]. The resulting hematopoietic progenitor cells are further differentiated into various hematopoietic lineages such as erythroid, megakaryocytic and myeloid cells. By optimizing the cytokine setting, monocytic cells including macrophages and dendritic cells can also be obtained efficiently [Citation11] and used to model autoinflammatory disorders [Citation12].
Figure 1. A principle for directed differentiation of human PSCs to hematopoietic cells. Human PSCs can be differentiated into hematopoietic progenitor cells by a defined monolayer condition. Hematopoietic progenitor cells are then committed to each hematopoietic lineage by optimizing the cytokine setting.
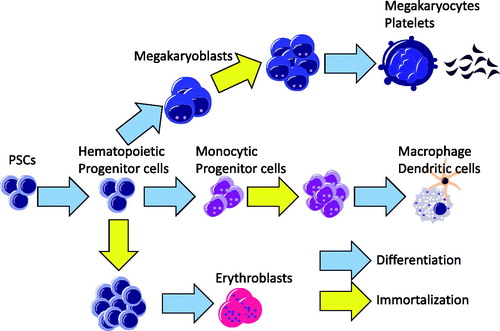
Although these cellular progenies can be used for functional analysis, one of the obstacles facing disease-associated iPSC studies is that direct differentiation from human PSCs is often time- and labor-intensive. Furthermore, the differentiation efficiency and the results of the functional analysis can often show high variance. To overcome these issues and stably obtain a large number of mature hematopoietic cells from human PSCs, researchers have tried to immortalize PSC-derived hematopoietic progenitors by introducing several genetic factors and differentiating the cells towards terminally differentiated hematopoietic progenies () [Citation13–17]. We have used this approach to obtain proliferating myeloid cell lines (iPS-MLs). The Satoru Senju Laboratory was the first to establish iPS-MLs from iPSC-derived monocytes by introducing three transgenes: cMYC, BMI1 and MDM2 [Citation16,Citation17]. iPS-MLs can be expanded in the presence of M-CSF and show an immature monocytic appearance with the expression of CD33 and CD14. Importantly, iPS-MLs can be differentiated into macrophages or dendritic cells and used for various functional analyses. By combining our robust differentiation protocol [Citation9,Citation11] and myeloid immortalization, we can obtain an unlimited number of functional myeloid cells established from the iPSCs of patients with immunological disorders.
Figure 2. Immortalization of hematopoietic progenitor cells from human PSCs. Hematopoietic progenitor cells can be immortalized by introducing appropriate transcription factors.
3. Identification of somatic NLRC4 mutation as a cause of autoinflammation
Neonatal-onset multisystem inflammatory disease [NOMID; OMIM #607715, also known as chronic infantile neurological cutaneous and articular syndrome (CINCA)] is an autoinflammatory disease with severe inflammatory symptoms persisting from the neonatal period [Citation18,Citation19]. Approximately 90% of NOMID patients carry heterozygous gain-of-function mutations in the NLR family pyrin domain containing 3 (NLRP3) gene either as a constitutive mutation [Citation20,Citation21] or somatic mosaicism [Citation22,Citation23]. Mutant NLRP3 causes excessive amounts of the proinflammatory cytokine IL-1β, resulting in IL-1β-dependent autoinflammation. However, in the remaining 10% of NOMID patients, the responsible genetic factor(s) remains unidentified. To understand the underlying pathophysiological and genetic mechanism for these mutation-negative patients, we recruited one NOMID patient without known genetic etiology. The patient is a Japanese male with typical NOMID symptoms [Citation24,Citation25] but negative for any NLRP3 mutation even as low-frequency somatic mosaicism [Citation23,Citation26]. A whole-exome sequencing analysis using DNA samples from the patient and his parents identified 26 genes as candidates, but the candidate list could not be narrowed further. Interestingly, despite the absence of any NLRP3 mutation, the clinical phenotype of the patient was driven by IL-1 based on the response to anti-IL-1 treatment [Citation24]. We established iPSC clones from the fibroblasts of the patient and differentiated them to monocytes. IL-1β secretion under LPS stimulation was then evaluated, because monocytes derived from NOMID patient-specific iPSCs are known to show excessive IL-1β production without any secondary activating signals, such as ATP or silica [Citation12]. When multiple iPSC clones were differentiated and evaluated, their phenotype was clearly separated into two groups based on the IL-1β secretion (high and low). Since this inter-clonal phenotypic heterogeneity seemed associated with the genetic heterogeneity of the iPSC clones, we performed whole exome sequencing of representative iPSC clones to identify mutations exclusively shared among clones with the disease-associated phenotype. As a result, we identified a heterozygous NLRC4 mutation, p.T177A (NC_000002.11:g.32476404T > C, NM_021209.4:c.529A > G), as the cause. Because each iPSC clone is derived from a single somatic cell [Citation27], this finding indicated that the patient has the somatic NLRC4 mutation. Indeed, the identical mutation was identified from the patient’s primary blood cells. The frequency of the mutant allele was calculated as approximately 30–34%. Since the mutation is heterozygous, the estimated mosaic frequency is therefore calculated as about 60–68%.
