
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background: Cardiomyopathy is a major cause of death in patients with systemic transthyretin amyloidosis. Long term effect of therapy designed to inhibit hepatic production of the amyloid precursor has not been established in cardiomyopathy. The purpose of this study was to evaluate the long term safety and efficacy of transthyretin specific antisense oligonucleotide therapy, inotersen, in transthyretin cardiomyopathy.
Methods: Patients with hereditary or wildtype transthyretin cardiomyopathy (NYHA I-III) with an LV wall thickness 1.3 cm and clinical evidence of congestive heart failure were eligible for this single centre, open label protocol. Safety and cardiac structural and functional parameters were prospectively studied.
Results: As of October 2018, 33 subjects have entered the study. Twenty have completed 1 year, 16 have completed 2 years, and 14 have completed three years. At the 2 year time point, mean LV mass decreased by 8.4% as measured by MRI, and exercise tolerance increased by 20.2 metres as measured by 6 minute walk test. Further positive indicators were noted at 3 years, with LV mass decreasing by 11.4% and 6MWT increasing by 16.2 metres.
Conclusion: Long term treatment of amyloid cardiomyopathy with inotersen is safe and effective in inhibiting progression and potentially reversing amyloid burden.
Introduction
Cardiomyopathy is a major cause of death in patients with transthyretin (ATTR) amyloidosis. Fatal cardiac arrhythmias account for some deaths but the complications of congestive heart failure take the largest toll [Citation1]. As cardiac output decreases effective organ perfusion suffers to the point where function is insufficient to prolong life. This is especially true for renal function, and cardiovascular hemodynamics (e.g. blood pressure) usually preclude dialysis [Citation2]. Infarction of peripheral tissues is rare but can occur and lead to infection. Any cardiac stress related to trauma, major surgery, or infection can have fatal consequences.
Supportive treatments of the consequences of ATTR cardiomyopathy are in 2 main categories which certainly prolong life beyond the recognised 2.5 to 4.5 year mean survival after diagnosis of ATTR cardiomyopathy [Citation3,Citation4]. (1) Maintenance of a normal or controlled ventricular rate and rhythm to allow optimal diastolic filling of the left ventricle favours maintaining cardiac output and therefore tissue perfusion. Cardioversion for atrial fibrillation is usually effective in the early stages of ATTR cardiomyopathy but often fails with advanced atrial enlargement. Ventricular pacemakers may be needed for conduction system failure but mechanical defibrillators are usually not optimally effective in cardiac amyloidosis. (2) Medical control of the systemic manifestations of cardiovascular amyloidosis is imperative in the care of patients with ATTR congestive heart failure. Retention of electrolytes and water as a response to the failing heart usually reaches a point where cardiac function suffers more than it is helped. Any medication given to ease the load on the heart may lead to fluid retention and decline in effective tissue perfusion. Diuretic medications to reverse the negative response of the kidney are the mainstay of alleviating the effects of congestive heart failure. While life can certainly be prolonged by these non-specific therapeutic measures they do not affect the progressive ingravescent course of transthyretin cardiomyopathy. Therapy to slow or stop the progression of cardiac amyloid burden is needed and may be achievable.
In August 2014, we initiated a study of patients with ATTR cardiomyopathy treated with inotersen, an antisense oligonucleotide (ASO), specifically targeting TTR mRNA and shown in animal and human studies to effectively inhibit the hepatic production of the transthyretin amyloid precursor [Citation5,Citation6]. These patients, in general, did not have significant peripheral neuropathy and, therefore, were not eligible for ongoing studies of inotersen for the treatment of peripheral neuropathy due to systemic amyloidosis. We previously reported data for patients in this study who completed one year on the TTR specific ASO (inotersen) that showed favourable inhibition of cardiac amyloid progression when compared to historical studies [Citation7–9]. The present report presents and analyzes data for subjects in this study who have completed 2 and 3 years treatment with inotersen.
Materials and methods
Study design
Study inclusion criteria include biopsy proven ATTR amyloidosis (hereditary or wild-type) with clinical signs and symptoms of CHF (NYHA I-III), a left ventricular wall thickness 1.3 cm on transthoracic echocardiography, stable renal function (GFR > 35 ml/min) and stable thyroid function (TSH < 10 mcU/ml or normal serum T4).
