
Abstract
Background: The objective of the present study was to evaluate the long-term safety and efficacy of tafamidis in treating hereditary transthyretin amyloid polyneuropathy.
Methods: A prospectively planned interim analysis was conducted on an on-going, phase III, open-label extension study following an 18-month, randomized, controlled study and 12-month, open-label extension study in ATTRV30M patients and a single-arm, open-label study in non-ATTRV30M patients. Thirty-seven ATTRV30M patients received placebo for 18 months, then switched to tafamidis and 38 ATTRV30M patients and 18 non-ATTRV30M patients continuously received tafamidis from day 1, up to 6 years.
Results: Long-term tafamidis was associated with a favourable safety/tolerability profile, without any unexpected adverse events. Patients initiating tafamidis at the start of the randomized study had less polyneuropathy progression versus those switching to tafamidis following 18 months of placebo and were less likely to progress to the next ambulatory stage after up to 6 years follow-up. In the patients who switched from placebo to tafamidis, polyneuropathy progression and deterioration in quality of life slowed significantly during long-term tafamidis treatment as compared with the previous placebo treatment. In non-ATTRV30M patients, some polyneuropathy progression was observed across all efficacy measures.
Conclusions: These data provide evidence for the long-term (up to 6 years) safety and efficacy of tafamidis.ClinicalTrials.gov: NCT00925002
Introduction
Hereditary transthyretin amyloidosis (ATTR) with polyneuropathy, traditionally referred to as familial amyloid polyneuropathy (ATTR-FAP), is a rare, progressive and fatal hereditary disorder associated with mutations in the TTR gene. These mutations result in destabilization of the tetrameric structure of the TTR protein, causing it to dissociate, misfold and aggregate as amyloid in peripheral nerve tissues, the heart and other organs [Citation1,Citation2]. Accumulation of TTR-derived amyloid fibrils results in severe, disabling sensorimotor disturbances (loss of sensation, pain, pre-disposition to severe tissue damage from inadvertent injuries, muscle weakness and loss of ambulation) and varying degrees of autonomic, cardiovascular, gastrointestinal, renal, leptomeningeal and urogenital dysfunction [Citation1]. Patients with ATTR amyloidosis can also present with phenotypes that consist predominantly of cardiologic or leptomeningeal involvement [Citation1]. If untreated, death occurs, on average, approximately 10 years after ATTR-FAP disease onset [Citation1].
Presenting symptoms and disease course of ATTR-FAP are influenced by the underlying TTR mutation and by geographic location [Citation3–5]. The most common and widely studied TTR mutation worldwide is ATTRV30M (p.TTRV50M) [Citation1,Citation6]; non-ATTRV30M mutations are increasingly reported and often associated with a higher incidence of mixed phenotypes (neuropathy, cardiomyopathy and leptomeningeal complications) and with a more rapid and severe disease course [Citation5].
ATTR-FAP purportedly affects up to 10,000 people worldwide [Citation7]. However, its prevalence is widely believed to be under-estimated because diagnosis of ATTR-FAP is confounded by the non-specific nature of symptoms and a lack of disease awareness [Citation8,Citation9]. A more recent review suggests that ATTR-FAP prevalence may in fact range from 5526 to 38,468 people worldwide [Citation10]. Accurate, timely diagnosis of ATTR-FAP is critical for early initiation of treatment to improve clinical outcomes and ameliorate disease progression [Citation7,Citation9].
Orthotopic liver transplantation is the only non-pharmacologic disease-modifying intervention for ATTR-FAP. However, liver transplantation is limited by shortage of cadaveric grafts and resultant demand–supply imbalance for liver transplants [Citation11], restriction to carefully selected patients (typically those with ATTRV30M mutations) [Citation12,Citation13], considerable morbidity and mortality and inherent risks associated with lifelong immunosuppression.
Efforts to develop safe, effective medicines to halt or slow ATTR-FAP disease progression are underway. Several pharmacologic compounds are in various stages of development, including TTR gene silencers and amyloid fibril disrupters [Citation7,Citation14]. The non-specific TTR stabilizer diflunisal (a non-steroidal anti-inflammatory drug [NSAID]) has demonstrated effect in slowing ATTR-FAP progression [Citation15]; however, it is not approved to treat ATTR-FAP and safety is a concern given the risk of serious gastrointestinal and renal side effects associated with chronic NSAID use [Citation16]. Tafamidis, a highly specific TTR stabilizer administered orally once daily, is the only medicine approved to delay disease progression in ATTR-FAP, and is approved in the European Union and several South American and Asian countries [Citation17–20].
An 18-month, double-blind, randomized, placebo-controlled study of tafamidis in 128 ATTRV30M patients with early stage ATTR-FAP and its subsequent 12-month open-label extension study demonstrated the safety and efficacy of tafamidis in delaying the progression of polyneuropathy [Citation21,Citation22]. At the end of the extension study, neurologic function was better preserved among patients who continuously received tafamidis than among those who received 18 months of placebo followed by 12 months of tafamidis, and underscores the benefit of early intervention [Citation22,Citation23]. A 12-month open-label study in 21 non-ATTRV30M patients further demonstrated the generalizability of the TTR-stabilizing effect, and indicated clinical efficacy of tafamidis for delaying disease progression across several other TTR variants [Citation24]. Tafamidis was associated with a favourable safety profile across these studies [Citation21,Citation22,Citation24].
A second, on-going, open-label extension study was initiated to study the long-term safety and efficacy of tafamidis in ATTRV30M and non-ATTRV30M patients with ATTR-FAP who completed the above-described studies [Citation21,Citation22,Citation24]. This prospectively planned interim analysis was conducted to confirm the long-term safety of tafamidis and to evaluate its effects on polyneuropathy progression after up to 6 years of treatment for ATTR-FAP.
Design and methods
Study design and participants
Study B3461023 (ClinicalTrials.gov: NCT00925002) enrolled ATTRV30M amyloidosis patients who completed the original registration study (Study Fx-005) and 12-month open-label extension (Study Fx-006) [Citation21,Citation22], and non-ATTRV30M patients who completed a separate 12-month open-label study with tafamidis (Study Fx1A-201) [Citation24] (). The current, on-going, open-label extension study was initiated in August 2009 for a period of up to 10 years to obtain long-term safety and efficacy data and to continue to provide tafamidis to these patients until its availability by prescription in their respective countries. All participants received oral tafamidis meglumine (20 mg soft gelatin capsule once daily). Eligibility criteria of the parent studies are published elsewhere [Citation21,Citation24]; those of Study B3461023 are listed in Supplementary Box S1. The study was conducted at nine study sites in eight countries (Argentina, Brazil, France, Germany, Italy, Portugal [two sites], Sweden and the USA) [Citation21,Citation24]. The study was approved by local regulatory authorities and institutional review boards at each site and conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent.
