
ABSTRACT
EU laws have exerted a powerful influence on research, manufacturing, supply, sale and procurement of the products on which national health systems rely. Pre-Brexit, the UK was closely involved in the policy and operation of regulations affecting these goods. Since Brexit, ideological polarisation and the political salience of health during a global pandemic have driven a rhetoric of competitive divergence. However, active UK policy divergence to date is limited. It is unsettled whether the UK, as a small market in this global industry, genuinely seeks a higher risk, more industry-friendly regulatory paradigm. With regulatory and policy capacity also under strain, important decisions have been delayed. The position of Northern Ireland remains highly precarious, with negotiations ongoing on how to handle its unique partial status within the single market. The UK’s attempt to remain within the EU’s research funding programme has consequently been pushed into involuntary divergence.
Introduction
Few policy areas show the technical complexity and political challenges of Brexit as effectively as pharmaceutical and medical device regulation. EU laws have exerted a powerful influence for decades on research, manufacturing, supply, sale and procurement of the products on which national health systems rely. Each step across the whole life cycle of healthcare products, from clinical trials to NHS purchasing, was substantially covered by EU law during the period of UK membership, and in many cases by EU governance structures and institutions. While some funding measures discussed below also existed, the methods of policy in the sector generally exemplified the characterisation of the EU as a ‘regulatory state’ which uses rules, rather than fiscal measures or ownership, to achieve its ends (Majone, Citation1997).
The concept of regulation discussed in this article is encompassed by its definition by Black as ‘the intentional use of authority to affect behaviour of a different party according to set standards, involving instruments of information-gathering and behaviour modification’ (Black, Citation2022). This understanding of regulation includes ‘hard law’, ‘soft law’, social norms, standards and the market (for other understandings, see: Shleifer, Citation2005; Baldwin et al., Citation2010; Baldwin et al., Citation2012).
As Great Britain approaches the end of two years outside the single market, an important question for understanding these fields is how, and why, the UK government has responded to its ability to depart from EU laws. Most of these policies are now fully within the domestic powers of the UK to change. A broad exception is that most relevant EU laws continue to apply to Northern Ireland (NI) under the Protocol on Ireland/Northern Ireland (hereafter the Protocol) signed as part of the Withdrawal Agreement in 2019. However, a greater role for the UK system within NI is outlined in the EU-UK Windsor Framework agreed in February 2023 (UK Government, Citation2023).
Before and after Great Britain’s exit from the single market, competing political narratives counselled different approaches to this new policymaking freedom. Advocates of Brexit emphasised divergence from EU laws and policies. Industry representatives argued that continued alignment was more efficient, or the only approach immediately viable given the UK’s actual policymaking capacity.
We use a framework of three possible regulatory orientations for the UK after Brexit, and ask which the current situation best resembles. Are UK policies moving in parallel with those in the EU? Under this scenario, the UK makes deliberate choices to stay in step with the EU. Active changes are made, potentially under the political radar, to ensure that regulatory governance and content in the UK remain similar to or compatible with EU policies, as they continue to shift and evolve. Or is the UK position divergent, where policymakers make deliberate and different choices to those made within the EU? These could be in terms of regulatory governance (using UK institutions which inherit EU powers to make different individual choices, the ‘hardware’ of regulation (Armstrong, Citation2020), or in terms of regulatory content, where the UK actually changes the substance of its laws, the ‘software’ of regulation (Armstrong, Citation2020) away from those inherited).
Or is the UK, in fact, drifting? Here, UK politicians make few to no deliberate choices, resulting in temporary alignment, followed by divergence when the EU updates or replaces regulatory content, institutional structures or practices.
Whether the UK is acting in parallel to the EU, diverging from its policies, or drifting, is important because the current strategies pursued by UK policymakers limit the UK’s future degrees of freedom. Parallel choices, in certain instances, could involve the UK choosing bilaterally to retain mutual recognition or alignment by agreement with the EU, with mechanisms for evolution. Currently this is the case only in certain limited areas, but if a variant of membership of the European Economic Area were to return as a viable policy option, it would allow this robust form of continued alignment to apply across nearly all relevant fields.
In the following sections, we discuss the concepts of regulatory alignment, divergence and drift, then outline EU regulatory policies for healthcare products over the past decade and into the immediate future, to provide a baseline against which to understand and measure UK choices. We then examine the UK’s policy choices since EU law ceased to apply to Great Britain (GB) on 1 January 2021, also considering indications from successive UK governments about their priorities over this period. Thereafter, we evaluate constraints and pressures which have affected recent UK policymaking. Finally, we assess where these choices have fallen against our three scenarios (parallel, divergent, drifting), and what this indicates about the UK’s orientation, as it largely returns to holding full responsibility for healthcare product regulation for the first time in several decades.
Regulation: parallel, divergent or drifting?
When and why does Brexit cause institutional and policy change and along what lines? Regulatory alignment or divergence may be understood as arising through hierarchies, coordinating networks or more diffuse modes of governance (Armstrong, Citation2018). In ‘hierarchy’ mode, the reach of EU regulation outside EU Member States is explained as occurring through formal trade agreements (for example, with Switzerland, Vahl & Grolimund, Citation2006). Outside the single market, Great Britain enjoys at least theoretical freedom to diverge in many respects. In more diffuse (Börzel & Risse, Citation2012) conceptualisations of Europeanisation (Bradford, Citation2012; Lavenex & Schimmelfennig, Citation2009; Young, Citation2015), and ‘de-Europeanisation’ (Cygan, Citation2020; Kassim et al., Citation2021; Kassim et al., Citation2022), however, the reach of EU regulation is understood as operating in ways which may involve alignment irrespective of hierarchy. Alignment with the EU may be explained by the concept of ‘regulatory competition’, whereby countries seek ‘optimal’ regulatory structures so as to attract market actors (the much-disputed ‘Delaware effect’, Cary, Citation1974); practical ‘efficiency’ in regulatory capacity (Armstrong, Citation2020; De Maria et al., Citation2018); or a desire to increase (or sustain) trade with the EU (Bradford, Citation2012; Jancic, Citation2022; Prange-Gstöhl, Citation2009). The non-hierarchical or practical pull of EU alignment should not be overstated (Falkner & Gupta, Citation2009), but it is undeniable.
Regulatory drift occurs when policies are not updated in the face of societal change. Without reforms, existing policies, legal frameworks and/or institutions become a poor fit in the face of new challenges (Hacker, Citation2004; Streeck & Thelen, Citation2005). Regulatory drift can happen as a result of path dependency and institutional inertia (Esping-Andersen, Citation1999), or bureaucratic turf wars. It can occur as a result of differential legal jurisprudence (Vidigal, Citation2018). But it can also be politically driven (Hacker, Citation2004), with partisanship and ideological polarisation as significant contributing factors (Eisner, Citation2017).
In the case of UK health products regulation, disalignment is driven exogenously by the pace and timing of changes in EU laws and policies that are now outside of UK control. For healthcare products, underlying standards for safe and acceptable products and activity (‘regulatory content’) are largely internationally standardised in fields such as clinical trials and medical devices, though the demonstration of compliance varies, while in newer areas such as data governance, they may vary between major jurisdictions. The institutions and processes through which compliance is demonstrated and decided (‘regulatory governance’) vary widely and are subject to significant recent EU reforms. Outside the European Economic Area (EEA), by definition, the application and enforcement of EU standards by UK institutions and structures is excluded from being recognised by and within the EU. In effect, in the choice of form of Brexit taken in the Withdrawal Agreement and the EU-UK FTA, the UK was immediately forced to diverge in terms of regulatory governance.
By contrast, the internal legal form of the UK’s departure from the EU (the European Union (Withdrawal) Act 2018) meant all relevant law (‘regulatory content’) was generally retained as domestic statute, creating the immediate default result of remaining parallel to the EU. Yet, unless the UK took active measures to regain alignment, the active reform agenda of the EU meant drifting away would become the default setting.
Although politicians were forewarned about the likelihood of regulatory drift in the UK’s life sciences sector (Fahy et al., Citation2020; Health and Social Care Committee, Citation2018; Laurie, Citation2018), ideological polarisation in the UK between leavers and remainers has incentivised recent governments to emphasise active divergence from EU laws and policies in their political rhetoric. The Johnson, Truss and Sunak governments all stated that they hoped to use regulatory divergence to improve efficiency or to compete with the EU and other markets. In January 2022, the UK Government’s ‘Benefits of Brexit’ document emphasised an aggressive divergence agenda (Cabinet Office, Citation2022). Johnson stated that a benefit of his proposed ‘Brexit Freedoms Bill’ would be ‘enhancing our public health system by reforming clinical trials and medical devices legislation’ (Prime Minister’s Office, Citation2022). Upon becoming Prime Minister, Sunak promised to review or repeal all laws on the statute book by the end of 2023 (Parker, Citation2022).
This orientation towards divergence can be seen as rooted in a ‘policy frame’ (Schon & Rein, Citation1995) prevalent within circles supporting Brexit, emphasising considerations of sovereignty as a good in its own right, and rooted in a tradition of often heterodox economic and think tank analysis which sees reduced regulation as a strong determinant of economic performance. In contract, interest in alignment has been more prevalent among industry, civil society and factions within Government associated with the leadership of Cameron. It reflects a ‘policy frame’ focusing on facilitating trade and investment through meeting the demands of business, and on securing continued supplies for the NHS, reflecting the almost universal findings from the trade economics literature, and the analysis provided by the civil service.
Many aspects of regulatory governance for health products were immediately divergent post-Brexit, as Great Britain was excluded from the institutions and processes that carry out regulatory decision-making in the internal market. The EMA is the highest profile example, relocating to Amsterdam. GB-based ‘Notified Bodies’, private entities which provide quality and safety assurance for medical devices, were also no longer recognised as such by the EU. These changes meant that British and EU bodies could no longer take decisions across the same geographic scope as previously. MHRA, the UK regulator, took on power to approve a wide array of medicines previously reserved to the EU level, including new treatments for cancer.