Our data support the possibility that the patient has a disease-specific somatic NLRC4 mutation. To confirm the mutant NLRC4 is indeed disease causing, we knocked out the NLRC4 locus of a mutant iPSC clone using CRISPR/Cas9-based mutagenesis and established iPS-MLs both from the knocked-out clones and the original mutant clones. When the iPS-MLs from knockout clones were differentiated into macrophages, the excessive IL-1β secretion was completely abolished. Therefore, we concluded the somatic NLRC4 mutation found in the patient was indeed disease causing.
NLRC4 mutations were recently reported as causes of other autoinflammatory disorders [Citation28–30]. Interestingly, their phenotypes can be distinguished into two distinct entities: a very severe neonatal-onset phenotype and a much milder phenotype of cold-induced urticaria [Citation29]. The two extreme phenotypes of NLRC4-associated autoinflammatory disorders are in contrast to the phenotype of NLRP3-associated proinflammatory conditions, which show a clinical continuum with regard to the genotype-phenotype correlation [Citation18]. However, one recent clinical case showed intermediate clinical symptoms of NLRC4-associated autoinflammatory disorders, suggesting they too have a clinical continuum.
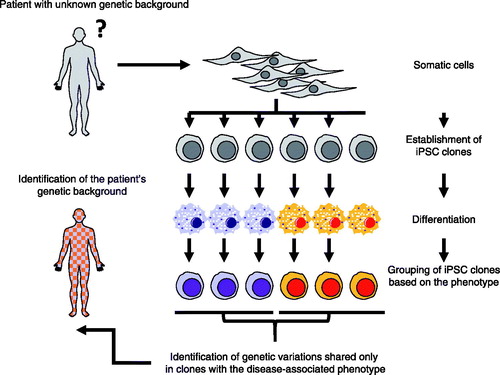
This above case demonstrates that iPSC-based forward genetic approaches can be used to identify the genetic etiology of rare sporadic cases of autoinflammatory syndrome (). Further, iPSC technology enables researchers to dissect the patient’s phenotype at the single cell level, which can be used to reveal genetic heterogeneity of the patient. To the best of our knowledge, this was the first report in which iPSC technology played an essential role in the identification of the precise genetic background of a patient. By connecting iPSC-based phenotype analysis with other technologies, such as next-generation sequencing and genome editing technology, iPSCs models can be used to investigate the detailed pathophysiology of rare immunological diseases, even when only a single patient is known.
Figure 3. Identification of somatic NLRC4 mutation from a patient with autoinflammation. The patient’s genetic background was identified through iPSC-based phenotype analysis.
4. Pluripotent stem cell model of Blau syndrome
Blau syndrome (OMIM: #186580) is a congenital monogenic granulomatois caused by mutations in Nucleotide-binding oligomerization domain-containing protein 2 (NOD2) gene [Citation31,Citation32]. Since the disease onset is usually before 4 years old, Blau syndrome was alternatively named as early-onset sarcoidosis. Patients with Blau syndrome develop intractable granulomas in the eye, skin and joints. Although inflammatory symptoms such as fever and rush are relatively mild, these granulomas cause severe complications such as joint contraction and blindness. Recently, anti-TNF therapy was reported effective for controlling the disease [Citation33,Citation34], but the long-term effectiveness is still unestablished. Therefore, specific curative therapeutic options are being considered.