Figure 1. Patient population. aClinicalTrials.gov: NCT00409175 [Citation21]. bOther reasons for screen failure included protocol non-compliance (n = 5), withdrawal of consent (n = 2), potential liver transplant (n = 1), site closure (n = 1), missing reason (n = 3). cThe patient discontinued due to an AE of increased nausea without a post-baseline efficacy assessment. dThe reasons were discontinuation due to an AE of worsening nausea, or due to a negative TTR ATTRV30M genotype, without post-baseline efficacy assessment. eThe three AEs leading to discontinuation were diarrhoea, urticaria and pregnancy (normal outcome). fThe two AEs leading to discontinuation were paraesthesia/fatigue and worsening cardiac amyloidosis. gClinicalTrials.gov: NCT00791492 [Citation22]. hTwo patients who completed the tafamidis arm of the Fx-005 study did not enrol into Study Fx-006; one was pregnant at the Fx-005 month 18 visit and the other wanted to become pregnant and declined to participate in Study Fx-006. iThree patients who completed the placebo arm of the Fx-005 study did not enrol into Study Fx-006; one underwent a liver transplant, two declined to participate (one due to an imminent liver transplant, one did not want to attend clinic visits). jClinicalTrials.gov: NCT00630864 [Citation24]. kIncludes one combined liver and heart transplant. lThe AE was a transient ischemic attack. mClinicalTrials.gov: NCT00925002. nIn total, two patients who completed study Fx-006 declined to participate in Study B3461023. oThe treatment-emergent AE resulting in discontinuation was renal impairment (considered treatment-related), and the causes of the deaths were cardiac failure and lymphoma (death occurred 10 days after permanent discontinuation from tafamidis). pThe treatment-emergent AE resulting in discontinuation was sepsis, and the cause of the patient death was ileus. qThe treatment-emergent AEs resulting in discontinuation were faecal incontinence (considered treatment related, patient died 2 years after study discontinuation of unknown cause) and gastrointestinal disorder. The causes of the four deaths were cardiac arrest, sepsis, amyloidosis disease progression and heart transplant (death occurred 18 days after permanent discontinuation of tafamidis). rA total of 56 patients completed Study B3461023 when tafamidis became accessible via prescription upon regulatory approval of tafamidis for treatment of ATTR-FAP in their country. AE: adverse event; ATTR-FAP: transthyretin familial amyloid polyneuropathy.
![Figure 1. Patient population. aClinicalTrials.gov: NCT00409175 [Citation21]. bOther reasons for screen failure included protocol non-compliance (n = 5), withdrawal of consent (n = 2), potential liver transplant (n = 1), site closure (n = 1), missing reason (n = 3). cThe patient discontinued due to an AE of increased nausea without a post-baseline efficacy assessment. dThe reasons were discontinuation due to an AE of worsening nausea, or due to a negative TTR ATTRV30M genotype, without post-baseline efficacy assessment. eThe three AEs leading to discontinuation were diarrhoea, urticaria and pregnancy (normal outcome). fThe two AEs leading to discontinuation were paraesthesia/fatigue and worsening cardiac amyloidosis. gClinicalTrials.gov: NCT00791492 [Citation22]. hTwo patients who completed the tafamidis arm of the Fx-005 study did not enrol into Study Fx-006; one was pregnant at the Fx-005 month 18 visit and the other wanted to become pregnant and declined to participate in Study Fx-006. iThree patients who completed the placebo arm of the Fx-005 study did not enrol into Study Fx-006; one underwent a liver transplant, two declined to participate (one due to an imminent liver transplant, one did not want to attend clinic visits). jClinicalTrials.gov: NCT00630864 [Citation24]. kIncludes one combined liver and heart transplant. lThe AE was a transient ischemic attack. mClinicalTrials.gov: NCT00925002. nIn total, two patients who completed study Fx-006 declined to participate in Study B3461023. oThe treatment-emergent AE resulting in discontinuation was renal impairment (considered treatment-related), and the causes of the deaths were cardiac failure and lymphoma (death occurred 10 days after permanent discontinuation from tafamidis). pThe treatment-emergent AE resulting in discontinuation was sepsis, and the cause of the patient death was ileus. qThe treatment-emergent AEs resulting in discontinuation were faecal incontinence (considered treatment related, patient died 2 years after study discontinuation of unknown cause) and gastrointestinal disorder. The causes of the four deaths were cardiac arrest, sepsis, amyloidosis disease progression and heart transplant (death occurred 18 days after permanent discontinuation of tafamidis). rA total of 56 patients completed Study B3461023 when tafamidis became accessible via prescription upon regulatory approval of tafamidis for treatment of ATTR-FAP in their country. AE: adverse event; ATTR-FAP: transthyretin familial amyloid polyneuropathy.](/cms/asset/5713f006-ad5a-4b6f-b3a2-93c50e82fbd4/iamy_a_1357545_f0001_b.jpg)
Randomization and blinding
The original 18-month registration study that provided source data for the current analysis was a randomized, double-blind, placebo-controlled study [Citation21]. All other studies contributing source data, including the on-going 10-year extension study, were open-label, single-treatment study designs [Citation22,Citation24].
Outcome measures
Neuropathy Impairment Score-Lower Limbs (NIS-LL) [Citation25], total quality of life (TQOL) as measured using the Norfolk Quality of Life-Diabetic Neuropathy questionnaire (Norfolk QOL-DN) [Citation26,Citation27], Karnofsky performance status (not assessed in the ATTRV30M parent studies), body mass index (BMI) and modified BMI (mBMI) [Citation28] were evaluated at 6-month intervals during the parent studies and at 12-month intervals during the on-going extension study. The NIS-LL and its three components (sensation, reflexes and muscle weakness) assess the severity of peripheral neuropathy in the lower limbs and are considered valid, reliable measures of disease severity [Citation29].
Ambulation assessment by modified Polyneuropathy Disability Score was added in a protocol amendment in June 2011. Available ambulatory assessment data, recorded as part of routine patient care prior to this amendment, were retrospectively collected from study sites and ambulation status was mapped to clinical staging of ATTR-FAP based on Coutinho et al. [Citation30] (Supplementary Table S1). Albumin testing, a pre-requisite for calculation of mBMI (included in the parent studies), was added to the B3461023 study protocol in the same amendment. Accordingly, some ambulation and mBMI results were missing. Due to high rates of missing data among non-ATTRV30M patients, ambulation data are not reported and BMI is reported instead of mBMI for that cohort (ambulation data missing for 6 of 18 patients, mBMI data missing for at least eight of 18 patients at months 24, 36 and 48).