Outside of these areas, however, implementing ‘divergence’ has proved difficult. Specific radical proposals floated in 2021 and 2022, e.g., fundamentally diverging from EU data protection rules, and overriding rules on the fair award of public contracts during a ‘crisis’, have been softened or dropped. Little actually happened after the publication of ‘Benefits of Brexit’. As of early 2023, the Retained EU Law Bill before Parliament would place a ‘sunset clause’ on all retained EU law at the end of the same year unless it is specifically adapted or retained by the actions of Ministers. However, no plans have been articulated to use it to remove or change regulations relevant to health products. These inconsistencies reflect reported tensions within the Conservative Party and the Government (Parker, Citation2022), where a policy frame oriented towards divergence clashes with politicians and civil servants who see evidence and mechanisms framing alignment as economically more beneficial.
Northern Ireland remains in a fundamentally different position, caught between alignment and being partially returned to GB’s largely static and responsive rules. The UK government’s prolonged push to reassimilate the NI medicines market with that of Great Britain, partly reflected in the 2023 Windsor Framework agreed with the EU, increases divergence from the EU in this part of the UK, as is the case across trade in goods (Murray & Robb, Citation2023). However, this represents a desire to recapture the status quo of easy movement of medical supplies before Brexit, rather than a radical new direction.
The following sections contrast changes in EU regulation with the UK’s regulatory response, highlighting areas where the UK remains in parallel alignment with the EU, has diverged from EU policies, or is exhibiting regulatory drift.
EU healthcare product regulation
The EU has considerable regulatory competence for almost every stage of the lifespan of a health product or innovation from invention to use in patient care. This competence applies through (i) research and clinical trials, including data protection, (ii) marketing authorisation, (iii) sale and supply, (iv) ‘health technology assessment’, determining whether products are efficient enough to be publicly funded, and (v) procurement or purchasing.
In all of these areas, regulatory competence is formally shared with Member States. EU law and practice, and the trade and institutions they create, have a powerful effect on healthcare and biomedical scientific innovation in EU (and EEA) Member States. The sector is an example of the ‘regulatory state’ paradigm (Majone, Citation1997) with this intensive regulation seeking to achieve a very wide range of policy objectives while production and supply remain almost entirely privately owned (although research and purchasing often occurs in the public sector). This section provides a brief overview of those changing laws and practices.
Research
EU law plays a central role in regulating clinical trials in Member States, while EU institutions and finance play an important part in biomedical research. This research role is delivered through Framework Programmes, seven-year initiatives which fund scientific work across a vast range of disciplines. Aligned with the EU’s Multiannual Financial Framework periods, the last such initiative, Horizon 2020, ran from 2014 to 2020: its successor, Horizon Europe, runs from 2021 to 2027. Costing €95.5 billion, Horizon Europe includes a cluster of research on health, as well as other initiatives widely used by life scientists, such as funding for junior researchers to work in other countries (European Commission, Citation2021c). While research priorities are set by the EU, non-Member States can reach agreements to join as associated members and their research institutions can bid for funds on the same basis. The EU’s ‘structural funds’, which promote development and education in less affluent regions, have also been a source of some investment in life sciences in the UK, particularly in Wales, where they have funded medical and life sciences centres such as the Brain Research Imaging Centre II in Cardiff (Dayan et al., Citation2021).
On 31 January 2022, after EU law had ceased to apply in the UK, the EU’s new Clinical Trial Regulation (European Union, Citation2014) entered into force, replacing the 2001 Clinical Trials Directive. The main goals of this new legislation are to provide higher standards, greater public transparency, a more favourable environment for conducting clinical research, and enhanced safety for clinical trial participants (EMA, Citation2022a). The Clinical Trial Regulation came after years of criticism of the previous system. Scientific and industry bodies criticised high administrative burdens and costs as delaying trials and making the EU a less attractive jurisdiction than the USA. Although the Directive was supposed to introduce a single set of regulations, in practice EU states implemented it in various ways (Hoey, Citation2007).
With this new system, the EU hopes to create a Clinical Trial process which is more efficient, more transparent, and more consistent throughout Europe. Synchronisation of assessment and supervision will take place via a new Clinical Trials Information System (CTIS), maintained by the European Medicines Agency (EMA) together with the EU Member States, EEA countries and the European Commission. All new trials have been required to use it from 31 January 2023. This will allow for single applications for trials run in different European countries and reduce administrative burdens for researchers and pharmaceutical companies. Other parts of the new Regulation aim to facilitate patient recruitment, allow patients to learn about trials in the EU, streamline safety reporting, and ensure coordinated/faster authorisation of clinical trials. The new system for clinical trials seeks to reproduce the European advantages of scale and mutual recognition, long offered for manufactured products, into the economic activity of research.
Clinical trials, like other research in the EU, must comply with EU data protection standards. These are primarily set out in the 2016 General Data Protection Regulation (European Union, 2016). This imposes many restrictions on the holding and processing of personal data, and in particular ‘sensitive’ personal data which includes information on physical and mental health. In most cases, the latter requires explicit consent for processing, although there is an exception for urgent interests of public health. The EU is considering amending its approach here, learning from Covid-19, to better support health ‘data altruism’ where patients or organisations give access to their information to benefit care or research (European Commission, Citation2022).
Authorisation and approval
The EU regulates medicines by approving pharmaceutical products through an extensive process of pre-market assessment, then regulating manufacture and import of the products approved using different mechanisms.
As is the established global practice, this process relies on clinical trials, followed by an application for marketing authorisation (licence) submitted to the regulator. The EU approach has a unique feature of different procedures working at cross-national and national levels. Products that use a new ingredient to treat certain major diseases, including cancer, and the ‘biological’ medicines made from living tissue which have played a dominant role in recent innovation, must be assessed for authorisation at the European level by the European Medicines Agency (EMA) through its Centralised Procedure. Products using existing ingredients and traditional pharmaceutical production are approved nationally, with each EU Member State having their own national authorisation procedure. New types of products for any disease may opt to use the Centralised Procedure.
Two other procedures, Mutual Recognition and the Decentralised Procedure, allow the duplication of national level authorisations across multiple EU member states (EMA, Citation2020). The Decentralised Procedure entails authorising medicines in more than one EU Member State in congruence. This procedure can be used for medicines that are not required to use the Centralised Procedure but that seek marketing authorisation in more than one EU Member State. The Mutual Recognition Procedure allows a marketing authorisation granted in one EU Member State to be recognised in other EU Member States.
Medical devices span a vast array of non-pharmaceutical but regulated health goods, from scalpels to pacemakers and robots. Some are disposable and cheap; others among the most sophisticated of manufactured products. Their authorisation is fundamentally different to medicines. As with clinical trials, Brexit occurred partway through a shift to newer models of EU regulation. Reforms were associated with widespread safety concerns, highlighted in a series of scandals, most notably regarding Poly Implant Prothèse silicone breast implants found to cause toxicity. These incidents accentuated long-held concerns that the EU was running a relatively forgiving system with a bias towards suppliers over patients. The EU Medical Device Regulation (MDR), adopted in May 2017 and entered into force in May 2021, seeks to address these concerns (EU 2017/245). An accompanying In-Vitro Diagnostics Regulation (IVDR) (EU 2017/746), covering products such as tests for infectious diseases, is being introduced in stages over the next five years. The new Regulations also seek to update policy to reflect the broader use of software in devices and the greater ability to use data to monitor products on sale.
As in other jurisdictions, the EU categorises medical devices into different classes depending on how high risk they are before assessing their conformity with requirements for reliability and safety. Higher risk products, which have important or long-term effects on patients’ bodies, need to be externally validated or audited to prove compliance. Conformity assessment is undertaken by one of over 70 privately owned ‘Notified Bodies’ in the different Member States on a fee-for-service basis. A positive assessment permits a manufacturer to stamp a device with the CE mark, signifying compliance with the rules of the single market.
However, clinical trials of the type used for medicines are not necessarily required. Changes under the MDR include broadening the range of products for which clinical trials are required, raising and reinforcing the criteria for the designation and oversight of Notified Bodies, and inclusion of certain aesthetic devices. Implementation of the MDR has been affected by the inability of Notified Bodies to cope with an increased workload. An industry group survey found that the majority of manufacturers intended not to transfer at least some products to the new system, presumably due to delays and fees: this would mean they are withdrawn from the EU market after 2024 (MedTech Europe, Citation2022).
The EU regulatory landscape is likely to evolve further. In 2021, the European Commission proposed a regulatory framework on artificial intelligence (AI) (European Commission, Citation2021b). This would categorise most uses of AI as a medical tool as ‘high risk’, requiring extensive transparency measures, risk assessment and human oversight.
Regulating for safety and compliance in the market
For approved medicines, processes largely defined by EU Directive 2001/83 aim to regulate manufacture and supply to ensure products are safe and that they comply with what has been approved. Crucial stages include batch testing, where a sample of medicine production is tested to make sure it is as it is supposed to be; Good Manufacturing Practice and other forms of good practice, guaranteed by inspection; and regulated Qualified Persons overseeing the release of medicines.
This approach allows the EU to reduce regulatory resources and offer suppliers access to a large market without duplicating work. There is almost no added regulatory cost to supplying across different Member States at the production and supply side. The system is, however, some way short of a truly single market in medicines due to the national level at which many authorisations are still defined, and the very different decisions taken at national level on which new medicines will be made available in the national health system at what point in time.
The 2011 EU Falsified Medicines Directive (2011/62/EU) introduces new dimensions of regulation at the wholesale and retail stages to prevent fraudulent medicines. Its key provisions include a unique identifier on each medicine box which is activated and deactivated in line with legislation to ensure traceability, and a device to prevent tampering. The Directive was not fully implemented until May 2021, after the UK had left the EU.