NOD2 is an intracellular pathogen recognition receptor and recognizes muramyl dipeptide (MDP), a substance common in bacteria [Citation35,Citation36]. NOD2 activates the nuclear factor kappa B (NF-κB) pathway in a ligand-dependent manner, which up-regulates proinflammatory cytokines and chemokines [Citation35–39]. On the other hand, the function of disease-associated mutant NOD2 is controversial. In several reports using HEK293 cells [Citation40] or in vitro protein reconstitution systems, ligand-independent enhanced NF-κB activity is reported, while a knock-in mouse model [Citation41] demonstrates a decreased proinflammatory response, indicating the mutations are loss-of-function. However, since the mouse model did not show any phenotype associated to Blau syndrome, a human model that is able to evaluate the unique function of mutant NOD2 in a strictly controlled condition is needed. Importantly, specific perturbations that evoke the proinflammatory response to NOD2 mutant cells remain unidentified.
To address these issues, we established iPSC clones from a Blau syndrome patient harboring heterozygous R334W (1000C > T) mutation and corrected the mutation by genome-editing to obtain an isogenic counterpart. By using the same targeting strategy, a knocked-in clone with heterozygous R334 mutation was generated from a wild-type iPSC clone. By using these two isogenic pairs for disease modeling, we confirmed consistency in the in vitro phenotype associated with the disease-associated NOD2 mutation using a strictly controlled genome background. Since NOD2 is mainly expressed in hematopoietic lineage cells, especially monocytic cells [Citation37], and the granulomas contain many macrophages, we next differentiated the iPSCs towards myelomonocytic lineage cells and sought a specific trigger that evokes the proinflammatory response to these cells. Interestingly, IFN-γ upregulated the expression of NOD2 in iPSCs-derived macrophages, as previously described [Citation42–44], indicating a priming role of IFN-γ in the NOD2-NF-κB signaling pathway. We established iPS-MLs from myeloid progenitors obtained from iPSCs and used iPS-ML-derived macrophages (iPS-MPs) for further analysis. Both wild-type and mutant iPS-MPs were able to react to MDP and activate the NF-κB pathway. IFN-γ increased the NF-κB reporter activity and facilitated the nuclear translocation of NF-κB p65 subunit in mutant iPS-MPs even in the absence of MDP. In line with this finding, IFN-γ treatment significantly increased the production of cytokines such as IL-6, IL-8 and TNF-α in mutant iPS-MPs without secondary MDP stimulation. Therefore, IFN-γ primes mutant iPS-MPs to activate the NF-κB pathway in a ligand-independent manner, resulting in the enhanced production of proinflammatory cytokines. This finding was supported by RNA-seq analysis using iPS-MPs, as IFN-γ treatment specifically enriched several inflammatory pathways in mutant iPS-MPs compared to their wild-type counterparts.
To confirm that these findings are indeed associated with patient cells, but not an artifact only recognized in iPS-MPs, we investigated the in vitro phenotypes of primary monocyte-derived macrophages obtained from another Blau patient. As expected, IFN-γ treated monocyte-derived macrophages from the patient exhibited an enhanced activation of the NF-κB pathway and production of proinflammatory cytokines, confirming the validity and usefulness of our iPSC-based model.