Safety evaluations included the assessment of treatment-emergent adverse events (AEs) and concomitant medications at 3-month intervals, clinical laboratory evaluation at 6-month intervals and physical examination and 12-lead electrocardiogram at 12-month intervals.
Statistical analysis
The cut-off date for this interim analysis was 31 December 2014. The efficacy analysis was based on the intent-to-treat (ITT) population, including all patients who received at least one dose of tafamidis in the on-going extension, and who had a baseline and at least one post-baseline NIS-LL assessments. The safety population comprised all enrolled patients who received at least one dose of tafamidis in the on-going extension study. Data were summarized separately for ATTRV30M and non-ATTRV30M patients. ATTRV30M patients were presented by treatment group as those who received tafamidis during both the registration study and open-label studies (tafamidis-to-tafamidis [T–T] group) and those who received placebo during the registration study and were switched to tafamidis on entry into the first open-label extension study (placebo-to-tafamidis [P–T] group).
Baseline was defined as the baseline of the respective parent study [Citation21,Citation24]. For ATTRV30M patients, data from baseline to month 18 are from the registration study; data from month 18 to 30 are from the first open-label extension study; and data beyond month 30 are from the on-going long-term extension study. For non-ATTRV30M patients, data from baseline to month 12 are from the initial 12-month study [Citation24] and data after month 12 are from the on-going long-term extension study.
For continuous efficacy outcomes, least squares mean (LSMean) and their standard error (SE) and 95% confidence interval (CI) were estimated for change from baseline for the ATTRV30M T–T, ATTRV30M P–T and non-ATTRV30M groups, and for the treatment group difference in change from baseline for ATTRV30M T–T versus P–T by repeated measures analysis of covariance with adjustment for baseline values (analysis models described in Supplementary Box S2). Categorical data were summarized as number, percentage and 95% CIs.
Kaplan–Meier estimates and their 95% CIs were computed for time to progression to next ambulatory stage from the first dose of study drug, and compared between treatment groups by the log-rank test. Patients who completed/discontinued the study and on-going patients without stage progression were censored at their last follow-up visit. Patients receiving a liver or heart transplant were censored at the time of transplant. If the date of a stage progression was missing, the mid-point date between the last known date of the previous stage and the first assessment date of the new stage was used.
The original, double-blind registration study in ATTRV30M patients and the subsequent open-label extension studies were conducted sequentially under separate protocols. In combination, these studies represent the structure of a delayed-start design [Citation31,Citation32] wherein patients in the original study were assigned randomly to either active treatment (tafamidis) or placebo and after a pre-specified delay of 18 months, those patients who initially received placebo were switched to active treatment (tafamidis) and those originally assigned to active treatment (tafamidis) continued on tafamidis. In an effort to better elucidate the disease-modifying effects of tafamidis in patients with an earlier versus later treatment start, additional slope analyses using a piecewise linear mixed-effects model with adjustment for the baseline covariate effect on the rate of progression were performed to assess the rate of disease progression and change in quality of life across the T–T and P–T study groups based on data collected for up to 66 months (analysis models described in Supplementary Box 2).
Results
Study population and baseline characteristics
A total of 93 patients were enrolled into the long-term open-label extension study: 38 ATTRV30M T–T patients, 37 ATTRV30M P–T patients and 18 non-ATTRV30M patients (). At the time of the data cut-off, 17 (18.3%) of the 93 enrolled patients were on-going in the study, 56 (60.2%) had completed the study when tafamidis became available to them by prescription and 20 (21.5%) had permanently discontinued the study. Reasons for discontinuation were death (n = 7), withdrawal of consent (n = 6), AE (n = 4) or other reasons (n = 3) (detailed below). Two patients had a liver transplant and one received a heart transplant. Per protocol, the liver-transplant patients continued in the study, but permanently discontinued tafamidis before undergoing surgery, and data from annual post-transplant visits were excluded from safety and efficacy analyses. The heart transplant patient was permanently discontinued from the study. Compared with ATTRV30M patients, non-ATTRV30M patients were older, had longer symptom duration, greater neurologic impairment and worse quality of life at baseline ().
Table 1. Baseline characteristics in the ITT population.
Treatment exposure
For patients who completed or discontinued the study, mean (SD) cumulative tafamidis exposure was 5.1 (0.8) years in the ATTRV30M T–T group, 3.5 (0.9) years in the ATTRV30M P–T group and 3.6 (1.0) years in the non-ATTRV30M group (Supplementary Table S2).
Safety
Tafamidis was generally well-tolerated, with no unexpected AEs. Eighty-six (92.5%) patients experienced at least one AE, 24 (25.8%) experienced at least one serious AE (SAE) and 21 (22.6%) had severe AEs (Supplementary Tables S3–S5). The most common SAEs included cardiac failure and chest pain (n = 3 [3.2%] each) and sepsis, urinary tract infection and transient ischemic attack (n = 2 [2.2%] each). One patient had treatment-related SAEs (pericardial effusion, cardiac disorder and aggravated renal function) and discontinued the study due to renal impairment. The most frequently reported severe AEs were cardiac failure, constipation and pleural effusion (n = 3 [3.2%] each); and sepsis, fall, transient ischemic attack and renal failure (n = 2 [2.2%] each). Of all severe AEs, one (pericardial effusion) was considered treatment-related. Twenty-three (24.7%) patients experienced at least one treatment-related AE, with headache, oedema peripheral and urinary tract infection (n = 2 [2.2%] each) most frequently reported (Supplementary Table S6).
In total, there were four (4.3%) discontinuations due to AEs (one faecal incontinence, one gastrointestinal disorder, one renal impairment [described under treatment-related SAEs above], one sepsis), two of which (faecal incontinence and renal impairment) were considered treatment-related. Another eight (8.6%) patients temporarily discontinued tafamidis due to AEs.
Deaths
Seven (7.5%) patients died during the study or within 30 days of completion/discontinuation (). Causes of death were cardiac arrest, cardiac failure, disease progression, heart transplant (18 days post-therapy), ileus, lymphoma (10 days post-therapy) and sepsis. One additional post-therapy death of unknown cause occurred 2 years after study discontinuation for faecal incontinence (listed among discontinuations due to AE). None of these eight deaths were treatment related; three patients were ATTRV30M and five patients were non-ATTRV30M.