Under both the previous and current EU approaches to medical devices regulation, the ‘conformity assessment’ process which allows a type of device to be placed on the market is also connected to controls for safety and compliance at the stage of production. The new Medical Devices Regulation and In-Vitro Diagnostic Device Regulation introduce further requirements at this stage, including a traceability system with unique device identifiers, introduction of an implant card, and strengthened post-market surveillance, with more requirements for firms to regularly report back data on the products they have placed on the market to a central EU system.
Purchasing and providing
Use and pricing of medicines within national health systems is conducted primarily by Member States, though EU legislative acts do apply standards for transparency (89/105/EEC) and support cooperation (2011/24/EU). Most countries conduct ‘health technology assessments’ which calculate whether a product improves survival or quality of life enough to be worth its price.
The recently adopted Regulation 2021/2282 will augment a very minimal EU transparency requirement (Directive 89/105/EEC) with a European approach while respecting ‘the exclusive national competence of Member States’ to decide on prices and purchasing. It will result in ‘joint clinical assessments’ at EU level to calculate the benefits of centrally approved medicines and the highest risk devices, starting with medicines from 2025. Member States must then ‘take account’ of these, along with the costs, when making their national decisions on whether a product is worth funding. The Commission will also support the exchange of information and reports between Member States to save time and bring more expertise to bear in the process (European Union, 2021).
The EU’s procurement framework for public contracts for goods and services is governed by the 2014 Public Contracts Directive (2014/24/EU). The Directive covers procurement of supplies and services through public contracts, where the state purchases, leases or otherwise enters into commercial contracts with or from ‘economic operators’ – private firms or other non-state entities. The Directive aims to open up public contracts to suppliers from other EU Member States, by setting procedural and transparency requirements which apply when public entities enter into contracts with private suppliers.
Health services are able to qualify for the Directive’s ‘light regime’ whereby a higher threshold (€750,000) and more flexible measures apply for competitive tendering open to all Member States. Alternatives to service procurement are provided for by the Directive. Individual Member States have successfully interpreted these rules to take some public services such as health out of the remit of procurement. First, if a public authority controls its provider, it then no longer qualifies as an ‘economic operator’ covered by the procurement rules (as might, for example, be the case in Scotland). Secondly, in a ‘qualified provider’ model, patients choose their care provider and money follows them (as is widely the case in France, or for some services in England).
The United Kingdom response
Research
Associate membership of Horizon Europe was a UK objective, with varying degrees of emphasis, from 2018 onwards. This means less input in setting priorities, but continued access on a similar basis for institutions and researchers. Research is perhaps the clearest area in which the firmly pro-Brexit Johnson government consistently accepted and even promoted the benefits of continued alignment, possibly because, unlike regulation, science funding was not seen in terms of sovereignty and competition. Upon signing the EU-UK Trade and Cooperation Agreement (TCA) with the EU in late 2020, the UK Government described association as ‘collaboration on scientific research, fulfilling the Government’s manifesto commitment to make the UK a science and research superpower’ (UK Government, Citation2020a).
This aspiration was blocked by the EU throughout 2022, collateral damage from rancorous negotiations over the Ireland/Northern Ireland Protocol. Successful UK applicants for European Research Council grants were given a two-month ultimatum in 2022 to move to the EU or lose their funding. UK institutions currently are not eligible to lead projects or receive funding. This situation has been widely criticised as a blow for UK science, partly due to the lost prestige and ability to attract staff and funders from these grants (Thompson, Citation2022). Despite this, the UK government adopted policies to replicate associate membership to some extent, committing to take overpaying successful applicants to Horizon Europe (UKRI, Citation2022). In February 2023, with the announcement of the Windsor Framework to update the Protocol, European Commission President von der Leyern stated that she would be happy to commence UK association ‘immediately’ following its implementation (Vaughan, Citation2023). However, briefing to the UK press subsequently suggested that the UK government was reconsidering its aim to accede, despite the views of science bodies, in favour of trying to continue with a separate system (Parker et al., Citation2023).
The UK has replaced the Structural Funds with a domestic UK Shared Prosperity Fund for regional development funding, which has a remit covering research although primarily in the context of supporting small business. There has been a commitment that devolved UK countries, including Wales which historically particularly benefitted from the Structural Funds, will get at least equivalent funding under the new approach, but the Welsh Government has argued that this pledge has been broken by miscounting EU funds across the spending period (Dayan et al., Citation2021).
Through 2022 the MHRA held a consultation on proposed reforms for the clinical trials system in Great Britain. These are largely a project of re-alignment with the reformed EU system, coupled with some specific responses to competitiveness challenges the UK now faces outside of the EU. Like the new EU measures, the MHRA’s proposals set out maximum timeframes for decisions and responses. They create lighter-touch regimes for less risky trials, and look to increase transparency by publishing trial data as a default. However, they adopt different strategies for competitiveness, reflecting an inability to reduce duplication across countries as the EU approach does. The MHRA seeks to make the UK a competitive place to launch trials by combining ethics and research approval. Documents published at the conclusion of the consultation emphasised somewhat shorter turnaround time commitments than those set out by the EU (MHRA, Citation2023). There is also a contrasting approach to safety notifications. Whereas the EU regulation imposes obligations to notify Member States and regulators on safety incidents, the MHRA proposal requires fewer reports and notifications when a safety incident occurs. The major steps here were reiterated as policy in the Government’s response to consultation responses, despite several measures being disagreed to by a majority of those who replied to the exercise (MHRA, Citation2023). Risk assessment for the ‘Good Clinical Practice’ stage is also to be amended to look at projects as a whole.
NI continues to follow the EU’s 2014 Clinical Trials Regulation under the Protocol.
Data protection offers one of the clearest routes for a non-EU member to seek alignment and mutual recognition, and the UK has so far taken advantage of this. In June 2021, following interim decisions, the European Commission agreed to grant ‘adequacy’ status (European Commission, Citation2021a). This status recognises other jurisdictions which meet standards of protection, rule of law, supervision and international commitments set out in the General Data Protection Regulation (EU 2016/679), and allows data to flow back and forth with the same ease, and under essentially the same protections and requirements, as it did during the period of EU membership. The grant of it to the UK reflects its continued use of the GDPR as retained law. However, the Commission took the unusual step of giving this status an expiry date of four years (European Commission, Citation2021b), possibly reflecting concerns raised about divergence in the UK and about its provisions allowing security services to access personal data.
Various parts of the UK government have at different times signalled an interest in divergence and begun processes to diverge, though with mixed messages about the size of the change and whether it would result in giving up adequacy status. The 2021 Taskforce on Innovation, Growth and Regulatory Reform (TIGRR, Citation2022) commissioned by Johnson suggested removing basic consent mechanisms of GDPR. However, the UK Data Protection and Digital Information Bill ultimately brought forward in 2022 under Johnson proposed a relatively specific set of changes. Government ministers reassured MPs that ‘the Bill seeks to retain our data adequacy decision so businesses can trade freely’ (UK Parliament, Citation2022). During his campaign for Conservative Party leadership, Sunak claimed ‘the EU’s Byzantine rules are preventing British tech companies from innovating’, and that he would ‘remove’ its ‘burdens’ (Sunak, Citation2022). Following the ascension of Truss as Prime Minister, shortly succeeded by Sunak in October 2022, the Bill was parked. At the time of writing its fate remained unclear.
Authorisation and approval
Exit from the single market enables the UK to diverge in medicines authorisation with one significant limit: Northern Ireland remains for now subject to the EU system, with some allowances and alterations, under the Protocol on Ireland/Northern Ireland included in the Withdrawal Agreement. Even this would be altered under Article 4 of the new EU Regulation proposed as part of the 2023 Windsor Framework, which would mean that within a continuing framework of EU law, UK authorisations would determine medicine availability within NI (European Commission, Citation2023).
At present, Great Britain is still following inherited EU medicinal authorisation regulation and no clear proposals have yet emerged for changing this. MHRA operates a ‘reliance route’, accepting EMA decisions on cutting-edge and novel drugs covered by the centralised procedure. This approach is similar to that adopted by other regulators outside the large US and EU markets, such as Singapore’s Health Sciences Authority. MHRA continues to accept the decisions of individual EU member states through processes mirroring those of the EU.
While divergence in regulatory content remains limited, divergence in regulatory governance has occurred. MHRA is now discharging duties for Great Britain which previously sat with EMA, and as such it takes different decisions and is able to create different types of initiatives with other global and domestic bodies. MHRA has approved some products that have yet to be approved by EMA, and vice versa, such as COVID-19 vaccines. In the initial emergency use approval of Pfizer’s MRNA vaccine in 2020, MHRA approval occurred while the UK remained subject to EU law. Indeed, despite claims to the contrary by then Health Secretary Hancock, MHRA approval occurred under Regulation 174 of the 2012 Human Medicines Regulations, which make Article 5 of the 2004 EU Human Medicines Directive UK law (MHRA, Citation2020).
The later approval of the first bivalent COVID-19 booster vaccine in August 2022, however, did rest on divergent regulatory governance, with the UK approving this before the EU – at least for Great Britain. Northern Ireland again received authorisation through the emergency mechanism compatible with EU law, to which it remains subject (MHRA, Citation2022b). The EMA has subsequently approved the vaccine.
The UK is not included in the EU’s HTA Regulation, and has made no comparable attempts to conduct joint assessments with other countries. The UK introduced the Innovative Licensing and Access Pathway (ILAP) in March 2021. This initiative brings together MHRA and the bodies that conduct Health Technology Assessment (HTA) to decide whether medicines are cost-effective enough for the National Health Services in Scotland, England and Wales. The aim of the ILAP is to speed up licencing (‘authorisation’) and access to medicines, by carrying out each process contemporaneously.