Transthoracic echocardiograms are performed at baseline and serially every 6-months after starting the ASO. The echocardiograms are performed in one facility by 2 trained technicians. Global left ventricular longitudinal systolic strain is performed on General Electric (Chicago, Illinois) machines using a semi-automated method. The strain values are listed as an absolute number. Cardiac MRI is obtained at baseline and every 12-months after start of ASO treatment. Interventricular septum (IVS) and left posterior wall thickness (LVPW) are measured on transthoracic echocardiograms as recommended by chamber quantification guidelines [Citation10]. The truncated ellipsoid method is used to calculated left ventricular mass (LVM). MRI is not done if the patient has a pacemaker or other implanted cardiac device that is not MR-conditional or other contraindication (ex. irregular rhythm).
MRI is performed with a 1.5-T whole-body scanner (Magnetom Avanto, Siemens Medical Solutions, Erlangen, Germany) with a 45-mT/m gradient system using a 6-channel anterior and a 6-channel posterior torso coil. Images are acquired during breath hold during inspiration using electrocardiographic gating. Evaluation of the left ventricle is performed using steady state free precession cine sequences in standard 2-chamber and 4-chamber views followed by contiguous short-axis slices from the atrioventricular ring to the apex.
Circle Cardiovascular Imaging (cvi), commercial software is used for assessment of left ventricular structure and mass. Left ventricular mass calculation was performed using semi-automated tracings of the endocardium and epicardium in end-diastole. Major papillary muscles were included in the volume calculations.
Study oversight
The study was approved by the Indiana University Institutional Review Board and by the FDA. The standards of Good Clinical Practice and the Declaration of Helsinki were followed. All subjects signed a written informed consent prior to participation.
Study drug
Inotersen (Tegsedi™, Ionis Pharmacetuicals and Akcea Therapeutics), is a 2nd-generation 2'-O-(2-methoxyethyl) modified antisense oligonucleotide that targets transthyretin messenger RNA (mRNA). Inotersen is complementary to a region in the 3’ untranslated region of the transthyretin mRNA and binds by Watson and Crick base pairing. This hybridisation leads to a RNase H1-mediated degradation of transthyretin mRNA, and resultant inhibition of translation of the TTR protein [Citation6,Citation11].
Inotersen is manufactured by Ionis Pharmaceuticals, Inc., (Carlsbad, CA, USA) and Akcea Therapeutics (Boston, MA, USA) in accordance with the standards of Good Manufacturing Practices. Inotersen is supplied in stoppered glass vials or pre-filled syringes at a concentration of 200 mg/mL solution. Inotersen is administered as 300 mg/1.5 ml subcutaneously once a week without a loading dose with modification of total dose and variations in time intervals at the discretion of the investigator. CBC with platelet count is obtained weekly and renal function (urine protein and quantitative serum creatinine concentration) at least every 6 weeks.
Efficacy measures
6-min walk test (6MWT), beta-type natriuretic peptide (BNP), and transthoracic echocardiogram are performed at baseline and repeated every 6 months throughout the trial. Cardiac MRI, when not contradicted, is obtained at baseline and every year throughout the study.
Statistics
Both structural (MRI, IVS) and functional (systolic strain, 6 MWT, BNP) parameters followed normality by a Shapiro-Wilk test (p-value > 0.05 for normality) and were eligible to use a mixed effect model. We then used a mixed effect model for repeated measurement of 4 times (baseline, 1 year, 2 year, and 3 year) to test for time effect and group effect (wild type versus hereditary) on the structural and functional parameters. The mixed effect model captured the unstructured correlation across the repeated measurements of 4 time points within a subject and the fixed effects were modelled to test for the time and the group effect. Each analysis adjusted for only age since all subjects except one were male.
