
Abstract
Background: Tafamidis is approved in over 40 countries to delay neurologic progression in patients with transthyretin amyloid polyneuropathy (ATTR-PN). A comprehensive, integrated analysis of safety data from interventional, observational and surveillance studies of tafamidis in ATTR-PN patients was conducted.
Methods: Safety data from all sponsored, completed, or ongoing, Phase 2/3 studies of tafamidis in ATTR-PN patients as of 3 January 2017 were pooled. Also assessed were safety data from the ongoing Transthyretin Amyloidosis Outcomes Survey (THAOS) as of 3 January 2017 and post-marketing surveillance reports as of 31 March 2017.
Results: There were 137 patients in Phase 2/3 studies (mean duration of tafamidis exposure, 44.2 months), with 134 (97.8%) experiencing ≥1 treatment-emergent adverse event (TEAE) and 46 (33.6%) ≥1 treatment-emergent serious adverse event (TESAE). The most common TEAEs were diarrhoea (26.3%), urinary tract infection (UTI; 25.5%) and influenza (21.2%). In THAOS, 661 subjects had tafamidis exposure (mean duration, 27.6 months), with 250 (37.8%) experiencing ≥1 TEAE and 96 (14.5%) ≥1 TESAE. The most common TEAE was UTI (6.1%). Post-marketing surveillance reports generally reflected the known safety profile of tafamidis.
Conclusions: This analysis did not reveal any significant new safety findings; tafamidis was generally safe and well tolerated in ATTR-PN patients.
Trial registration: ClinicalTrials.gov identifier: NCT00409175.
Trial registration: ClinicalTrials.gov identifier: NCT00791492.
Trial registration: ClinicalTrials.gov identifier: NCT00630864.
Trial registration: ClinicalTrials.gov identifier: NCT01435655.
Trial registration: ClinicalTrials.gov identifier: NCT00925002.
Trial registration: ClinicalTrials.gov identifier: NCT00628745.
Introduction
Hereditary transthyretin amyloidosis (ATTR amyloidosis) is a progressive, rare disease caused by deposition of amyloid fibrils in peripheral nerves and vital organs ultimately leading to advanced transthyretin amyloid polyneuropathy (ATTR-PN) and/or restrictive transthyretin amyloid cardiomyopathy (ATTR-CM) [Citation1–3]. There are more than 100 known pathogenic mutations in the transthyretin gene (TTR) giving rise to hereditary ATTR amyloidosis (ATTRm) [Citation4,Citation5]. ATTR-PN affects at least 10,000, and perhaps as many as 40,000, individuals worldwide [Citation6]. ATTR-CM also includes an acquired form of the disease due to deposition of wild-type transthyretin protein in the heart [Citation1].
Tafamidis is a selective TTR stabilizer and the only oral medicine, approved in over 40 countries worldwide, to delay neurologic progression in adult patients with early-stage ATTR-PN [Citation7,Citation8]. Tafamidis is also under investigation for the treatment of ATTR-CM in which it was well tolerated and demonstrated efficacy [Citation9–11]. Published clinical trials, together with approximately 3508 patient-years of post-marketing experience as of 15 May 2017, have provided a consistent safety profile of tafamidis [Citation10,Citation12–16]. The established safety profile of tafamidis in patients with ATTR-PN was based on observations from the initial Phase 3 randomized, double-blind, placebo-controlled study [Citation12]. This trial identified four adverse events (AEs) with a possible causal association to tafamidis: diarrhoea, urinary tract infection (UTI), upper abdominal pain and vaginal infection [Citation12,Citation17].
Nonclinical safety data have also informed the safety profile of tafamidis. For example, as tafamidis is metabolized by Phase 2 glucuronidation [Citation7,Citation17], with acyl-glucuronide the only identified metabolite in humans, there exists a potential risk of idiosyncratic drug reactions, including immune responses and hepatic toxicity. In initial, nonclinical studies, tafamidis-associated hepatic alterations were observed but only at doses higher than 15 times those used in humans. These findings were consistent with hepatocellular enzyme induction and were considered to represent an adaptive response to high doses rather than any direct effect of treatment, and were not considered adverse. Nevertheless, pharmacovigilance activities include the ongoing monitoring of potential hypersensitivity and hepatic events.