This sort of cooperation is compatible with EU law, under which a previously existing Early Access to Medicines Scheme operates permitting the use of innovative medicines ahead of authorisation. The EMA also operates the PRIME scheme for rapid assessment of medicines targeting unmet needs (EMA, Citation2022b). However, ILAP does do something that would be hard to achieve for the EU, where approval processes are largely led by EMA and cost-effectiveness processes by national authorities. In this specific area, the UK has used divergence in regulatory governance to create a different, potentially improved, process.
In March 2023, the UK Chancellor of the Exchequer announced that MHRA ‘is exploring partnerships with trusted international agencies, such as in the US, Europe and Japan, to provide simple, rapid approvals for medicines and technologies that have received their approval from 2024’ (HM Treasury, Citation2023). This appears to signal an acceleration and expansion of the existing policy of accepting EU approvals. The announcement was in response to a formal letter from the UK Government Chief Scientific Advisor stating that ‘access to innovative medicines in the UK can contribute directly to poor health outcomes including survival rates for certain types of cancer, and there are areas where the UK is slower than the EU average’ (Chief Scientific Advisor, Citation2023). We found similar signs across innovative medicine generally in a recent report (McCarey et al., Citation2022). The Chancellor at the same time promised a ‘swift approval process’ from 2024 for specific fields, mentioning cancer vaccines and AI therapeutics.
GB has effectively been lagging in regulation for medical devices in comparison to the EU (Dobson, Citation2021). Current Great Britain legislation reflects the EU’s old Medical Devices Directive (MDD 93/42/EEC) and the In-Vitro Diagnostic Medical Devices Directive (IVDD 98/79/EC), which represented alignment only at the moment Great Britain left the single market. The UK is consequently running in essence an old version of EU legislation, without the updates to increase safety and consistency, or the reclassification of new and higher risk products as discussed above. The UK currently continues to accept EU conformity assessments.
Recently, MHRA announced a response to improving regulation regarding medical devices in the UK (MHRA, Citation2022a). EU CE marks under the new MDR will continue to be accepted for five years. This is the outcome of significant ongoing policy and political negotiation and tension. The UK government initially refused on general principle to countenance extension for medical devices or other products, with a minister telling the House of Commons that ‘there are no plans to extend the recognition of CE marking on the GB market, as this would mean recognising EU regulations, even where there is divergence’ (Hansard, Citation2021). The industry body ABHI campaigned against this, warning that ‘This deadline if applied, runs the risk of reducing or limiting medical product availability in the UK, and as a consequence, potentially increasing associated patient safety issues’ (ABHI, Citation2021). As in other areas, industry interests and the short-term needs of patients and the NHS won the day.
MHRA also set out a UK agenda for regulation reflecting an earlier consultation: as of November 2022, the statutory introduction of these appears to have been delayed. Close reading and analysis of the proposals suggests that the UK is aligning the new medical device regulation for Scotland, England and Wales very closely with the EU’s new regulation. The primary direction of any aspects of divergence is in allowing alignment with USA regulatory governance and content, rather than taking an independent direction.
Medical devices will be reclassified, largely aligned with the EU Regulations. Strengthened post-market surveillance requirements will be introduced, including timelines and the principle of sporadic safety reports. Traceability of medical devices will also be improved, via introducing Unique Device Identification and registering of devices, as under the EU Regulations. Further there will be a requirement for UK manufacturers and responsible persons for non-UK manufacturers to have a ‘Qualified Person’ meeting standards for regulatory compliance, analogous to the Person Responsible for Regulatory Compliance created in the EU Regulations.
Multiple other analogous measures mean that new medical device regulation in the UK will very closely resemble the EU (Van Ramsdonk & Hill, Citation2022). However, there are some differences. Classification will be based on global standards rather than those in the EU. An accelerated approval route will allow the UK regulator to accept approvals from different regulators globally, not only the EU. MHRA will also offer certificates for the Medical Device Single Audit programme, designed to transfer approvals across a set of non-EU countries including the USA. Perhaps most significant is a pre-approval route for innovative devices such as software as a medical device or devices using artificial intelligence or machine learning. This is analogous to conditional approval for medicines (Dennis, Citation2022), and is intended to offer a fast and competitive route to launch products in the UK, even as the evidence normal approval would require is gathered.
NI, meanwhile, is following the EU system as described above, including the 2017 Regulations which never applied in Great Britain. The recent agreement will not alter this (UK Government, Citation2023). MHRA acts as the regulator within Northern Ireland, but under Article 7 of the Protocol, a Northern Irish device destined to be shipped across the EU needs to obtain a CE mark from a regulator in an EEA country (UK Government, Citation2020b).
Regulating for safety and compliance in the market
The UK system for medicines on the market, like those elsewhere including the EU, requires suppliers to follow good manufacturing practice (GMP) or analogues for imports, based on inspections, and to carry out batch testing, which involves examining samples of medicines to ensure they comply with what has actually been approved. On the whole these processes are globally standardised, which means that there can be mutual recognition between, for instance, the USA and the EU. Under the EU-UK TCA, GMP continues to be mutually recognised, but batch testing is not. The UK continues to accept EU batch testing, but the EU does not accept Great Britain batch testing, despite there being no divergence in regulatory content. Consequently, batch testing of medicines in Great Britain has become less attractive for manufacturers.
Although medicines are not subject to tariffs, medicines moving between Great Britain and Northern Ireland face several regulatory barriers under the Protocol. Because NI is effectively part of the single market for nearly all medicines regulations, it is not notionally permitted to batch test products in Great Britain for sale there. Along with the divergence in authorisation discussed above, this is of particular concern to the NHS in Northern Ireland, since around 80 per cent of medicines used there come from Great Britain. Many of those working in the health sector in Northern Ireland see it as ‘absolutely unacceptable’ for a medicine to be available in Great Britain, but not Northern Ireland (McCarey et al., Citation2022b).
While medical product regulation has not been an emphasis of political rhetoric in Great Britain, the question of medicines trade between Great Britain and Northern Ireland has been intensely politicised. Prime Minister Johnson reportedly cited this specific issue during a G7 speech as a reason to take action suspending elements of the Protocol (Phillips, Citation2021), a threat later supported by legislation and resulting in a prolonged crisis in EU-UK relations. The EU attempted to ease movement of medicines between Great Britain and Northern Ireland by unilaterally adopting a Directive and a Regulation in April 2022 (European Union, 2022). Under these rules, batch testing in Great Britain can be accepted for medicines for the Northern Ireland market, but only so long as the UK ensures that medicines tested are not allowed to enter the EU’s single market, especially Ireland. The 2023 Windsor Framework, yet to be fully adopted, sets out more radical changes, including a permanent exemption on batch testing (European Commission, Citation2023).
The control of the sale of fraudulent medicines is an area of divergence. Since the 2011 EU Falsified Medicines Directive was not fully, formally implemented until Great Britain left the single market, it does not apply there. It does apply in Northern Ireland where the threat of having to deactivate and reapply unique identifiers and anti-tampering devices on each medicine box when shipping via Great Britain was viewed with concern by industry and health service officials (Dayan et al., Citation2022). Patients in Great Britain are thus considerably less well protected against fraudulent medicines than in the EU. This may in future extend to Northern Ireland as well, as it would be fully exempted from the FMD under the 2023 regulation proposed as part of the Windsor Framework (European Commission, Citation2023).
Purchasing and providing
The UK is currently aligned with the EU for procurement of goods and services. The 2015 Public Contracts Regulations implement and mostly transpose the 2014 EU Directive (Public Contracts Regulations, 2015) including the light-touch regime for services. Health-related goods are procured both through central agencies in Wales, Scotland, Northern Ireland and England, and by providers and pharmacists. Other goods, and services such as cleaning, are procured by bodies providing healthcare, e.g., NHS Trusts in England.
However, in 2019, the UK Government announced it would reform and streamline this framework, arguing that Brexit provided the opportunity to cut back on cumbersome and costly competition procedures (UK Cabinet Office, Citation2020). There is potential for improvement in this area as the heavy use of contract awards within the NHS, combined with the theoretical independence of NHS trusts, brought a particularly large proportion of health system activity under EU procurement rules compared to other EU countries. This represents an unusual area in which specific interest in a changed relationship with EU regulation was quite widespread even before Brexit within the NHS. The King’s Fund in a 2015 paper noted that ‘the opposition is not alone in wishing to tear up the current rule book’. It concluded that a different approach to managing the English NHS could have altered this picture within existing EU law (Collins, Citation2015).
The appeal of legislative divergence may primarily be that it enables clinical NHS services to be removed from these procurement requirements without the embarrassment, for a Conservative government, of unpicking some of the trappings of marketisation which give it the appearance of being a competitive market (Dayan et al., Citation2021).
A Procurement Bill, applying only to England and Wales, was introduced to the House of Lords in June 2022, containing both the ‘light-touch regime’ (s.8) and the power to be disapplied by Ministers via regulation, where health services or goods procurement is concerned (s.111). Meanwhile, the 2022 Health and Care Act disapplied the relevant section of previous legislation, and carved out a power for the Secretary of State to create standalone regulations and accompanying guidance for the procurement of specific NHS services and ‘other goods or services that are procured together with those health care services’ (s.79.1), ostensibly to improve integrated commissioning. These regulations would be the subject of a new ‘Provider Selection Regime’ (Health and Care Act, 2022) revoking the current 2013 National Health Service Patient Choice, Procurement and Competition Regulations, n.2. This proposed divergence has raised concerns about excessive discretion and poor accountability, because of the lack of clarity in proposals, the resulting potential for subjective judgement in awarding contracts, and the removal of existing avenues for legal redress. In the absence of clear, new options for legal redress and removal of redress options at EU level, judicial review would be the only available recourse; the hurdle to prove procedural unfairness or excessive use of an authority’s powers is very high. A substantive amount of detail on the mechanism for award decisions as well as for legal challenge and notice periods is left to statutory guidance – as opposed to future regulations – that has yet to be produced. This runs the risk of making challenge harder (especially for smaller providers) and reducing accountability for poor performance. It does not appear that the final legislation and regulation will be in force before well into 2023.