The most common laboratory abnormalities in ATTRV30M patients were increased neutrophils (T–T, n = 8/38 [21.1%]; P–T, n = 6/37 [16.2%] P–T), decreased lymphocytes (T–T, n = 7/38 [18.4%]; P–T, n = 5/37 [13.5%] P–T), increased gamma glutamyl transferase (T–T, n = 4/38 [10.5%]; P–T, n = 4/37 [10.8%]) and decreased thyrotropin (T–T, n = 4/38 [10.5%]; P–T, n = 2/37 [5.4%]) (Supplementary Table S7). For non-ATTRV30M patients, the most common laboratory abnormalities were increased prothrombin time (n = 6/11 [54.5%]) and increased blood urea nitrogen (n = 3/18 [16.7%]). Four (10.5%) patients in the ATTRV30M T–T group, one (2.7%) patient in the ATTRV30M P–T group and four (22.2%) non-ATTRV30M patients had a Fridericia-corrected QT interval >500 ms.
Efficacy
ATTRV30M patients who were treated continuously with tafamidis (i.e. the ATTRV30M T–T group) experienced numerically less deterioration in neurologic function throughout continued long-term follow-up compared with those who initiated tafamidis 18 months later (i.e. ATTRV30M P–T group) (, Supplementary Table S8). Thus, the LSMean (SE) increase (i.e. worsening) from baseline in NIS-LL was consistently smaller in the ATTRV30M T–T group than in the ATTRV30M P–T group (7.4 [2.1] versus 12.2 [2.2] at month 66), and the separation between treatment groups observed at the end of the initial 18-month, double-blind, placebo-controlled phase persisted (baseline-adjusted LSMean [SE; 95% CI] difference [T–T minus P–T]: –2.8 [1.5; –5.9, 0.3] at month 18, –4.9 [3.0; –10.9, 1.2] at month 66). The reduction in NIS-LL increase in the T–T group compared with the P–T group primarily reflected slower progression of muscle weakness (). For non-ATTRV30M patients, the mean (SD) baseline NIS-LL was 31.1 (24.4) and the LSMean (SE) increase from baseline to month 48 was 14.2 (2.9) ().
Figure 2. Intent-to-treat Repeated-measures Analysis of Covariance of Efficacy Endpoints in ATTRV30M Patients for LSMean (SE) Change from Baseline. The placebo-to-tafamidis (P–T) group received placebo for 18 months then switched to tafamidis 20 mg/day. The tafamidis-to-tafamidis (T–T) group received tafamidis 20 mg/day continuously from day 1. (A) NIS-LL: Score ranges from 0 to 88 points, with higher scores reflecting greater neurologic impairment. Mean (SD) baseline scores were 6.8 (10.8) in the T–T and 11.6 (14.1) in the P–T groups. LSMean (SE) change from baseline at month 66 was 7.4 (2.1) in the T–T and 12.2 (2.2) in the P–T groups, with an LSMean (SE) treatment group difference of −4.9 (3.0) points (95% CI: −10.9, 1.2) after adjustment for baseline value. (B) NIS-LL Muscle Weakness: This subscore ranges from 0 to 64 points. Mean (SD) baseline scores were 2.1 (6.4) in the T–T and 4.2 (9.6) in the P–T groups. LSMean (SE) change from baseline at month 66 was 3.6 (1.6) in the T–T and 8.0 (1.6) in the P–T groups, with an LSMean (SE) treatment group difference of −4.4 (2.3) points (95% CI: −8.9, 0.1) after adjustment for baseline value. (C) NIS-LL Reflexes: This subscore ranges from 0 to 8 points. Mean (SD) baseline scores were 0.8 (1.8) in the T–T and 1.8 (2.4) in the P–T groups. LSMean (SE) change from baseline at month 66 was 1.2 (0.3) in the T–T and 1.4 (0.3) in the P–T groups, with an LSMean (SE) treatment group difference of −0.2 (0.4) points (95% CI: −1.1, 0.7) after adjustment for baseline value. (D) NIS-LL Sensation: This subscore ranges from 0 to 16 points. Mean (SD) baseline scores were 3.9 (3.7) in the T–T and 5.6 (3.8) in the P–T groups. LSMean (SE) change from baseline at month 66 was 2.2 (0.6) in the T–T and 2.8 (0.7) in the P–T groups, with an LSMean (SE) treatment group difference of −0.5 (0.9) points (95% CI: −2.4, 1.3) after adjustment for baseline value. (E) Norfolk QOL-DN TQOL: The score ranges from –2 to 138 points, with higher scores indicating worse QOL. Mean (SD) baseline scores were 24.1 (26.3) in the T–T and 29.9 (30.1) in the P–T groups. LSMean (SE) change from baseline at month 66 was 4.0 (4.0) in the T–T and 6.0 (4.2) in the P–T groups, with an LSMean (SE) treatment group difference of −2.1 (5.8) points (95% CI: −13.7, 9.5) after adjustment for baseline value. (F) mBMI: mBMI, which is the product of serum albumin (g/L) and BMI (weight [kg] divided by squared height [m]), compensates for oedema formation. Mean (SD) baseline scores were 1027.6 (174.6) in the T–T and 1060.4 (227.7) g/L × kg/m2 in the in the P–T groups. LSMean (SE) change from baseline at month 66 was –2.7 (20.0) in the T–T and 50.6 (21.0) in the P–T groups, with an LSMean (SE) treatment group difference of −53.3 (29.0) g/L × kg/m2 (95% CI: −111.0, 4.5) after adjustment for baseline value. *The dashed horizontal lines represent the tafamidis treatment period of the placebo-to-tafamidis group. CI: confidence interval; LSMean: least squares mean; mBMI: modified body mass index; NIS-LL: Neuropathy Impairment Score for the Lower Limbs; QOL: quality of life; QOL-DN: Quality of Life-Diabetic Neuropathy; SD: standard deviation; SE: standard error; TQOL: total quality of life score.