While safety data from completed (and some ongoing) clinical trials have been published, there are less data available on the real-world use of tafamidis and there has been no combined analysis of all ATTR-PN patients who have received tafamidis. This report describes a comprehensive, integrated safety analysis of all available patient-level safety data from sponsored interventional and observational studies of tafamidis in patients with ATTR-PN conducted as of 3 January 2017; spanning 6 years following the approval of tafamidis for the treatment of patients with ATTR-PN in Europe [Citation17].
Methods
Source data
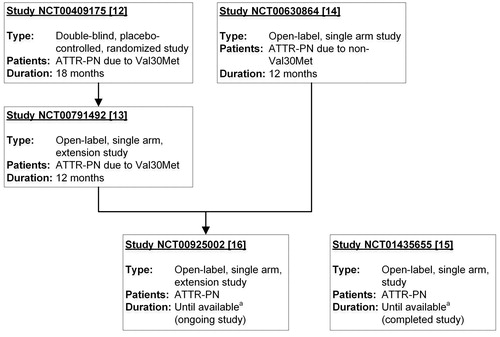
All clinical trials of tafamidis in patients with ATTR-PN sponsored by Pfizer were included in this analysis. Completed and ongoing interventional clinical trials (i.e. excluding registry/survey and surveillance data) were analysed with data collected as of 3 January 2017. Data from four completed trials (one of which was placebo-controlled; the others open-label) and one ongoing open-label trial in patients with ATTR-PN [Citation12–16] were pooled for this analysis to provide a larger data-set over a longer period of time to allow for a more robust assessment of safety (). Those patients receiving liver transplant discontinued tafamidis treatment prior to the procedure but safety data continued to be collected for those who returned for follow-up assessment.
Figure 1. Interventional studies in patients with ATTR-PN. Summary of clinical studies in patients with ATTR-PN that were pooled in this analysis. Tafamidis dose was 20 mg once daily in each study. NCT00791492 is an extension study of NCT00409175. NCT00925002 is an extension study of NCT00791492 and NCT00630864. aStudies with duration “until available” are ongoing until tafamidis is commercially available for the patient in patient’s country. ATTR-PN: transthyretin amyloid polyneuropathy.
Data from all subjects treated with tafamidis in the ongoing, global, longitudinal and observational Transthyretin Amyloidosis Outcomes Survey (THAOS; NCT00628745) [Citation18] were analysed separately with a data cut-off of 3 January 2017. Data from a post-marketing surveillance study in Japan, NCT02146378, and with data from the worldwide spontaneous post-marketing safety database, were analysed separately with a data cut-off of 31 March 2017. In Japan, a Pharmaceuticals and Medical Devices Agency (PMDA) mandated post-marketing surveillance study does not require signed patient consent forms; therefore, in order to ensure patient anonymity, only a limited amount of safety information is made available to the sponsor.
Pfizer’s safety database contains AEs reported spontaneously to Pfizer by consumers, the general public, health care providers and health authorities, cases published in the medical literature and from Pfizer-sponsored marketing programmes, and cases of serious AEs reported from clinical and non-interventional studies regardless of causality. The dose of tafamidis for all patients in this analysis was 20 mg of tafamidis meglumine once daily.
Definition of adverse events
A treatment-emergent adverse event (TEAE) was defined as any untoward medical occurrence in a subject administered tafamidis regardless of whether it was considered to be tafamidis-related or not. This included any newly occurring TEAE or a previous condition/event that worsened in severity or frequency following initiation of tafamidis. A treatment-emergent serious adverse event (TESAE) was any TEAE which resulted in death; was life-threatening; required hospitalization; resulted in a persistent or significant disability/incapacity; or was an important medical event. TEAEs were considered to be related to study medication if reported by the investigator as “Yes” related or where relatedness was recorded as “Unknown”.