Original proposals for the Bill included an option for direct awards of contracts in times of ‘crisis’ that was the subject of criticism from transparency advocates (Transparency International, Citation2021). During the COVID-19 pandemic, some UK procurement contracts were seen to be untransparent, ineffective, and potentially dangerous, notoriously for purchasing PPE (NAO, Citation2020). Following criticism (Hansard, Citation2022), the Procurement Bill now states that the award must be necessary to ‘(a) protect human, animal or plant life or health, or (b) protect public order or safety’, and that this must be justified by [strict necessity] for reasons of ‘extreme and unavoidable urgency’, where a competitive tender cannot take place (s.39, Sch.5,13).
The House of Lords also amended the Bill to explicitly include the NHS in its remit, defeating Government. Reflecting the debate discussed above, peers voiced concerns that detail was lacking on what the Provider Selection Regime and the Bill would apply to and how, and that no impact assessment had been conducted on the interplay of these two concurrent sets of rules for the NHS, as well as about the still-unspecified nature of emergency exemptions from competitive tenders (HL Deb, 28 November 2022, vol.725, col.1576 −1600). A new clause reintroducing a Ministerial power to selectively disapply the Bill with respect to procurement by NHS England through Regulation was added at Committee Stage (Procurement Bill Deb, 31 January 2023, col.1–10). It is intended to enable and clarify the interaction between the Bill and the new Provider Selection Regime. The Bill will continue to be debated and potentially amended.
Finally, the new regime could potentially regain some control from larger private providers with stronger legal teams. However, it could also fragment the procurement landscape and further complicate it. The Competition and Markets Authority raised the risk of bid-rigging – another costly practice (OECD, Citation2012) – if the new system were poorly designed (CMA, Citation2021).
Parallel, divergent or drifting?
The UK’s policy response as described above varies by policy area, containing elements of parallel alignment with the EU, some limited divergence, and much drifting. In each case, the complexity of healthcare products regulation will make it difficult for outside observers to keep track of changes in anything more than a superficial way.
Some of the UK policies described above show a parallel regulatory orientation. On data protection, the UK has sought and accepted data adequacy status, recognising EU data protection standards. Prospects for radical divergence in the foreseeable future have been held out but appear limited. Regarding clinical trials, UK policy for a long-term system creates the appearance of divergence but substantively, proposals under consultation seek to align with new EU standards. There is micro-level tweaking of regulatory standards, seeking to be marginally quicker and easier, but on a broader level, the direction of travel is essentially aligned with the EU approach.
For medicine authorisations, the ‘reliance route’ reflects a conscious choice to remain aligned by accepting EU approvals. There appears to be a current commitment to even more rapid automatic transposition of EU decisions. The UK applies the same standards and processes as the EU but tries to apply them quicker and more efficiently – with success only in specific areas. It is aligned in regulatory content but operationally seeks competitiveness advantages through changes in governance which have shifted powers to the national level. Of course, there is no benefit in terms of access to the EU market: the EU does not accept UK authorisations. On medicine supply, the UK chose continued alignment with the EU on good manufacturing practice, with mutual recognition supported by a joint committee to deal with issues and divergence, and to some extent on batch testing where it recognises EU tests even without any reciprocity.
For medical devices, the UK will continue to recognise CE marking for at least a further five years. This is described as short term only, perhaps for ideological reasons, but the MHRA’s reform proposal is deliberate parallelism in the medium term. Some tweaks appear to superficially sound divergent, for example emphasising ‘global’ not ‘EU’ standards when these are essentially the same. But there are also some elements of intentional competitiveness enhancing divergence (see below). And in 2019, the UK agreed with the EU a path of very extensive continued alignment for Northern Ireland across nearly every relevant area, although grace periods, unilateral EU actions and potentially the recent Windsor Framework place it increasingly under divergent UK approaches.
Some choices demonstrate divergence. In medicines licencing, ILAP is trying to link MHRA and NICE requirements. In this regard, it is an active divergence strategy taking advantage of the concentration of competencies at a single level. The EU may be seen as exploring the same concept by trying to shift responsibilities for technology assessment to an EU level, though this is a very slow process. The announcement of a new ‘swift approval’ route for the UK in early 2023 on the face of it implies further divergence, but with detail lacking it may simply involve an attempt to address the MHRA’s problems with understaffing and underfunding following Brexit.
In procurement, the UK currently retains EU law, but new reforms are on the horizon. Although a new Bill has been introduced, many key details will be left for later guidance, and it is unclear whether the new regime claimed as ‘divergence’ for clinical services contracts would have been essentially possible anyway under the EU system through changing definitions.
In clinical trials, while the MHRA can be seen as aligning more with the new EU regulation in many respects, its proposals do contain some areas of genuine divergence. The proposals for a UK medical devices regulation also contain some areas of meaningful divergence, including the proposed condensed trial process. Most substantive may be the UK’s expanded participation in the multinational MDSAP programme, and its proposals to cooperate globally with other jurisdictions. These reflect divergence primarily in the direction of alignment and cooperation with other trading powers, led by the USA, rather than an independent path for the UK. The Procurement Bill currently before Parliament would introduce some different approaches to the inherited EU law. However, one of the more radical proposals around powers during ‘crisis’ has been considerably softened following criticism.
For Northern Ireland, insofar as certain elements of divergence have occurred in medicines licencing and may do in medical devices, the UK Government’s preferred option of UK regulation optionally applying in Northern Ireland, partially realised in the Windsor Framework, would create divergence from the EU approach which it currently applies, albeit with many exemptions and grace periods.
Other areas show regulatory drift as EU policies change more rapidly than UK decision-making. Pending the changes proposed by MHRA, Great Britain continues to use the previous system of clinical trials. Ironically, moving away from this, as the EU has now done, was perhaps the instance of divergence in health regulation which was most consistently raised as an opportunity from Brexit. The UK has not passed any new law on falsified medicines, similar to the EU FMD or otherwise.
In some areas policy is unclear because the UK has either not acted or has not fleshed out the details of new policy. Great Britain currently operates under the old model of the EU’s approach to authorising medical devices, and will until new legislation is passed domestically. As a consequence of negotiations on the Northern Ireland Protocol and thus no UK participation in Horizon Europe, the UK has been involuntarily forced into attempting to continue funding streams and processes domestically. And the UK currently has no equivalent to the Falsified Medicines Directive system. In Great Britain, this was partially implemented before being removed. In batch testing and other aspects of regulatory compliance, the UK continues to use retained EU law without meaningful changes in content, and to recognise EU tests. The EU has also not moved away from this position, but does not recognise UK tests.
Finally, some areas have the potential for drift, but drift has not occurred because EU policies have not changed. The EU’s role in health technology assessment is a good example. The UK is not required to use the single set of criteria set out in the new regulation to assess evidence, but no policy changes have been made in the UK in terms of what the national health services are willing to purchase in comparison to the EU, or vice versa. There is not meaningful divergence here, but also not really drift: the EU has not moved away from where the UK is because of the nature of the EU regulation.
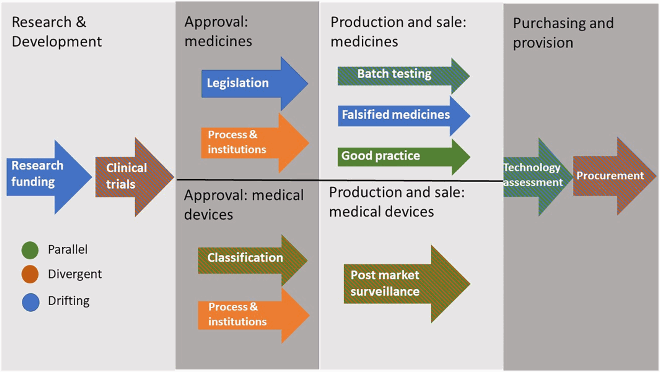
The diagram below illustrates, for medicines and medical devices in Great Britain, the current position of divergence, drift and parallelism across the regulatory pathway. The wide variety in different orientations can be clearly seen, as well as the limited degree of divergence. Fields where the UK has published policy documents indicating an intention to diverge or to remain parallel to the EU, but has not yet done so, are shown as mixed.
Discussion: constraints on UK policymaking potential
On paper, if we think of regulation through hierarchy, policymakers in London operate with considerable control. They now have few international commitments for most areas of healthcare product regulation. With a centralised state and a Parliamentary majority, the means for a radical overhaul are at hand. There is no legal or procedural barrier to prevent the Government in Westminster from entirely redesigning or eliminating almost any medicine or medical devices regulation applying to Great Britain. An aggressive and proactive strategy of competing by removing requirements nearly every developed country has – or indeed a strategy of imposing significantly more stringent requirements – could have been pursued. Alternatively, the UK could have reacted to each change in EU law to dynamically mirror its larger neighbour.
Yet in reality, and thinking of regulation as operating through more diffuse mechanisms as the complex picture above suggests, the UK government operates with far from a free hand. Ministers and officials are caught between several competing priorities, differ among themselves in ideology, assumption, and policy framing, and work in a world of competing policy actors whose grievances in many cases predate leaving the EU.