![Figure 2. Intent-to-treat Repeated-measures Analysis of Covariance of Efficacy Endpoints in ATTRV30M Patients for LSMean (SE) Change from Baseline. The placebo-to-tafamidis (P–T) group received placebo for 18 months then switched to tafamidis 20 mg/day. The tafamidis-to-tafamidis (T–T) group received tafamidis 20 mg/day continuously from day 1. (A) NIS-LL: Score ranges from 0 to 88 points, with higher scores reflecting greater neurologic impairment. Mean (SD) baseline scores were 6.8 (10.8) in the T–T and 11.6 (14.1) in the P–T groups. LSMean (SE) change from baseline at month 66 was 7.4 (2.1) in the T–T and 12.2 (2.2) in the P–T groups, with an LSMean (SE) treatment group difference of −4.9 (3.0) points (95% CI: −10.9, 1.2) after adjustment for baseline value. (B) NIS-LL Muscle Weakness: This subscore ranges from 0 to 64 points. Mean (SD) baseline scores were 2.1 (6.4) in the T–T and 4.2 (9.6) in the P–T groups. LSMean (SE) change from baseline at month 66 was 3.6 (1.6) in the T–T and 8.0 (1.6) in the P–T groups, with an LSMean (SE) treatment group difference of −4.4 (2.3) points (95% CI: −8.9, 0.1) after adjustment for baseline value. (C) NIS-LL Reflexes: This subscore ranges from 0 to 8 points. Mean (SD) baseline scores were 0.8 (1.8) in the T–T and 1.8 (2.4) in the P–T groups. LSMean (SE) change from baseline at month 66 was 1.2 (0.3) in the T–T and 1.4 (0.3) in the P–T groups, with an LSMean (SE) treatment group difference of −0.2 (0.4) points (95% CI: −1.1, 0.7) after adjustment for baseline value. (D) NIS-LL Sensation: This subscore ranges from 0 to 16 points. Mean (SD) baseline scores were 3.9 (3.7) in the T–T and 5.6 (3.8) in the P–T groups. LSMean (SE) change from baseline at month 66 was 2.2 (0.6) in the T–T and 2.8 (0.7) in the P–T groups, with an LSMean (SE) treatment group difference of −0.5 (0.9) points (95% CI: −2.4, 1.3) after adjustment for baseline value. (E) Norfolk QOL-DN TQOL: The score ranges from –2 to 138 points, with higher scores indicating worse QOL. Mean (SD) baseline scores were 24.1 (26.3) in the T–T and 29.9 (30.1) in the P–T groups. LSMean (SE) change from baseline at month 66 was 4.0 (4.0) in the T–T and 6.0 (4.2) in the P–T groups, with an LSMean (SE) treatment group difference of −2.1 (5.8) points (95% CI: −13.7, 9.5) after adjustment for baseline value. (F) mBMI: mBMI, which is the product of serum albumin (g/L) and BMI (weight [kg] divided by squared height [m]), compensates for oedema formation. Mean (SD) baseline scores were 1027.6 (174.6) in the T–T and 1060.4 (227.7) g/L × kg/m2 in the in the P–T groups. LSMean (SE) change from baseline at month 66 was –2.7 (20.0) in the T–T and 50.6 (21.0) in the P–T groups, with an LSMean (SE) treatment group difference of −53.3 (29.0) g/L × kg/m2 (95% CI: −111.0, 4.5) after adjustment for baseline value. *The dashed horizontal lines represent the tafamidis treatment period of the placebo-to-tafamidis group. CI: confidence interval; LSMean: least squares mean; mBMI: modified body mass index; NIS-LL: Neuropathy Impairment Score for the Lower Limbs; QOL: quality of life; QOL-DN: Quality of Life-Diabetic Neuropathy; SD: standard deviation; SE: standard error; TQOL: total quality of life score.](/cms/asset/fed25b21-2017-472e-95ea-dc9a7d7cb3a0/iamy_a_1357545_f0002_b.jpg)
Figure 3. Intent-to-treat Repeated-measures Analysis of Efficacy Endpoints in non-ATTRV30M Patients. Least squares mean (LSMean) (SE) change from baseline: (A) NIS-LL: The mean (SD) baseline score was 31.1 (24.4) points. The LSMean (SE) change from baseline at month 48 was 14.2 (2.9) points (95% CI: 8.0–20.4). (B) NIS-LL Muscle Weakness: The mean (SD) baseline score was 16.6 (16.9) points. The LSMean (SE) change from baseline at month 48 was 8.9 (2.3) points (95% CI: 4.1–13.7). (C) NIS-LL Reflexes: The mean (SD) baseline score was 5.6 (3.4) points. The LSMean change from baseline at month 48 is not available because the repeated measures analysis model did not converge for this time point. The LSMean (SE) change from baseline at month 36 (the preceding time point) was 1.3 (0.6) points (95% CI: 0.1–2.6). (D) NIS-LL Sensation: The mean (SD) baseline score was 8.9 (5.4) points. The LSMean (SE) change from baseline at month 48 was 2.6 (0.8) points (95% CI: 0.9–4.2). (E) Norfolk QOL-DN TQOL: The mean (SD) baseline score was 53.9 (34.2) points. The LSMean (SE) change from baseline at month 48 was 24.8 (8.4) points (95% CI: 7.0–42.7). (F) Karnofsky performance status index: Scores range from 0 to 100, with lower scores indicating greater functional impairment. The mean (SD) baseline score was 72.2 (13.5) points. The LSMean (SE) change from baseline at month 48 was −13.4 (3.3) points (95% CI: –20.4, –6.4). (G) BMI: The mean (SD) baseline score was 25.2 (3.6) kg/m2. The LSMean (SE) change from baseline at month 48 was –1.0 (0.6) kg/m2 (95% CI: –2.2, 0.2). BMI: body mass index; CI: confidence interval; LSMean: least squares mean; NIS-LL: Neuropathy Impairment Score for the Lower Limbs; QOL-DN: Quality of Life-Diabetic Neuropathy; SD: standard deviation; SE: standard error; TQOL: total quality of life score.
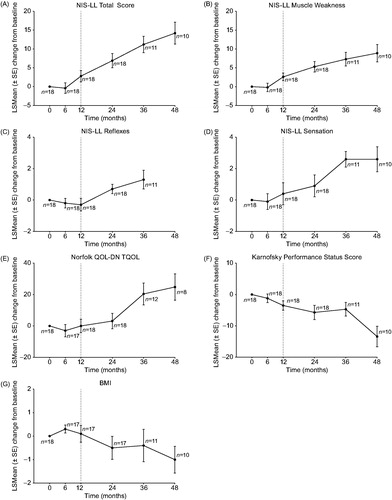
LSMean changes from baseline in TQOL for ATTRV30M patients showed variability, but generally increased (i.e. worsened) over time (, Supplementary Table S8). The most marked increase over time occurred during placebo treatment in the P–T group (LSMean [SE] increase from baseline to month 18: 6.5 [3.2]). By month 66, LSMean (SE) increases from baseline were similar in the T–T and P–T groups (4.0 [4.0] versus 6.0 [4.2]). For non-ATTRV30M patients, the mean (SD) baseline TQOL was 53.9 (34.2) and the LSMean (SE) increase from baseline to month 48 was 24.8 (8.4) ().