Results
As of October 2018, 33 subjects with ATTR cardiomyopathy (10 hereditary, 23 wild type) have entered this open label study (). The mean age of the hereditary patients at the time of study entry was 63.4 years and the mean age of the wild-type patients at the time of study entry was 76.2 years. At the time of study entry, in the hereditary group (n = 10) the interventricular septum thickness ranged from 1.3 to 2.2 cm with a mean of 1.85 cm, LVEF was ≥ 50% (n = 9) in the majority of patients, and the absolute value of the left ventricular global longitudinal strain ranged from 11.8 to 19.1 with a mean of 13.3. In the wild-type group (n = 23) at the time of study entry interventricular septal thickness ranged from 1.4 to 2.8 cm, with a mean of 2 cm, the left ventricular ejection fraction was ≤ 50% majority of the patients (n = 16), and the absolute value of the global longitudinal systolic strain ranged from 5.8 to 13 with a mean of 9.2. Nine subjects have left the study for the following reasons: 1) voluntary withdrawal (n = 6), 2) non-compliance (n = 2), and 3) nondrug related death (n = 1).
Table 1. Baseline characteristics of patients at study entry.
Structural studies
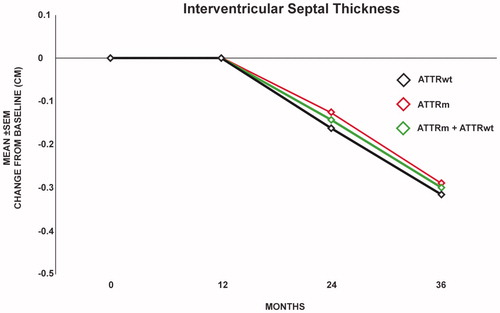
Mean left ventricular mass for all subjects calculated by MRI showed a progressive decline at one year (0.54%, n = 10), two years (8.5%, n = 10), and 3 years (11.5%, n = 8) (). This decrease in LVM was seen in both hereditary (n = 7) ATTR and wild type cardiomyopathy (n = 3) (). Only 2 patients showed an increase in LVM over the 3 year time period of observation and both with less than 10 percent change. Subsequent MRI imaging in these 2 patients showed a decline in LVM on MRI, suggesting that these increases fell within the expected margin of error with LVM calculations and were not significant. While calculation of LVM by echo gave variable results, monitoring of left ventricular wall thickness generally agreed with the decrease in LVM calculated by MRI (). In the hereditary group at 2 years the interventricular septal thickness ranged from 1.1 to 2.1 cm with a mean of 1.7 cm (n = 8) and at 3 years it ranged from 1 to 1.9 cm with a mean of 1.5 cm (n = 8). In the wild-type group at 3 years the interventricular septal thickness ranged from 1.5 to 2.4 cm with a mean of 2 cm (n = 8) and at 3 years ranged from 1.4 to 1.9 cm with a mean of 1.9 cm (n = 6). In both the hereditary and wild-type patients there was a decrease in left ventricular septal thickness over time ().
Figure 1. Left ventricular Mass on MRI. Mean decrease in left ventricular mass compared to baseline was 0.54% (n = 10), 8.5% (n = 10), and 11.5% (n = 8) at 1, 2, and 3 years respectively.
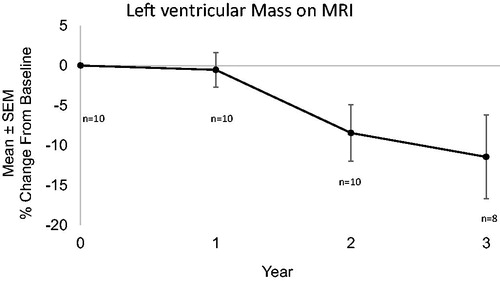
Figure 2. Echocardiogram and MRI of a subject with hereditary ATTR cardiomyopathy: Top (Echocardiogram): IVS decreased from 1.8 cm (left) at baseline to 1.4 cm at 2.5 years (right). Bottom (MRI): LVM decreased from 331 grams at baseline (left) to 149 grams at 3 years (right).
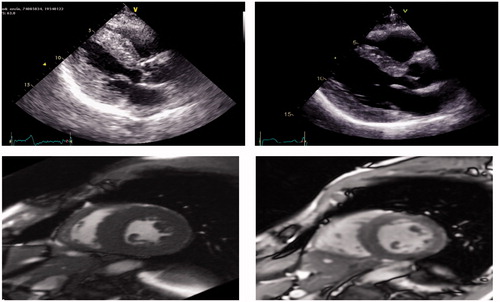
Figure 3. Interventricular septal thickness on Echo compared to baseline (n = 20) was stable at one year (n = 20) and decreased by 0.14 cm (n = 16) at 2 years and 0.3 cm (n = 14) at 3 years (p = .019) in all patients (hereditary and wild-type) as shown by the center line. The decrease in wall thickness was similar in the wild-type patients (bottom line) and the hereditary patients (top line).