Since Brexit, several political divisions on these issues have been created both by clashes of objective interests, and, overlapping these, by differences of ideology and understanding. One source of conflict relates to the UK’s competitiveness as a jurisdiction in which to operate and supply medical products. The Protocol means that Northern Ireland is at least for now a different jurisdiction for medical products. It lies significantly outwith the ability of UK policymakers to take policy decisions, and yet the UK government retains moral and political accountability for outcomes in medicine supply and in economic competitiveness. Northern Ireland faces costs from remaining in the single market while its main source of pharmaceuticals, Great Britain, has left. The Windsor Framework brings it into partial alignment with Great Britain to resolve this. But it may also raise the costs of any further divergence in the rest of the UK, as in order to meet the requirements of the agreement, UK processes must be compatible with EU law.
Even if the UK remains completely aligned to the EU market in regulatory content, the end of mutual recognition at multiple stages of regulation means in the immediate term higher costs and greater burden on researchers, producers and importers. The need to go through a different process for access to the UK market, because different bodies are responsible, makes it less attractive and the UK a smaller global player. Simple drift at least does not worsen the situation, and, where it involves sticking with less demanding standards or simply accepting EU approvals and tests, it keeps costs low. But it makes the UK a passive rule-taker, and at the same time increases the difference between the UK and EU rules as EU regulation evolves and the EU finds new opportunities to streamline processes in fields such as clinical trials, leading to an administrative burden of operating in the UK.
Key policy actors here include the Government and ideological networks around it, and sophisticated representative and civil society bodies around science, medicines, and devices, such as the Association of the British Pharmaceutical Industry, Cancer Research UK and Wellcome Trust. These bodies, which the civil servants we interviewed for our Health and International Relations Monitor project (Dayan et al., Citation2021) see as powerful within the policy apparatus, were in general openly or implicitly concerned by Brexit and sought a high degree of alignment during negotiations with the EU. They embrace particular, targeted divergence around accelerated approvals, and in some cases lower demands from UK regulatory processes, but otherwise broadly favour continued parallel alignment as is seen in consultation responses to the MHRA processes on clinical trials and devices.
Another cleavage lies between actors pushing for better safety and quality and those who want more innovation. The dominant narratives of competitive divergence in authorisation currently emphasise accelerating clinical trials and increasing opportunities to place medicines and devices on the market before they complete the full traditional process. But this intrinsically risks increasing the ability for ineffective or even dangerous products to reach patients.
Pre-existing tension exists between the UK pharmaceutical industry and the publicly funded, perennially cash-strapped National Health Service. The former has long chafed under strict cost controls and the sluggish rate at which the NHS rolls out new, innovative, and profitable products. These constraints are the same for every country: what has changed is that the UK is now a small regulatory space in a global market. By making the NHS a less appealing place to do research and supply products, Brexit has potentially weakened the NHS bargaining position.
Beyond these concrete economic trade-offs and the interest groups representing them, policy tension and incoherence reflect the different understandings, priorities and positions of different political groups. An informal but closely connected coalition of MPs, think tanks, and professional advisors form a clear interest group committed in principle to divergence from the EU. They made up much of the personnel and leaders of the Leave campaign in 2016, and much of a parliamentary wing that repeatedly undermined May’s proposed Withdrawal Agreement with the European Union until 2019. The governments of Johnson, Truss, and Sunak have all represented their views sporadically. They are a significant constituency in governments flirting with ending data adequacy and setting end dates for the acceptance of CE marks for medical devices earlier.
However, this group took power with few specific ideas about applying the over-arching policy frame of competitive divergence and sovereignty within this sector. The only point relating to health product regulation in the campaign materials and proposals of Vote Leave was heavy criticism of the Clinical Trials Directive, described as a ‘stupid rule’ (Vote Leave, Citation2016). This was already being replaced by the point of UK exit. No particular proposals for fundamentally altering it were ever laid out by the Leave campaign figureheads who later became Prime Ministers and senior ministers, either before or during their time in government. It is illustrative that the UK’s eventual cautious proposals for reform were in fact left to a regulatory agency, with limited overt political engagement.
Meanwhile, there are indications that civil servants and even many government ministers continued to see these issues through a policy framing in which divergence was a burden, reflecting the general findings of analysis by the Treasury, Bank of England and Office for Budget Responsibility (HMT, Citation2018), and the messages received from influential industry groups and corporations. This was reflected perhaps most clearly in the UK’s choice during 2021 to actually seek continued full mutual recognition for medicines batch testing (Van Arnum, Citation2020) and good practice inspections, the latter successfully, and its consistent attempts to join Horizon Europe.
The ‘policy advisory system’ in the UK has been described as increasingly complex, disaggregated and potentially unstable (Diamond, Citation2020) and the cleavages on health products reflect this. The competing forces of these different groups formed around ideology and interests, and the resulting fluctuations in strategic direction, reflect this picture although the outcome has often been stasis rather than spectacular blunders.
The convoluted course of the UK’s intention to join the Horizon Europe science funding scheme from 2021 to 2023 illustrates several of these dynamics. Without an obvious agenda from groups favouring divergence, the overwhelming preference of the UK’s science establishment for alignment was reflected in a consistent UK desire to join as an associate member. However, after this was repeatedly delayed, reports suggested the UK government potentially moving away from accession. This was apparently less for positive reasons than because of a path-dependent effect where benefits had potentially been eroded over a prolonged period of being left out against their will.
Additionally, regulatory and political capacity is an important constraint (Diamond, Citation2021) following the UK’s split from an EU ecosystem where policy was previously made at multiple levels. The EU has continued to change and expand its regulation of health products rapidly, introducing long-planned systems for controlling falsified medicines, streamlining clinical trials regulation and legislating for greater cooperation in assessing which products ought to be purchased and provided by health systems.
Meanwhile, the UK has reduced the budget and capacity of the MHRA considerably, unwilling to replace the funding lost now that the regulator no longer receives contracts from the EMA. The impact of this is implicitly recognised in the commitment of new funding in early 2023 following a warning from the UK Chief Scientific Advisor that the slow approval of drugs might be worsening mortality. The UK Treasury in announcing this emphasised ‘allowing the regulator to maximise its use of Brexit freedoms’, reflecting a discourse of competitive divergence, but in parallel to this also promised faster acceptance of EU authorisation, driven by a pragmatic need to make the drifting regulatory system capable of keeping up.
A final important change directly related to Brexit, creating expanded regulatory potential rather than constraint, is that the UK – or at least GB – now has a more comprehensive set of powers at a single tier of government, while in the EU these are distributed widely because of separation of competences. This may create the potential for a subtly different form of ‘regulatory state’ (Levi-Faur, Citation2014; Majone, Citation2004) in the UK, one which is connected closely to executive and operational priorities rather than serving only as the impassive arbiter of neoliberal competition traditionally described in the literature. There was previously a bifurcation between areas of shared or even exclusive EU competence, and areas of national competence: for example, between centralised EMA medicines approvals, and NHS decisions on whether to fund products at a national level. Now the UK has the ability to link up the full scope of the regulatory life cycle, which the EU cannot easily do because of the division of competences. Whether the UK, or Great Britain, will be able to fulfil this potential remains to be seen.
Conclusion
There is an unresolved tension in UK politics as to whether the country’s ambition is to try to compete via regulatory change, accepting the costs of increased divergence and possibly risks to safety, or simply to manage the disruption of Brexit by alignment and unilateral recognition of EU regulation. While Brexit has vastly expanded the options to change how healthcare products are regulated in Great Britain, the UK is currently taking only small steps outside the box of possibilities to which it was confined as an EU member. This is juxtaposed with sporadic lunges, often symbolic, towards a more radical approach, from actors close to and inside recent governments whose enthusiasm for divergence is a matter of ideology, rather than being built around specific priorities for this sector.
Our account shows that the simple distinction between ‘divergence or not’ is not a meaningful question given the complexity of regulation in this area. Vitally, current political discourse around this issue incorrectly assumes a stationary EU, and fails to reflect the important changes which have resulted simply from the lack of continued mutual recognition in areas such as batch testing, and the inheritance of previous EU competencies by UK bodies. As we show above, the UK is running on a parallel course to the EU in some areas, although this may prove to be a temporary situation in many cases. It is divergent in some others. Often the divergence that has actually occurred to date reflects a change in regulatory governance, with new processes and choices becoming possible because new UK bodies have taken over the roles EU bodies used to hold. Action has been slow to non-existent, meanwhile, in changing regulatory content, which requires the difficult and complex process of actually rewriting inherited EU laws. In many areas, the UK is demonstrating regulatory drift, and will continue to drift for as long as the EU implements reforms while the UK stands still.
This conclusion is important because standing still is not a cost neutral option. Over time, drift matters because it is likely to impact investment decisions, research focus, and product availability within the NHS. Given the sums of money involved and the very long development cycles for pharmaceuticals and other novel technologies, there is a real risk that investors and suppliers seek to manage the uncertainty associated with drift by moving their activities elsewhere.
Lack of a consistent approach and widespread drifting away from the EU by default demonstrates that the UK lacks a discernible strategic direction for Great Britain – or even a way of having a public discussion about what the strategic direction should be. The third consecutive government to be nominally committed to an economic strategy of success through divergence remains, like its predecessors, divided as to where and whether this is actually possible in practice. The Retained EU Law Bill, with its general regulatory sunset clause accompanied by no new specific proposals for health product regulation, encapsulates the bind between the perceived political imperative to diverge and the difficulty in finding and constructing viable alternatives. Observers of the health policy process in the UK, and its accountability and transparency, should be worried about the lack of consistent, strategic regulatory action which results.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes on contributors
Mark Dayan
Mark Dayan is Brexit and Trade Programme Lead at the Nuffield Trust.
Tamara Hervey
Tamara Hervey is Jean Monnet Professor of EU Law ad personam at City, University of London.
Nick Fahy
Nicholas Fahy is the research group director for health and well-being at RAND Europe.
Elizabeth Vlachakis
Elizabeth Vlachakis is currently completing her MPH in Health Management & Policy at the University of Michigan and worked with the Health and International Relations Monitor programme based at the Nuffield Trust in 2022.