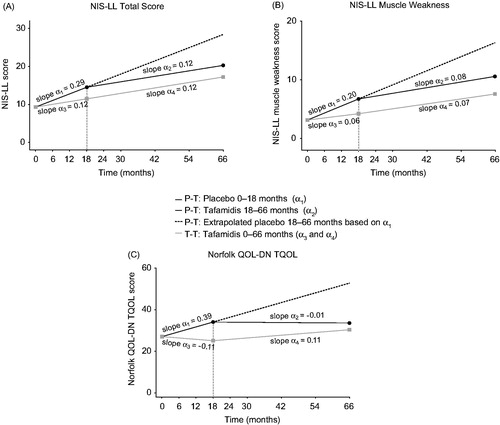
To assess the efficacy of tafamidis as a function of earlier versus delayed treatment start, a post-hoc slope analysis was conducted to compare the rate of disease progression in NIS-LL, NIS-LL muscle weakness and TQOL within and between the T–T and P–T groups during the double-blind, placebo-controlled phase (months 0–18) and open-label phases (months 18–66) (). The T–T group had significantly slower progression rates on NIS-LL and NIS-LL muscle weakness compared with the P-T group during months 0–18 (baseline-adjusted mean [95% CI] slope difference [tafamidis minus placebo]: –0.17 [–0.31, –0.03] points/month, p = .019 for NIS-LL; –0.13 [–0.25, –0.02] points/month, p = .024 for NIS-LL muscle weakness; , Supplementary Table S9). Similar findings were observed for TQOL (–0.49 [–0.84, –0.14] points/month, p = .006; , Supplementary Table S9). After switching to tafamidis, the P-T group showed a significant reduction in the rate of change in NIS-LL, NIS-LL muscle weakness and TQOL during months 18–66 as compared to months 0–18 (baseline-adjusted mean [95% CI] slope difference [tafamidis minus placebo]: –0.17 [–0.29, –0.06] points/month, p = .004 for NIS-LL; –0.11 [–0.21, –0.02] points/month, p = .02 for NIS-LL muscle weakness; –0.39 [–0.67, –0.12] points/month, p = .006 for TQOL; ; Supplementary Table S9). The rate of change in disease progression upon switching from placebo to tafamidis became similar to that observed following continued use of tafamidis, as evidenced by the comparable slopes observed during the open-label phases (baseline-adjusted mean [95% CI] slope difference: 0.006 [–0.08, 0.09] points/month, p = .890 for NIS-LL; –0.01 [–0.08, 0.05] points/month, p = .718 for NIS-LL muscle weakness; 0.12 [–0.05, 0.29] points/month, p = .179 for TQOL; , Supplementary Table S9).
Figure 4. Intent-to-treat Slope Analysis of Efficacy Endpoints in ATTRV30M Patients (A) NIS-LL; (B) NIS-LL Muscle Weakness; (C) Norfolk QOL-DN TQOL). The placebo-to-tafamidis (P–T) group (black solid line) received placebo for 18 months then switched to tafamidis 20 mg/day. The tafamidis-to-tafamidis (T–T) group (grey solid line) received tafamidis 20 mg/day continuously from day 1. According to the analytic model and shown here, the slopes are adjusted at mean baseline value of the two treatment groups. The black dashed line extending from months 18 to 66 of the P–T group is the extrapolated projection of disease progression had the patients remained on placebo. For further details on the slope analysis and definitions of α1–α4, see Supplementary Box S2. NIS-LL: Neuropathy Impairment Score for the Lower Limbs; QOL-DN: Quality of Life-Diabetic Neuropathy; TQOL: total quality of life score.
Karnofsky performance status was not included in the double-blind study and first extension study in ATTRV30M patients (i.e. the first 30 months) [Citation21,Citation22], precluding an assessment of LSMean change from baseline. The mean (SD) Karnofsky performance status scores at month 30 (start of on-going extension study) and month 66, respectively, were 83.8 (13.0) and 85.9 (10.8) in the ATTRV30M T-T group and 80.3 (11.8) and 78.7 (17.7) in the ATTRV30M P-T group. For non-ATTRV30M patients, the mean (SD) Karnofsky performance status score was 72.2 (13.5) at baseline of the parent study and gradually decreased (worsened) over time with an LSMean (SE) change from baseline to month 48 of –13.4 (3.3) ().
Regarding nutritional status, ATTRV30M T–T patients showed minor numerical decreases (worsening) in mBMI relative to baseline at months 54 and 66 (LSMean [SE] change in g/L × kg/m2: –7.1 [20.3] at month 54, –2.7 [20.0] at month 66) (, Supplementary Table S8). ATTRV30M P–T patients showed steady worsening in mBMI relative to baseline during placebo treatment and an improvement (increase from baseline) after switching to tafamidis, persisting through month 66 (LSMean [SE] change in g/L × kg/m2: –31.8 [13.5] at month 18 and 50.6 [21.0] at month 66). Non-ATTRV30M patients showed some degree of worsening in BMI relative to baseline at months 24–48 ().
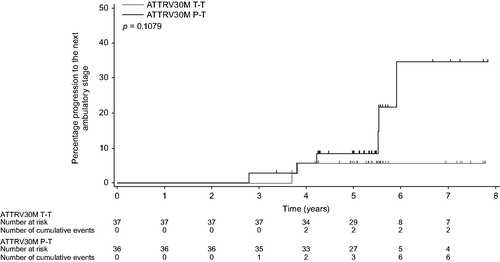
Two patients in the ATTRV30M T–T group versus six patients in the ATTRV30M P–T group progressed to the next ambulatory stage by year 6; the 6-year progression rates (95% CI) from the first dose of study drug were estimated as 5.5% (1.4–20.2%) versus 34.6% (14.4–68.8%), respectively ().
Figure 5. Kaplan–Meier Analyses of Progression to the Next Ambulatory Stage in ATTRV30M Patients. Kaplan–Meier plot of the time from the first drug dose in Study Fx-005 to progression to the next ambulatory stage. Two patients with missing baseline assessment and Stage 3 disease at the initial ambulatory assessment were excluded. Vertical lines indicate censored observations. P–T: placebo-to-tafamidis group received placebo for 18 months and then switched to tafamidis 20 mg/day; T–T: tafamidis-to-tafamidis group received tafamidis 20 mg/day continuously from day 1.
Discussion
These data represent the longest (up to 6 years) longitudinal evaluation to date of a medicine for treatment of ATTR-FAP and support the long-term safety and efficacy of tafamidis in delaying neurologic disease progression for ATTRV30M and non-ATTRV30M patients. Extending the observations of the double-blind study of tafamidis in ATTRV30M patients [Citation21] and the initial 12-month open-label extension study [Citation22] and the open-label 12-month study in non-ATTRV30M patients [Citation24] by 3 additional years, the current analysis found that tafamidis had a favourable safety/tolerability profile over the long-term, with no deaths associated with its use. The type and incidence of AEs, concomitant medications and results of clinical laboratory evaluations, vital signs, electrocardiograms and physical examinations in this interim analysis reflected the underlying disease of ATTR-FAP and the known safety profile of tafamidis [Citation21,Citation22,Citation24].