Functional studies
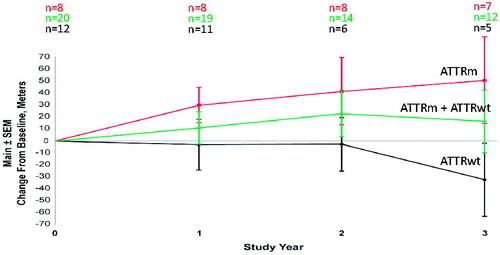
After 2 or 3 years of therapy with inotersen, the left ventricular ejection fraction remained stable in most patients. In the hereditary patients that completed 2 or 3 years the LVEF was ≥50% in all patients (n = 8). In the wild-type patients, at baseline 16 of 23 had an LVEF ≤ 50%. Of the 8 wild-type patients that completed 2 or 3 years, 4 had an ≤ 50% at 2 years and 5 had an LVEF ≤ 50% at 3 years. Mean 6MWT distance for all subjects increased progressively for 2 years observation then stabilised. This was largely due to changes for the hereditary patients, whereas the wild type ATTR patients maintained a relatively steady state (). Global longitudinal systolic strain improved by 0.3 ± 0.9 in all patients at 3 years (n = 11). The improvement was mainly in the hereditary patients who showed improvement of 1.3 compared to baseline at 3 years (n = 7), whereas global longitudinal declined by 1.4 in the wild-type patients (n = 4). When controlling for age, statistical analysis of global longitudinal strain showed significant difference in hereditary and wild type patients (p = .004).
Figure 4. Center line: 6MWT in patients tested at 1, 2, and 3 years. Mean 6MWT increased by 10.63 (n = 19), 22.2 (n = 14) and 16.5 (n = 12) meters compared to baseline at 1, 2, and 3 years respectively. Top line: 6-MWT in ATTRm patients. Mean 6-MWT increased by 29.3 (n = 8), 40.9 (n = 8) and 50.2 (n = 7) meters at 1, 2, and 3 years respectively. Bottom line: 6-MWT in ATTRwt patients. Mean 6MWT decreased by 3 (n = 11), 2.6 (n = 6), and 31.7 (n = 5) metres compared to baseline at 1, 2, and 3 years respectively.
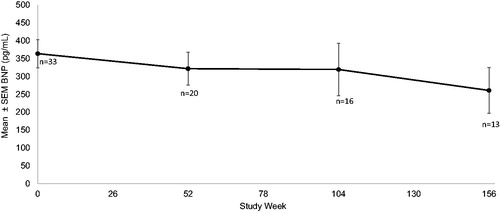
BNP showed a steady decline in the mean value for all subjects (). As with 6MWT distances greater changes were evident for the hereditary patients. Transthyretin serum concentrations varied from 50% to 90% suppression in this study. Without a loading dose in the first week maximal TTR suppression often did not occur until 6 weeks of weekly dose administration. Comparison of TTR levels to either functional or structural changes did not reveal a significant correlation.
Figure 5. Mean BNP decreased from 353.8 pg/dl (n = 33) at baseline to 321.7 pg/dl (n = 20), 319.4 pg/dl (n = 16) and to 260.8 pg/dl (n = 14) at 1, 2, and 3 years respectively.
Statistical analysis of structural (MRI, IVS) and functional (systolic stain, 6 MWT, BNP) parameters showed significant differences for wild type versus hereditary patients for MRI LVM (p = .006), echo longitudinal systolic strain (p = .004) and IVS (p = .014). In addition, the time effect on IVS (p = .019) was observed. Despite trends as noted in and changes in 6MWT and BNP did not meet statistical significance.
Safety and tolerability
Safety
Analysis of blood platelet levels in patients who have completed 1, 2, and 3 years reveals average decrease in platelet level of 14%, 15%, and 15% respectively compared to baseline. The lowest platelet count of each individual subject noted at any clinic visit revealed an average nadir of 27%, 30%, and 35% during years 1, 2, and 3 respectively. In the present study of patients with ATTR cardiomyopathy, many patients are on oral anticoagulation for atrial arrhythmias and some on antiplatelet agents for coronary artery disease. Even with concomitant use of anticoagulant or antiplatelet therapy, no significant bleeding episode or severe thrombocytopenia has been observed in any subject.