Martha McCarey
Martha McCarey is a Researcher at the Nuffield Trust.
Mark Flear
Mark Flear is a Reader at the School of Law, Queen’s University Belfast.
Scott Greer
Scott L. Greer, PhD is Professor of Health Management and Policy, Global Public Health, and Political Science (by courtesy) at the University of Michigan and is also Senior Expert Advisor on Health Governance for the European Observatory on Health Systems and Policies.
Holly Jarman
Holly Jarman, PhD is an Associate Professor at the Department of Health Management and Policy at the University of Michigan School of Public Health, and a Senior Social Scientist working with the US Army Corps of Engineers’ Engineer Research Development Center.
References
Legislation
UK
- Health and Social Care Act, (2012). https://www.legislation.gov.uk/ukpga/2012/7/section/75/enacted
- Procurement Bill [HL], Grand Committee, (26 October 2022). https://bills.parliament.uk/publications/48269/documents/2396
- The Public Contracts Regulations, Statutory Instrument n.102, (2015). https://www.legislation.gov.uk/uksi/2015/102/mad
- The National Health Service (Procurement, Patient Choice and Competition) (No. 2) Regulations, Statutory Instrument 500, (2013). https://www.legislation.gov.uk/uksi/2013/500/made
- Health and Care Act, (2022). https://www.legislation.gov.uk/ukpga/2022/31/enacted
- Retained EU Law (Revocation and Reform) Bill (2022). https://bills.parliament.uk/bills/3340/publications
EU
- Directive 2014/24/EU of the European Parliament and of the Council of 26 February 2014 on public procurement and repealing Directive 2004/18/EC Text with EEA relevance. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32014L0024
- Directive 93/42/EEC of 14 June 1993 concerning medical devices. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=uriserv%3AOJ.L_.2016.119.01.0001.01.ENG&toc=OJ%3AL%3A2016%3A119%3ATOC
- Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices. https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=celex%3A31998L0079
- Directive 2011/62/EU of the European Parliament and of the Council of 8 June 2011 amending Directive 2001/83/EC on the Community code relating to medicinal products for human use. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32011L0062
- Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02017R0745-20170505
- Regulation (EU) 2017.746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices. https://eur-lex.europa.eu/eli/reg/2017/746/oj
- Regulation (EU) 2016/679 of the European Parliament and of the Council of 27 April 2016 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=uriserv%3AOJ.L_.2016.119.01.0001.01.ENG&toc=OJ%3AL%3A2016%3A119%3ATOC
- Council Directive 89/105/EEC of 21 December 1988 relating to the transparency of measures regulating the prices of medicinal products for human use and their inclusion in the scope of national health insurance systems. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A31989L0105
- Directive 2011/24/EU of the European Parliament and of the Council of 9 March 2011 on the application of patients’ rights in cross border healthcare. https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2011:088:0045:0065:en:PDF
- European Union (Eur-Lex, 2021) Regulation (EU) 2021/2282 of the European Parliament and of the Council of 15 December 2021 on health technology assessment. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32021R2282
Literature
- Armstrong, K. A. (2018). Regulatory alignment and divergence after Brexit. Journal of European Public Policy, 25(8), 1099–1117. https://doi.org/10.1080/13501763.2018.1467956
- Armstrong, K. A. (2020). Regulatory autonomy after EU membership: Alignment, Divergence and the Discipline of Law. European Law Review, 45(2), 207–221. https://doi.org/10.2139/ssrn.3593482
- Association of British HealthTech Industries. (2021). How realistic is the 1st July 2023 deadline? https://www.abhi.org.uk/membership/members-area/monthly-reports-spotlights/2021/october/how-realistic-is-the-1st-july-2023-deadline/
- Baldwin, R., Cave, M., & Lodge, M. (2010). Introduction: Regulation, the field and the developing agenda. In R. Baldwin, M. Cave, & M. Lodge (Eds.), The Oxford handbook on regulation (pp. 3–16). Oxford University Press.
- Baldwin, R., Cave, M., & Lodge, M. (2012). Understanding regulation: Theory, strategy, and practice (2nd edn.). Oxford University Press.
- Black, J. (2002). Critical reflections on regulation. Australian Journal of Legal Philosophy, 27, 1–35.
- Börzel, T. A., & Risse, T. (2012). From Europeanisation to diffusion: Introduction. West European Politics, 35(1), 1–19. https://doi.org/10.4324/9781003061373
- Bradford, A. (2012). The Brussels effect. Northwestern University Law Review, 107(1), 1–67.
- Cabinet Office. (2020). Transforming Public Procurement. Government Publishing Service. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/943946/Transforming_public_procurement.pdf
- Cabinet Office (UK Government. (2022). The benefits of Brexit. https://www.gov.uk/government/publications/the-benefits-of-brexit
- Cary, W. L. (1974). Federalism and Corporate Law: Reflections upon Delaware. The Yale Law Journal, 83(4), 663–705. https://doi.org/10.2307/795524
- Chief Scientific Advisor. (2023). Letter from Sir Patrick Vallance to the Chancellor. HM Treasury. https://www.gov.uk/government/publications/pro-innovation-regulation-of-technologies-review-life-sciences-interim-report/letter-from-sir-patrick-vallance-to-the-chancellor#:~:text = Pro%2DInnovation%20Regulation%20of%20Technologies%20Review,-I%20recently%20wrote&text = The%20MHRA%20is%20recognised%20as,the%20UK%20system%20can%20act
- Collins, B. (2015). Procurement and competition rules: Can the NHS be exempted? King’s Fund. https://www.kingsfund.org.uk/sites/default/files/field/field_publication_file/procurement-competition-rules-kings-fund-mar-2015.pdf
- Competition and Markets Authority. (2021). Transforming Public Procurement: Response from the Competition and Markets Authority. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/974861/CMA_Response_to_Transforming_Public_Procurement_Green_Paper_.pdf
- Cygan, A. (2020). De-Europeanisation of UK regulatory governance and the future UK-EU trade relationship. ERA Forum 20, 509–529. https://doi.org/10.1007/s12027-020-00599-6
- Dayan, M., Hervey, T., Flear, M., Jarman, H., McCarey, M., Fahy, N., & Greer, S. L. (2022). Protocol politics mean hard times ahead for health in Northern Ireland. Nuffield Trust. https://www.nuffieldtrust.org.uk/news-item/protocol-politics-mean-hard-times-ahead-for-health-in-northern-ireland
- Dayan, M., McCarey, M., Hervey, T., Fahy, N., Greer, S. L., Jarman, H., Stewart, E., & Bristow, D. (2021). Going it alone: Health and Brexit in the UK. Nuffield Trust. https://www.nuffieldtrust.org.uk/research/going-it-alone-health-and-brexit-in-the-uk.
- De Maria, C., Di Pietro, L., Díaz Lantada, A., Madete, J., Ngaju Makobore, P., Mridha, M., Ravizza, A., Torop, J., & Ahluwalia, A. (2018). Safe innovation: On medical device legislation in Europe and Africa. Health Policy and Technology, 7(2), 156–165. https://doi.org/10.1016/j.hlpt.2018.01.012
- Dennis, A. (2022). Analysis: MHRA response to consultation on UK regulation of medical devices. Taylor Wessing. https://www.taylorwessing.com/en/insights-and-events/insights/2022/08/mhra-response-to-consultation-on-uk-regulation-of-medical-devices
- Diamond, P. (2020). Polycentric governance and policy advice: Lessons from Whitehall policy advisory systems. Politics & Policy, 48. https://doi.org/10.1332/030557320X15870482509817
- Diamond, P. (2021). Cummings’s evidence reinforces the impression that ineptitude over COVID-19 reflected errors made by individual ministers. That’s only part of the story. LSE Politics & Policy Blog. https://blogs.lse.ac.uk/politicsandpolicy/covid19-cummings-public-administration/
- Dobson, S.-J. (2021). The legislation and regulation of medical devices. In P. Feldschreiber (Ed.), The law and regulation of medicines and medical devices. Oxford University Press. https://doi.org/10.1093/oso/9780192847546.003.0007
- Eisner, M. A. (2017). Regulatory politics in an age of polarization and drift: Beyond deregulation. Routledge.
- Esping-Andersen, G. (1999). Social foundations of postindustrial economies. Oxford University Press.
- European Commission. (2021a). Data protection: Commission adopts adequacy decisions for the UK. European Commission. https://ec.europa.eu/commission/presscorner/detail/en/ip_21_3183
- European Commission. (2021b). Regulatory framework proposal on artificial intelligence. European Commission. https://digital-strategy.ec.europa.eu/en/policies/regulatory-framework-ai
- European Commission. (2021c). Horizon Europe: The EU research and innovation programme. Publications Office of the European Union. https://op.europa.eu/en/web/eu-law-and-publications/publication-detail/-/publication/93de16a0-821d-11eb-9ac9-01aa75ed71a1
- European Commission. (2022). Proposal for a Regulation of the European Parliament and of the Council on the European Health Data Space. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri = celex%3A52022PC0197
- European Commission. (2023). Proposal for a regulation of the European Parliament and of the Council on specific rules relating to medicinal products for human use intended to be placed on the market of Northern Ireland. https://commission.europa.eu/system/files/2023-02/COM_2023_122_1_EN_ACT_part1_v2.pdf
- European Medicines Agency. (2020). Authorisation of medicines. https://www.ema.europa.eu/en/about-us/what-we-do/authorisation-medicines
- European Medicines Agency. (2022a). Clinical Trials Regulation. https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials/clinical-trials-regulation
- European Medicines Agency. (2022b). PRIME: Priority Medicines. https://www.ema.europa.eu/en/human-regulatory/research-development/prime-priority-medicines
- European Union. (2014). Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC Text with EEA relevance. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32014R0536
- Fahy, N., Hervey N., Dayan, M., Flear, M., Galsworthy, M., Greer, S., Jarman, H., & McKee, M. (2020). Assessing the potential impact on health of the UK's future relationship agreement with the EU: Analysis of the negotiating positions. Health Economics, Policy and Law, 16(3), 290–307. http://doi.org/10.1017/S1744133120000171
- Falkner, R., & Gupta, A. (2009). The limits of regulatory convergence: Globalization and GMO politics in the south. Int Environ Agreements, 9, 113–133. https://doi.org/10.1007/S10784-009-9094-X
- Hacker, J. S. (2004). Privatizing risk without privatizing the welfare state: The hidden politics of social policy retrenchment in the United States. American Political Science Review, 98(2), 243–260. https://doi.org/10.1017/S0003055404001121
- Hansard. (2021). Department for Business Written Questions: House of Commons 1st March 2021.