Coding and statistical analysis of adverse events
In clinical trials, TEAEs were either originally coded using, or subsequently updated to, the Medical Dictionary for Regulatory Activities (MedDRA) version 20.0 and used similar methods to capture safety data; therefore pooling of these data is appropriate. Data presented from clinical trials are from each safety population, defined as all patients receiving ≥1 dose of study medication. As NCT00791492 was an extension study of NCT00409175, and NCT00925002 was an extension study of NCT00791492 and NCT00630864, the trials had patients in common; however, each patient receiving tafamidis was counted only once in this analysis. Integrated data are presented using descriptive statistics: including number, mean, SD, median, interquartile range (IQR) and range (minimum to maximum) for continuous variables; and number and percentage for categorical variables. Data are shown for all tafamidis-treated patients together, and for patients with the Val30Met (p.Val50Met) mutation compared with patients with any other mutation (non-Val30Met).
Non-interventional studies were coded using MedDRA version 19.1. In THAOS (NCT00628745), the safety population included all subjects with any tafamidis treatment exposure from the time they were enrolled in THAOS, but excluded their time during any interventional clinical trial participation.
Results
Demographics and safety outcomes in clinical studies in patients with ATTR-PN
There were 137 patients with ATTR-PN treated with tafamidis in Phase 2/3 studies (115 Val30Met, 22 non-Val30Met) (). Study NCT00409175 [Citation12] included 65 patients treated with tafamidis and 63 treated with placebo. The mean (SD) duration of exposure to tafamidis in all patients was 44.2 (27.0) months (). However, a greater proportion of Val30Met patients had a duration of exposure ≥60 months (37 patients, 32.2%) than non-Val30Met patients (4, 18.2%). Medical histories of Val30Met and non-Val30Met patients treated with tafamidis were broadly similar ().
Table 1. Demographic and baseline characteristics in all completed and ongoing studies in patients with ATTR-PN.
Table 2. Duration of exposure to tafamidis in completed and ongoing studies in patients with ATTR-PN and in the non-interventional THAOS registry (NCT00628745).
Table 3. Summary of most commonTable Footnotea medical history in all completed and ongoing studies in patients with ATTR-PN.
Overall incidence of TEAEs was similar across the four open-label studies with differences in incidence of individual TEAEs generally not considered clinically meaningful. Overall, ≥1 TEAE was reported in 134 (97.8%) tafamidis-treated (), and 61 (96.8%) placebo-treated patients. The most common TEAEs reported with tafamidis were diarrhoea (26.3%), UTI (25.5%) and influenza (21.2%) (), while the most common TEAEs reported with placebo were headache (19.0%), diarrhoea (17.5%) and neuralgia (17.5%). Data comparing the demographics and most common TEAEs reported with tafamidis and placebo in study NCT00409175 have been published previously [Citation12].
Table 4. Summary of most common TEAEs in all completed and ongoing studies in patients with ATTR-PN.
There was a higher proportion of treatment-related TEAEs in Val30Met patients (70 patients with ≥1 event, 60.9%) than non-Val30Met patients (10 patients, 45.5%). With the exception of UTI (8.0% of patients), each of the most common treatment-related TEAEs, which also included headache (8.8%), diarrhoea (7.3%) and pain in extremity (7.3%) in tafamidis-treated patients occurred with similar (or higher) incidence in placebo-treated patients.
Overall, 12 (8.8%) tafamidis-treated, and 3 (4.8%) placebo-treated patients with ATTR-PN experienced a TEAE resulting in discontinuation. TESAEs were more common in non-Val30Met patients than Val30Met patients (). TESAEs occurring in ≥2 patients in total were: UTI (5 patients, 3.6%); cardiac failure, sepsis and vomiting (4, 2.9%); and chest pain, fall, pneumonia, bacterial pneumonia and transient ischaemic attack (3, 2.2%). In study NCT00409175, there were 7 (10.8%) patients with ≥1 TESAE with tafamidis and 7 (11.1%) with placebo.
There were 9 (6.6%) tafamidis-treated patients (6 Val30Met, 3 non-Val30Met) with ≥1 TESAE that was considered treatment-related, these were: 1 patient with cardiac failure, pericardial effusion and renal impairment; 1 patient with malaise and urinary retention; and 1 patient each with ankle fracture, lymphoma, meningitis, sudden death, transient ischaemic attack, UTI and urticaria. In study NCT00409175, there were 2 TESAEs considered treatment-related with tafamidis (3.1%) and 2 with placebo (3.2%).