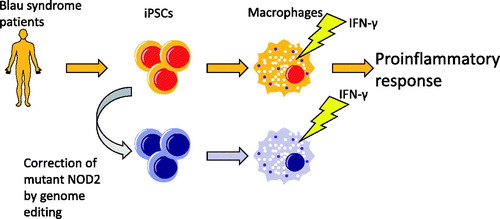
Using iPSCs, we identified IFN-γ as a specific trigger that induces the pro-inflammatory response seen in Blau syndrome (). Substantial evidence has already indicated a pathophysiological role of IFN-γ in the pathophysiology of the disease. For example, IFN-γ-expressing cells were reported positive in and around the granulomas of Blau syndrome patients [Citation45]. Also in some patients, BCG vaccination, which evokes an INF-γ response, was sometimes associated with the onset of the disease [Citation46,Citation47]. Furthermore, humans lacking IFN-γ receptors such as IFN-γR1 or IFN-γR2 do not display any mature granulomas [Citation48], indicating an indispensable role of IFN-γ in the formation of granulomas. However, it was through an iPSC-based disease model that we could reveal a previously undefined IFN-γ-dependent pathway that activates NOD2-mutated macrophages in Blau syndrome.
Figure 4. Establishment of a PSC-based disease model for Blau syndrome. Establishment of genome-edited isogenic counterparts enables precise phenotypic comparisons to study disease-associated mutations.
5. Conclusions
Here, we provide some examples of using iPSC technology for studying immunological disorders. When combined with other novel technologies such as next generation sequencing and genome editing, these models provide a phenotypic analysis that can identify extremely rare genetic conditions, showing the future possibility of iPSC-based personalized diagnosis in rare diseases. The development of therapeutic approaches for immunological disorders using iPSC technology are also anticipated.
Acknowledgements
I thank Dr Peter Karagiannis (CiRA, Kyoto University) for reading the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
References
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147.
- Keller G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev. 2005;19:1129–1155.
- Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872.
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676.
- Pessach IM, Ordovas-Montanes J, Zhang SY, et al. Induced pluripotent stem cells: a novel frontier in the study of human primary immunodeficiencies. J Allergy Clin Immunol. 2011;127:1400.e4–1407.e4.
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–144.
- Masters SL, Simon A, Aksentijevich I, et al. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. 2009;27:621–668.
- Goldbach-Mansky R. Immunology in clinic review series; focus on autoinflammatory diseases: update on monogenic autoinflammatory diseases: the role of interleukin (IL)-1 and an emerging role for cytokines beyond IL-1. Clin Exp Immunol. 2012;167:391–404.
- Niwa A, Heike T, Umeda K, et al. A novel serum-free monolayer culture for orderly hematopoietic differentiation of human pluripotent cells via mesodermal progenitors. PLoS One. 2011;6:e22261.
- Ohta R, Niwa A, Taniguchi Y, et al. Laminin-guided highly efficient endothelial commitment from human pluripotent stem cells. Sci Rep. 2016;6:35680.
- Yanagimachi MD, Niwa A, Tanaka T, et al. Robust and highly-efficient differentiation of functional monocytic cells from human pluripotent stem cells under serum- and feeder cell-free conditions. PLoS One. 2013;8:e59243.
- Tanaka T, Takahashi K, Yamane M, et al. Induced pluripotent stem cells from CINCA syndrome patients as a model for dissecting somatic mosaicism and drug discovery. Blood. 2012;120:1299–1308.
- Nakamura S, Takayama N, Hirata S, et al. Expandable megakaryocyte cell lines enable clinically applicable generation of platelets from human induced pluripotent stem cells. Cell Stem Cell. 2014;14:535–548.
- Doulatov S, Vo LT, Chou SS, et al. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell. 2013;13:459–470.
- Doulatov S, Vo LT, Macari ER, et al. Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci Transl Med. 2017;9:eaah5645.
- Haruta M, Tomita Y, Yuno A, et al. TAP-deficient human iPS cell-derived myeloid cell lines as unlimited cell source for dendritic cell-like antigen-presenting cells. Gene Ther. 2013;20:504–513.
- Haga E, Endo Y, Haruta M, et al. Therapy of peritoneally disseminated colon cancer by TAP-deficient embryonic stem cell-derived macrophages in allogeneic recipients. JI. 2014;193:2024–2033.
- Aksentijevich I, CDP, Remmers EF, Mueller JL, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56:1273–1285.
- Prieur AM, Griscelli C, Lampert F, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl. 1987;66:57–68.
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46:3340–3348.