Among the ATTRV30M patients, those who continuously received tafamidis experienced numerically less deterioration of neurologic function from baseline compared with those initially treated with placebo and then switched to tafamidis 18 months later. In fact, patients who received tafamidis from the start of the pivotal study were less likely to progress to the next ambulatory stage by 6 years than patients who started tafamidis later. These observations highlight the value of early tafamidis treatment to preserve neurologic functioning. Although earlier treatment is advantageous, patients benefitted from tafamidis even when initiated later in the course of their disease. This is supported by the findings that patients who switched from placebo to tafamidis showed a reduction in the rate of neurologic progression over the next 4 years of open-label tafamidis treatment that was comparable to the rate of disease progression observed in patients who continuously received tafamidis over 5.5 years.
Similar findings were observed for TQOL. Compared with the worsening observed during the 18-month placebo phase, TQOL remained relatively stable during long-term tafamidis treatment in ATTRV30M patients and was comparable to that observed in the patients who had received tafamidis from study start. Nutritional status as assessed by mBMI remained relatively stable during long-term tafamidis treatment, which is consistent with published analyses by Suhr et al. [Citation33], and lends further support for the long-term efficacy of tafamidis in slowing disease progression.
The results of the slope analysis support the disease modifying effects of tafamidis. During the first 18 months of treatment, patients receiving tafamidis had significantly slower progression rates on NIS-LL, NIS-LL muscle weakness and TQOL compared with those receiving placebo. Following the switch to tafamidis, patients originally receiving placebo exhibited a slowing of disease progression and at a rate significantly less than that observed during placebo treatment. Further, following the switch to tafamidis, disease progression rates were comparable to those observed in the patients who had received tafamidis continuously.
Additional support for the disease modifying effects of tafamidis comes from a subgroup analysis of the on-going extension study wherein ATTRV30M patients with very mild ATTR-FAP (NIS-LL ≤10) at the start of tafamidis treatment showed minimal neurologic disease progression over time with a mean (95% CI) NIS-LL change from baseline to 5.5 years of 5.3 (1.6, 9.1) points [Citation23]. These findings are particularly salient when considering that reported disease progression rates in untreated heterogeneous ATTR-FAP populations range from ∼4 to 6 points per year in NIS-LL [Citation15,Citation22].
Non-ATTRV30M patients, who were older and had more advanced disease than the ATTRV30M patients, demonstrated worsening in efficacy endpoints throughout 4 years of treatment. As patients with greater impairment at baseline tend to experience greater changes in disease progression over time, as measured by NIS-LL [Citation34], the progressive worsening observed may be related to the advanced disease in these patients. The absence of a comparator in the non-ATTRV30M group of the present study poses a challenge in interpreting these data. However, a recent independent study in 61 Italian patients found tafamidis slowed neurologic progression after the first 6 months of treatment in patients with advanced disease and in those with non-ATTRV30M mutations [Citation35]. Furthermore, an observational study of a heterogeneous group of 43 tafamidis-treated ATTR-FAP patients (at various stages of illness and comprising ATTRV30M and non-ATTRV30M genotypes) demonstrated a slowing of disease progression in about one-third of the patients who were followed for 3 years [Citation36].
This interim analysis shares limitations typically encountered in studies on rare diseases, including limited statistical power due to a relatively small sample size and lack of a control group for non-ATTRV30M patients initially (first 12 months) and for both genotype groups during the extension studies. Decreases in patient numbers over time further reduced the precision of some estimates of treatment effect. Retrospective collection of ambulatory data resulted in missing assessments. The open-label design presents a possibility of bias [Citation22], and the slope analyses to examine the disease progression associated with the delayed-start effect were conducted post-hoc. Finally, the results may differ among patients with early- versus late-onset ATTR-FAP and this was not explored in the present study. Notwithstanding these limitations, these results provide important evidence for the long-term safety and efficacy of tafamidis in slowing ATTR-FAP disease progression, and support an enduring benefit of earlier treatment initiation.
Conclusions
This long-term analysis out to 6 years, which draws on multiple studies from the tafamidis clinical development program, expands the understanding of the tafamidis treatment experience for ATTR-FAP. The results confirm and extend the favourable safety and efficacy profile of tafamidis reported previously [Citation21,Citation22,Citation24,Citation35,Citation37]. Overall, the findings underscore the sustainability of tafamidis in preserving neurologic function over the long-term, and add to the growing body of evidence supporting early initiation of tafamidis to slow disease progression in patients with ATTR-FAP [Citation21,Citation22,Citation24,Citation35,Citation37].
| Abbreviations | ||
| AE | = | adverse event |
| ATTR-FAP | = | transthyretin familial amyloid polyneuropathy |
| CI | = | confidence interval |
| ITT | = | intent-to-treat |
| LS | = | least squares |
| mBMI | = | modified body mass index |
| NIS-LL | = | Neuropathy Impairment Score–Lower Limbs |
| Norfolk QOL-DN | = | Norfolk Quality of Life–Diabetic Neuropathy questionnaire |
| NSAID | = | non-steroidal anti-inflammatory drug |
| P-T | = | placebo-to-tafamidis |
| SAE | = | serious adverse event |
| SD | = | standard deviation |
| SE | = | standard error |
| TQOL | = | total quality of life |
| T-T | = | tafamidis-to-tafamidis |
| TTR | = | transthyretin |
Supplemental_Materials_7-13-17.docx
Download MS Word (62.5 KB)Acknowledgements
Medical writing support was provided by Shuang Li, PhD, Susanne Vidot, PhD, and Diane Hoffman, PhD, at Engage Scientific Solutions and was funded by Pfizer. The authors thank Donna R. Grogan, MD (at FoldRx Pharmaceuticals, Cambridge, MA, USA at the time of the study) for her contribution to study concept and design, and also thank Michael Gaffney, PhD, from Global Statistics at Pfizer for his contributions to the post-hoc slope analytic model. We further wish to thank all the B3461023 study investigators for their support and assistance with these studies: Teresa Coelho, MD (Hospital Santo Antonio, Porto, Portugal), Isabel Conceição, MD (Hospital de Santa Maria, Lisbon, Portugal), Giampaolo Merlini, MD (Amyloid Research and Treatment Center, IRCCS Fondazione Policlinico, San Matteo, University of Pavia, Italy), Violaine Planté-Bordeneuve, MD (CHU de Bicêtre Service de Neurologie, Le Kremlin Bicêtre, France), Hartmut H.-J. Schmidt, MD (Universitaetsklinikum Muenster, Muenster, Germany), Ole B. Suhr, MD (Umeå University Hospital, Umeå, Sweden) and Márcia Waddington-Cruz, MD (Hospital Universitário Clementino Fraga Filho, Rio de Janeiro, Brazil). We also thank Martín Nogués, MD, and Florencia Picone (both Raul Carrea Institute for Neurological Research, FLENI, Buenos Aires, Argentina) for their contributions. Finally, we express our sincere gratitude to all participating patients without whom this research would not have been possible.