- Hansard. (2022). Procurement Bill: House of Lords Volume 825, Monday 28th November 2022.
- Health and Social Care Committee. (2018). Brexit: Medicines, medical devices, and substances of human origin. House of Commons.
- Her Majesty’s Treasury. (2018). EU Exit Long-term economic analysis. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/760484/28_November_EU_Exit_-_Long-term_economic_analysis__1_.pdf
- Her Majesty’s Treasury. (2023). Spring Budget 2023. https://www.gov.uk/government/publications/spring-budget-2023/spring-budget-2023-html
- Hoey, R. (2007). Lancet. The EU Clinical Trials Directive: 3 years on thelancet.com/journals/lancet/article/PIIS0140-6736(07)60797-1/fulltext
- Jancic, D. (2022). Regulatory strings that bind and the UK Parliament after Brexit. Comp Eur Polit, 20, 566–584. https://doi.org/10.1057/s41295-022-00296-3
- Kassim, H., Davis, C., Ennis, S., & Jordan, A. (2021). UK Regulation after Brexit. UK in a Changing Europe.
- Kassim, H., Davis, C., Ennis, S., & Jordan, A. (2022). UK Regulation after Brexit revisited. UK in a Changing Europe.
- Laurie, G. (2018). How do we make sense of chaos? Navigating health research regulation through the liminality of the Brexit process. Medical Law International, 18(2–3), 110–134. https://journals.sagepub.com/doi/full/10.11770968533218799533#fn72-0968533218799533
- Lavenex, S., & Schimmelfennig, F. (2009). EU rules beyond EU borders: Theorizing external governance in European politics. Journal of European Public Policy, 16(6), 791–812. https://doi.org/10.1080/13501760903087696
- Levi-Faur, D. (2014). The odyssey of the regulatory state: From a ‘thin’ monomorphic concept to a ‘thick’ and polymorphic concept. Law and Policy, 35. https://doi.org/10.1111/lapo.12000
- Majone, G. (1997). From the positive to the regulatory state. Journal of Public Policy, 17(2). https://doi.org/10.1017/S0143814X00003524
- Majone, G. (2004). Regulating Europe. Routledge.
- McCarey, M., Dayan, M., Hervey, T., Flear, M., Jarman, H., Fahy, N., Greer, S. L., & Bristow, D. (2022). Brexit: Six years on. Nuffield Trust. https://www.nuffieldtrust.org.uk/research/health-and-brexit-six-years-on
- MedTech Europe. (2022). MedTech Europe Survey Report analysing the availability of Medical Devices in 2022 in connection to the Medical Device Regulation (MDR) implementation. MedTech Europe. https://www.medtecheurope.org/wp-content/uploads/2022/07/medtech-europe-survey-report-analysing-the-availability-of-medical-devices-in-2022-in-connection-to-the-medical-device-regulation-mdr-implementation.pdf
- MHRA. (2020). Regulatory approval of Pfizer/BioNTech vaccine for Covid-19. MHRA. https://www.gov.uk/government/publications/regulatory-approval-of-pfizer-biontech-vaccine-for-covid-19
- MHRA. (2022a). Government response to consultation on the future regulation of medical devices in the United Kingdom. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1085333/Government_response_to_consultation_on_the_future_regulation_of_medical_devices_in_the_United_Kingdom.pdf
- MHRA. (2022b). First bivalent covid-19 booster vaccine approved by UK medicines regulator. MHRA. https://www.gov.uk/government/news/first-bivalent-covid-19-booster-vaccine-approved-by-uk-medicines-regulator
- MHRA. (2023). Government response to consultation on legislative proposals for clinical trials. MHRA. https://www.gov.uk/government/consultations/consultation-on-proposals-for-legislative-changes-for-clinical-trials/outcome/government-response-to-consultation-on-legislative-proposals-for-clinical-trials
- Murray, C. R. G., & Robb, N. (2023). From the Protocol to the Windsor Framework. Northern Ireland Legal Quarterly.
- National Audit Office. (2020). The supply of personal protective equipment during the Covid-19 Pandemic. NAO. https://www.nao.org.uk/reports/supplying-the-nhs-and-adult-social-care-sector-with-personal-protective-equipment-ppe/
- OECD. (2012). Fighting Bid-Rigging in Public Procurement. https://www.oecd.org/competition/cartels/fightingbidrigginginpublicprocurement.htm
- Parker, G. (2022). UK plan to scrap all EU laws suffers new setback. Financial Times. https://www.ft.com/content/0c0593a3-19f1-45fe-aad1-2ed25e30b5f8
- Parker, G., Staton, B., & Bounds, A. (2023). Rishi Sunak holds back on rejoining Horizon after Brexit breakthrough. Financial Times. https://www.ft.com/content/409bccc7-8cb4-4f44-87c4-340d15147c55
- Phillips, A. (2021). Boris Johnson: 30 medicines unavailable in NI due to EU protocol. Chemist & Druggist. https://www.chemistanddruggist.co.uk/CD007244/Boris-Johnson-30-medicines-unavailable-in-NI-due-to-EU-protocol
- Prange-Gstöhl, H. (2009). Enlarging the EU’s internal energy market: Why would third countries accept EU rule export? Energy Policy, 37(12), 5296–5303. https://doi.org/10.1016/J.ENPOL.2009.07.070
- Prime Minister’s Office. (2022). Prime Minister pledges Brexit Freedoms Bill to cut red tape. Number 10 Downing Street. https://www.gov.uk/government/news/prime-minister-pledges-brexit-freedoms-bill-to-cut-eu-red-tape
- Schon, D. A., & Rein, M. (1995). Frame reflection: Towards the resolution of intractable policy controversies. Little, Brown.
- Shleifer, A. (2005). Understanding regulation. European Financial Management, 11(4), 439–451. https://scholar.harvard.edu/files/shleifer/files/02_eufm00121.pdf
- Streeck, W., & Thelen, K. (2005). Beyond continuity: Institutional change in advanced political economies. Oxford University Press.
- Sunak, R. (2022). We got Brexit done – now we must capitalise on the freedoms it gave us. Daily Telegraph.
- Taskforce for Innovation. Growth and Regulatory Reform (UK Government, 2022). Final Report https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/994125/FINAL_TIGRR_REPORT__1_.pdf
- Thompson, B. (2022). Horizon Europe unites scientists – let’s keep it separate from debates over the Northern Ireland protocol. Wellcome Trust. https://wellcome.org/news/horizon-europe-unites-scientists-northern-ireland-protocol-debates
- Transparency International. (2021). Concern over corruption red flags in 20% of UK PPE procurement. https://www.transparency.org.uk/track-and-trace-uk-PPE-procurement-corruption-risk-VIP-lane
- UK Government. (2020a). UK-EU Trade and Cooperation Agreement Summary. Government Publishing Service. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/962125/TCA_SUMMARY_PDF_V1-.pdf
- UK Government. (2020b). Agreement on the withdrawal of the United Kingdom of Great Britain and Northern Ireland from the European Union and the European Atomic Energy Community. National Archives. https://www.legislation.gov.uk/eut/withdrawal-agreement/contents/adopted
- UK Government. (2023). The Windsor Framework: A new way forward. HM Stationery Office. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1138989/The_Windsor_Framework_a_new_way_forward.pdf
- UK Parliament. (2022). Written Question and Answer 63308. https://questions-statements.parliament.uk/written-questions/detail/2022-10-14/63308
- UKRI. (2022). Horizon Europe funding: Help for UK applicants. UKRI. https://www.ukri.org/apply-for-funding/horizon-europe/
- Vahl, M., & Grolimund, N. (2006). Integration without membership: Switzerland’s bilateral agreements with the European union. Centre for European Policy Studies.
- Van Arnum, P. (2020). The UK and the Pharma Industry in a post-Brexit world. DCAT VCI. https://www.dcatvci.org/features/uk-and-pharma-in-a-post-brexit-world/
- Van Ramsdonk, A., & Hill, K. (2022). What will the new UK MHRA consultation mean in terms of medical device and IVD regulations? Emergo. https://www.emergobyul.com/blog/2022/07/what-will-uk-mhra-consultation-bring-terms-medical-device-and-ivd-regulations
- Vaughan, R. (2023). Windsor Framework NI protocol deal ‘immediately’ paves way for UK to rejoin £84bn Horizon Europe scheme. The Independent. https://inews.co.uk/news/politics/windsor-framework-ni-protocol-deal-immediately-paves-way-uk-rejoin-eu-84bn-horizon-research-scheme-2176626
- Vidigal, G. (2018). From the board: The nativist turn one year on: Is the system holding? Legal Issues of Economic Integration, 45(2), 111–119. https://doi.org/10.54648/leie2018006
- Vote Leave. (2016). Briefing: Leave is the best choice for science and technology. http://www.voteleavetakecontrol.org/briefing_science.html
- Young, A. R. (2015). The European Union as a global regulator? Context and comparison. Journal of European Public Policy, 22(9), 1233–1252. https://doi.org/10.1080/13501763.2015.1046902