A total of 11 deaths were reported in patients with ATTR-PN treated with tafamidis. In study NCT00409175, there were a total of 5 deaths reported, 2 in patients treated with tafamidis and 3 in patients treated with placebo [Citation12]. All deaths occurred post-liver transplantation and none were considered related to tafamidis treatment. Of the 2 deaths reported in NCT01435655, 1 was not considered related to treatment; however, a causal relationship could not be excluded in 1 case of sudden death in a 73-year-old male. There were 7 deaths reported in NCT00925002, none considered related to treatment.
Demographics and safety outcomes in the non-interventional THAOS registry (NCT00628745)
A total of 739 subjects with ATTR amyloidosis with some tafamidis treatment exposure have enrolled in THAOS (NCT00628745). The safety population (n = 661) included all subjects with any tafamidis treatment exposure from the time they were enrolled in THAOS, but excluded their time during participation in other clinical trials. As of 3 January 2017, the safety population included 355 males and 306 females, with a mean age of 48.2 years and a mean age at onset of disease of 42.6 years. For the majority of subjects (62.5%) race was not available; 32.8% were Caucasian. The mean duration of disease at baseline was 5.6 years and subjects had a mean (SD) duration of tafamidis exposure of 2.3 (1.5) years ().
There were a total of 559 TEAEs, with 250 (37.8%) subjects experiencing at least 1 TEAE (). In total, 52 (7.9%) subjects had a TEAE considered by the investigator to be related to treatment. The most common of these were: UTI (5 events, 0.8%), and upper abdominal pain, hepatotoxicity, nausea, edema peripheral, renal impairment, skin lesion and vomiting (2 each, 0.3%).
Table 5. Summary of most common TEAEs in the non-interventional THAOS registry (NCT00628745).
A total of 21 subjects experienced a TEAE leading to discontinuation. The most common of these were disease progression (5 subjects), hereditary neuropathic amyloidosis (4 subjects) and liver transplant (2 subjects). In total, 191 TESAEs were reported in 96 (14.5%) subjects with the most common being cardiac failure (1.4%), vomiting (1.1%), UTI (0.8%), hereditary neuropathic amyloidosis (0.8%), pyelonephritis (0.6%), and congestive cardiac failure, urosepsis, and syncope (0.5% each). There were 4 TESAEs considered treatment-related in 4 subjects: 1 incident each of spontaneous abortion, toxic hepatitis, pyelonephritis and acute pyelonephritis.
There were 25 TEAEs associated with a fatal outcome, all of which were judged to be unrelated to treatment. The cause of death, where known, was typically related to disease progression or a concurrent medical condition consistent with ATTR amyloidosis.
Safety outcomes from post-marketing experience
Post-marketing study in Japan
Data on subject demographics are not available from the post-marketing surveillance study in Japan (NCT02146378). As of 31 March 2017, 322 subjects have been enrolled in the study with a total of 135 TESAEs reported in at least 58 subjects. The most common of these were condition aggravated (14 subjects), cardiac failure (9 subjects) and hereditary neuropathic amyloidosis (8 subjects). There were 6 TESAEs in 3 subjects considered by the investigator to be possibly related to tafamidis treatment: 1 subject with bacteraemia, pneumonia and sepsis; 1 subject with pancreatic carcinoma; and 1 subject with hereditary neuropathic amyloidosis and disease progression. A total of 25 deaths have been reported. Of these, 2 were considered by the investigator to be possibly related to tafamidis treatment: sepsis in a subject aged ≥80 years; and hereditary neuropathic amyloidosis and disease progression in a subject aged ≥70 years.
Spontaneous safety reporting
There were 134 spontaneously reported cases generally reflecting the known safety profile of tafamidis and/or underlying diseases/age of the population. These cases were from 75 males and 42 females (with 17 unknown), and ranged in age from 24 to 87 years (mean, 58 years). The most common preferred terms reported were drug ineffective (15 cases), diarrhoea (14 cases) and vomiting (13 cases) (). Of the reported cases, 65 were considered serious cases and 69 were considered non-serious cases. There were 9 deaths unrelated to tafamidis and 3 inconclusive due to lack of information.
Table 6. Summary of most common TEAEs reported in the spontaneous safety reporting database.