- Feldmann J, Prieur AM, Quartier P, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71:198–203.
- Tanaka N, Izawa K, Saito MK, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum. 2011;63:3625–3632.
- Saito M, Nishikomori R, Kambe N, et al. Disease-associated CIAS1 mutations induce monocyte death, revealing low-level mosaicism in mutation-negative cryopyrin-associated periodic syndrome patients. Blood. 2008;111:2132–2141.
- Kashiwagi Y, Kawashima H, Nishimata S, et al. Extreme efficiency of anti-interleukin 1 agent (anakinra) in a Japanese case of CINCA syndrome. Clin Rheumatol. 2008;27:277–279.
- Kawashima H, Sato A, Nishimata S, et al. A case report of neonatal onset multisystemic inflammatory disease in Japan treated with continuous hemodiafiltration and steroid pulse therapy. Ther Apher Dial. 2007;11:232–234.
- Izawa K, Hijikata A, Tanaka N, et al. Detection of base substitution-type somatic mosaicism of the NLRP3 gene with >99.9% statistical confidence by massively parallel sequencing. DNA Res. 2012;19:143–152.
- Hanna J, Markoulaki S, Schorderet P, et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell. 2008;133:250–264.
- Romberg N, Al Moussawi K, Nelson-Williams C, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet. 2014;46:1135–1139.
- Kitamura A, Sasaki Y, Abe T, et al. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med. 2014;211:2385–2396.
- Canna SW, de Jesus AA, Gouni S, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46:1140–1146.
- Blau EB. Familial granulomatous arthritis, iritis, and rash. J Pediatr. 1985;107:689–693.
- Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29:19–20.
- Milman N, Andersen CB, Hansen A, et al. Favourable effect of TNF-alpha inhibitor (infliximab) on Blau syndrome in monozygotic twins with a de novo CARD15 mutation. APMIS. 2006;114:912–919.
- Otsubo Y, Okafuji I, Shimizu T, et al. A long-term follow-up of Japanese mother and her daughter with Blau syndrome: effective treatment of anti-TNF inhibitors and useful diagnostic tool of joint ultrasound examination. Modern Rheumatol. 2017;27:169–173.
- Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872.
- Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. 2003;278:5509–5512.
- Ogura Y, Inohara N, Benito A, et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818.
- Inohara N, Koseki T, Lin J, et al. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–27831.
- Abbott DW, Wilkins A, Asara JM, et al. The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol. 2004;14:2217–2227.
- Kanazawa N, Okafuji I, Kambe N, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105:1195–1197.
- Dugan J, Griffiths E, Snow P, et al. Blau syndrome-associated Nod2 mutation alters expression of full-length NOD2 and limits responses to muramyl dipeptide in knock-in mice. J Immunol. 2015;194:349–357.
- Rosenstiel P, Fantini M, Brautigam K, et al. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology. 2003;124:1001–1009.
- Totemeyer S, Sheppard M, Lloyd A, et al. IFN-gamma enhances production of nitric oxide from macrophages via a mechanism that depends on nucleotide oligomerization domain-2. J Immunol. 2006;176:4804–4810.
- Lee KH, Biswas A, Liu YJ, et al. Proteasomal degradation of Nod2 protein mediates tolerance to bacterial cell wall components. J Biol Chem. 2012;287:39800–39811.
- Janssen CE, Rose CD, De Hertogh G, et al. Morphologic and immunohistochemical characterization of granulomas in the nucleotide oligomerization domain 2-related disorders Blau syndrome and Crohn disease . J Allergy Clin Immunol. 2012;129:1076–1084.
- Osborne GE, Mallon E, Mayou SC. Juvenile sarcoidosis after BCG vaccination. J Am Acad Dermatol. 2003;48:S99–S102.
- Okafuji I, Nishikomori R, Kanazawa N, et al. Role of the NOD2 genotype in the clinical phenotype of Blau syndrome and early-onset sarcoidosis. Arthritis Rheum. 2009;60:242–250.
- Bustamante J, Boisson-Dupuis S, Abel L, et al. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol. 2014;26:454–470.