Disclosure statement
F. Barroso has received funding from Pfizer for scientific meeting expenses (travel and registration), and serves on the Transthyretin Amyloidosis Outcomes Survey (THAOS) Scientific Board, which is sponsored by Pfizer. D. Judge has received research support from Pfizer and has served as a scientific advisor for Pfizer, Invitae, MyoKardia and Glaxo Smith Kline. B. Ebede, H. Li, M. Stewart, L. Amass and M. Sultan are full-time employees of Pfizer and hold stock and/or stock options.
Additional information
Funding
Related Research Data
References
- Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31–49.
- Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86:1036–1043.
- Coelho T, Maurer MS, Suhr OB. THAOS - The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29:63–76.
- Adams D, Coelho T, Obici L, et al. Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology. 2015;85:675–682.
- Mariani LL, Lozeron P, Théaudin M, et al. Genotype-phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Ann Neurol. 2015;78:901–916.
- Rowczenio DM, Noor I, Gillmore JD, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35:E2403–24E2412.
- Plante-Bordeneuve V. Update in the diagnosis and management of transthyretin familial amyloid polyneuropathy. J Neurol. 2014;261:1227–1233.
- Adams D, Théaudin M, Cauquil C, et al. FAP neuropathy and emerging treatments. Curr Neurol Neurosci Rep. 2014;14:1–12.
- Conceição IM, González-Duarte A, Obici L, et al. Red-flag symptom clusters in transthyretin familial amyloid polyneuropathy” . J Peripher Nerv Syst. 2016;21:5–9.
- Schmidt H, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2017; submitted
- Azoulay D, Samuel D, Castaing D, et al. Domino liver transplants for metabolic disorders: experience with familial amyloidotic polyneuropathy. J Am Coll Surg. 1999;189:584–593.
- Ericzon BG, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99:1847–1854.
- Suhr OB, Larsson M, Ericzon BG, et al. Survival after transplantation in patients with mutations other than Val30Met: extracts from the FAP World Transplant Registry. Transplantation. 2016;100:373–381.
- Adams D. Recent advances in the treatment of familial amyloid polyneuropathy. Ther Adv Neurol Disord. 2013;6:129–139.
- Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658–2667.
- MedlinePlus. Diflunisal. [Internet]. Bethesda (MD): US National Library of Medicine; 2015 Sep 15 [cited 2017 July 7]. Available from: https://www.nlm.nih.gov/medlineplus/druginfo/meds/a684037.html.
- European Medicines Agency. Vyndaqel (tafamidis): European Public Assessment Report (EPAR). [Internet]. London (England): European Medicines Agency; 2016 Feb 8 [cited 2017 July 7]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002294/human_med_001498.jsp&mid=WC0b01ac058001d124.
- Japan Pharmaceuticals and Medical Devices Agency. Vyndaqel (tafamidis): report on the deliberation results of the regulatory review. [Internet]. Tokyo (Japan): Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour and Welfare 22 August 2013 [cited 2017 July 7]. Available from: http://www.pmda.go.jp/files/000153750.pdf#page=2&r=s&r=s.
- Waddington Cruz M, Benson MD. A review of tafamidis for the treatment of transthyretin-related amyloidosis. Neurol Ther. 2015;4:61–79.
- Coelho T, Merlini G, Bulawa CE, et al. Mechanism of action and clinical application of tafamidis in hereditary transthyretin amyloidosis. Neurol Ther. 2016;5:1–25.
- Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012;79:785–792.
- Coelho T, Maia LF, da Silva AM, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260:2802–2814.
- Waddington Cruz M, Amass L, Keohane D, et al. Early intervention with tafamidis provides long-term (5.5-year) delay of neurologic progression in transthyretin hereditary amyloid polyneuropathy. Amyloid 2016;23:178–183.
- Merlini G, Plante-Bordeneuve V, Judge DP, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Trans Res. 2013;6:1011–1020.
- Dyck PJ, Litchy WJ, Lehman KA, et al. Variables influencing neuropathic endpoints: the Rochester Diabetic Neuropathy Study of Healthy Subjects. Neurology 1995;45:1115–1121.
- Vinik EJ, Hayes RP, Oglesby A, et al. The development and validation of the Norfolk QOL-DN, a new measure of patients' perception of the effects of diabetes and diabetic neuropathy. Diabetes Technol Ther. 2005;7:497–508.
- Vinik EJ, Vinik AI, Paulson JF, et al. Norfolk QOL-DN: validation of a patient reported outcome measure in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2014;19:104–114.
- Suhr O, Danielsson A, Holmgren G, et al. Malnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathy. J Intern Med. 1994;235479–485.
- Coelho T, Vinik A, Vinik EJ, et al. Clinical measures in transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2017;55:323–332.
- Coutinho P, Martins da Silva A, Lopes Lima J, et al. Forty years of experience with type I amyloid neuropathy. Review of 483 cases. In: Glenner GG, Pinho e Costa P, Falcao de Freitas A, editors. Amyloid and amyloidosis. Amsterdam (Netherlands): Excerpta Medica; 1980. p. 88–98.
- D'Agostino RB. Sr. The delayed-start study design. N Engl J Med. 2009;361:1304–1306.
- Liu-Seifert H, Andersen SW, Lipkovich I, et al. A novel approach to delayed-start analyses for demonstrating disease-modifying effects in Alzheimer's disease. PLoS One. 2015;10:1–16.
- Suhr OB, Conceicao IM, Karayal ON, et al. Post hoc analysis of nutritional status in patients with transthyretin familial amyloid polyneuropathy: impact of tafamidis. Neurol Ther. 2014;3:101–112.
- Li H, Gundapaneni B, Schwartz J, et al. Impact of baseline neurologic score on disease progression in transthyretin familial amyloid polyneuropathy. International Society of Amyloidosis–XV International Symposium on Amyloidosis; 3–7 July 2016; Uppsala, Sweden, Abstract No. PB2, p. 222.
- Cortese A, Vita G, Luigetti M, et al. Monitoring effectiveness and safety of Tafamidis in transthyretin amyloidosis in Italy: a longitudinal multicenter study in a non-endemic area. J Neurol. 2016;263:916–924.
- Plante-Bordeneuve V, Gorram F, Salhi H, et al. Long-term treatment of transthyretin familial amyloid polyneuropathy with tafamidis: a clinical and neurophysiological study. J Neurol. 2017;264:268–276.
- Ando Y, Sekijima Y, Obayashi K, et al. Effects of tafamidis treatment on transthyretin (TTR) stabilization, efficacy, and safety in Japanese patients with familial amyloid polyneuropathy (TTR-FAP) with Val30Met and non-Val30Met: a phase III, open-label study. J Neurol Sci. 2016;362:266–271.