Discussion
This comprehensive, integrated safety summary of data from tafamidis-treated patients with ATTR-PN in clinical, observational and post-marketing trials, and post-marketing surveillance did not reveal any significant new safety findings, with data being consistent with the known safety profile of tafamidis as previously described [Citation10,Citation12–17]. Tafamidis (20 mg/day) was generally well tolerated in patients in clinical and observational trials and patients receiving treatment in clinical practice, with this analysis covering 6 years of tafamidis exposure since it was first approved to treat patients with ATTR-PN in Europe [Citation17].
In the first double-blind, placebo-controlled, clinical trial in patients with ATTR-PN (NCT00409175) [Citation12], there were 4 adverse reactions identified: diarrhoea, UTI, upper abdominal pain and vaginal infection. Since that trial was completed, the population of patients with ATTR-PN treated with tafamidis in completed and ongoing open-label studies [Citation13–16] has expanded from 127 to 137 patients, and the mean duration of exposure has expanded from 17.7 months to 44.2 months. These data from additional patients, as well as patients with longer exposure to tafamidis, did not reveal any significant new safety findings. While non-Val30Met patients had a higher incidence of TESAEs than Val30Met patients, this was likely due to underlying differences in patient characteristics. Specifically, non-Val30Met patients were older (median age, 63.0 years vs. 35.0 years), had longer established disease (median time since symptom onset, 42.1 months vs. 22.3 months), and had more severe disease (median NIS-LL score, 18.5 vs. 4.0) than Val30Met patients.
There are limited data on the safety of tafamidis during pregnancy, and women of childbearing potential are advised to use appropriate contraception while taking tafamidis, and for one month after stopping treatment with tafamidis [Citation17]. In this analysis, there were 18 cases of exposure to tafamidis during or within one month prior to pregnancy (either maternal exposure or exposure via father) and there was one incident of spontaneous abortion. The outcomes of the other pregnancies were: 12 normal newborns, 1 medical termination, and 3 with the outcome pending (including one set of twins).
As there may be a potential risk of hypersensitivity and hepatic toxicity with tafamidis, due to its metabolism by glucuronidation [Citation7,Citation17] and the observation in nonclinical studies of tafamidis-associated hepatic alterations at extremely high doses, ongoing monitoring of potential hypersensitivity reactions and hepatic events is part of the established pharmacovigilance activities. However, both this analysis and ongoing pharmacovigilance across the tafamidis clinical programme, including post-marketing experience, have not identified a safety signal associated with either hypersensitivity reactions or hepatic events with tafamidis treatment. In addition, there have not been any cases of tafamidis overdose reported in patients with ATTR-PN. Single doses of up to 480 mg have been administered to healthy subjects and did not identify any safety concerns.
Limitations
While there can be challenges in pooling data from different trials conducted over many sites, integrating data from several trials provides a larger data-set with exposure to drug over a longer period of time and allows for a more comprehensive safety assessment. However, it should be noted that a number of the studies in this analysis remain ongoing. The incidence of AEs in THAOS, which was lower than in the clinical trials, was potentially influenced by the limitations of data collection in an observational survey. In THAOS, data on medication use and AEs are collected at enrollment and on the recommended annual return visits [Citation18]. There are also inherent limitations in post-marketing surveillance and AE reporting systems [Citation19,Citation20]. Reports are voluntarily submitted and, as a consequence, the magnitude and impact of underreporting may not be known [Citation21]. Factors that may influence the reporting of an AE include the length of time the drug has been available; how widely adopted the drug is; the seriousness of the AE; and overall awareness by health professionals and patients of events associated with the drug and of the AE reporting system. As a result of these external factors that influence whether or not an AE is reported, a spontaneous reporting system will yield reporting proportions, not incidence rates. As such, it is generally not appropriate to make direct comparisons with other treatments using these data.
Furthermore, in many AE reports, clinical information (such as medical history, validation of diagnosis, time to onset of the AE, dose, and use of concomitant treatments) is missing or incomplete, with any follow-up information unavailable. At the same time, an accumulation of AE reports will not necessarily indicate that any particular AE was caused by the treatment; rather, the event may be due to underlying disease, other comorbid conditions, or concomitant medication. Ultimately, the spontaneous AE reporting system should be used only for signal detection, rather than for specific hypothesis testing [Citation20].
Despite these known limitations, the strengths of this analysis include a large number of patients evaluated across the various studies, the total duration of exposure to tafamidis, and the consistency between the pooled safety analysis and prior analyses reported in individual trials and related efforts [Citation12–17]. Overall, this analysis supports the currently understood safety profile of tafamidis as safe and well tolerated in patients with ATTR-PN.
| Abbreviations | ||
| AE | = | adverse event |
| ATTR-CM | = | transthyretin amyloid cardiomyopathy |
| ATTR-PN | = | transthyretin amyloid polyneuropathy |
| BMI | = | body mass index |
| IQR | = | interquartile range |
| MedDRA | = | Medical Dictionary for Regulatory Activities |
| NIS-LL | = | Neuropathy Impairment Score-Lower Limbs |
| PMDA | = | Pharmaceuticals and Medical Devices Agency |
| QOL | = | quality of life |
| TEAE | = | treatment-emergent adverse event |
| TESAE | = | treatment-emergent serious adverse event |
| THAOS | = | Transthyretin Amyloidosis Outcomes Survey |
| TTR | = | transthyretin |
| UTI | = | urinary tract infection |
Acknowledgements
We would like to thank all patients and investigators for their participation in these studies.
Disclosure statement
Alison Flynn, Marla Sultan, Huihua Li, Denise Rill, Ben Ebede, and Balarama Gundapaneni are full-time employees of Pfizer and hold stock options with Pfizer. At the time of this analysis Peter Huber and Jeffrey Schwartz were full-time employees of Pfizer, they hold stock options with Pfizer and are now retired.
Data availability
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the USA and/or EU or (2) in programmes that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Additional information
Funding
References
- Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31.
- Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337:898–909.
- Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086–1097.
- Connors LH, Lim A, Prokaeva T, et al. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 2003;10:160–184.
- Rowczenio DM, Noor I, Gillmore JD, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35:E2403–E2412.
- Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018;57:829–837.
- Coelho T, Merlini G, Bulawa CE, et al. Mechanism of action and clinical application of tafamidis in hereditary transthyretin amyloidosis. Neurol Ther. 2016;5:1–25.
- Waddington Cruz M, Benson MD. A review of tafamidis for the treatment of transthyretin-related amyloidosis. Neurol Ther. 2015;4:61–79.
- Maurer MS, Elliott P, Merlini G, et al. Design and rationale of the Phase 3 ATTR-ACT clinical trial (Tafamidis in transthyretin cardiomyopathy clinical trial). Circ Heart Fail. 2017;10. DOI:10.1161/CIRCHEARTFAILURE.116.003815
- Maurer MS, Grogan DR, Judge DP, et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail. 2015;8:519–526.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007–1016.
- Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79:785–792.
- Coelho T, Maia LF, da Silva AM, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260:2802–2814.
- Merlini G, Planté-Bordeneuve V, Judge DP, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Trans Res. 2013;6:1011–1020.
- Ando Y, Sekijima Y, Obayashi K, et al. Effects of tafamidis treatment on transthyretin (TTR) stabilization, efficacy, and safety in Japanese patients with familial amyloid polyneuropathy (TTR-FAP) with Val30Met and non-Val30Met: a phase III, open-label study. J Neurol Sci. 2016;362:266–271.
- Barroso FA, Judge DP, Ebede B, et al. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid. 2017;24:194–204.
- Pfizer Ltd. Vyndaqel® summary of product characteristics. [Internet]. Sandwich, UK; [cited 2019 Apr 23]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002294/WC500117862.pdf
- Planté-Bordeneuve V, Suhr OB, Maurer MS, et al. The Transthyretin Amyloidosis Outcomes Survey (THAOS) registry: design and methodology. Curr Med Res Opin. 2013;29:77–84.
- Goldman SA. Limitations and strengths of spontaneous reports data. Clin Ther. 1998;20:C40–C44.
- Brewer T, Colditz GA. Postmarketing surveillance and adverse drug reactions: current perspectives and future needs. JAMA. 1999;281:824–829.
- Hazell L, Shakir SA. Under-reporting of adverse drug reactions: a systematic review. Drug Saf. 2006;29:385–396.