Monitoring for adverse renal effects showed no significant proteinuria in any subject. Serum creatinine levels monitored at each clinic visit have shown fluctuations coincident with dosage of diuretics used for treatment of CHF. An entry criterion for the study was CrCl 35 cc/min and many patients have approached this level while on multiple diuretic agents, however, no patient has been dropped from the study due to renal insufficiency. Only one subject has died while on ASO medication, suffering sudden cardiac death after urgent cholecystectomy required for cholelithiasis related acute cholecystitis and pancreatitis.
Tolerability
Inflammation and induration associated with ASO subcutaneous injection, usually lower abdomen, were reported by approximately 25% of subjects. This has been transient with the use of alternating sites for injection. Injection of ASO is done by the patient or caregiver. Generalised “flu like” symptoms have occurred in a few patients and last for 24–48 h. This effect has been unpredictable, occurring after the first or second exposure to the ASO with resolution and no further episodes in some patients. Similar constitutional symptoms have been noted by a few patients later in their course of treatment. Only one patient has discontinued treatment due to this phenomenon.
Discussion
Until recently there has been no specific therapy for hereditary transthyretin amyloidosis. Progression of the disease has usually lead to death within 5–15 years whether due to the ravages of peripheral and autonomic neuropathy or cardiac arrhythmias or congestive heart failure [Citation12]. In addition the increasing presentation of wild type ATTR with predominately a clinical cardiac phenotype has emphasised the need for specific therapy to prevent or halt the continuous deposition of amyloid.
To date two lines of specific therapy for ATTR have been explored. Small molecule therapeutics (diflunisal and tafamidis) which affect the stability of the serum amyloid fibril precursor TTR have been shown to delay progression of amyloid neuropathy and tafamidis has now been found to alter the progression of ATTR cardiomyopathy as measured by all causes of death or hospitalisation [Citation12–15].
Therapy for hereditary ATTR amyloidosis by eliminating the TTR amyloid precursor by liver transplantation was first instituted by Holmgren et al. [Citation16]. This resulted in slowing systemic amyloid progression, most notably for patients with the Val30Met TTR mutation, but subsequent progression of the disease was due to continued amyloid formation and deposition enriched with the normal wild type TTR molecule [Citation17,Citation18].
More recently specific therapy for ATTR by parenteral administration of inhibitors of hepatic TTR production has shown highly significant inhibition of neuropathy progression in patients with hereditary ATTR amyloidosis [Citation19,Citation20]. Both antisense oligonucleotide and siRNA drugs specific for TTR mRNA have now gained FDA and EMA approval for treatment of patients with hereditary ATTR peripheral neuropathy. While not a primary objective of the controlled studies, analysis of cardiac parameters in these hereditary cohorts has suggested a beneficial effect on cardiomyopathy [Citation21]. It should be noted, that while inhibition of TTR production should have a similar response in patients with wild type ATTR cardiomyopathy, this has not been subject to controlled study.
The study, for which data are presented here, is not a controlled study. It was initiated to test the safety of the ASO inotersen in patients with moderately advanced cardiomyopathy with CHF. It also offered as open label treatment for hereditary ATTR patients who had cardiomyopathy but not peripheral neuropathy. Thus patients with wild type ATTR amyloidosis who do not generally have polyneuropathy have qualified for admission to this ongoing study. As of October 2018 16 patients have completed 2 years on study drug and 13 patients completed 3 years. Of note no patients have developed significant neuropathic symptoms or signs over the period of observation.
The results of this study suggest that therapy with inotersen has the potential to alter some of the structural changes of amyloid cardiomyopathy such as wall thickness and left ventricular mass. These structural changes may improve left ventricular stroke volume. It is hypothesised that improved stroke volume may account for the increase in 6MWT distance in the hereditary patients. The improvement in 6MWT distance is encouraging because previous studies have shown a decline in 6MWT in untreated patients and that stabiliser therapy may slow this decline, but does not result in improvement [Citation15,Citation22]. The mean decrease in BNP in this study is also promising, however, interpretation is limited by the fact that changes in BNP may result from specific therapy with inotersen as well as non-specific therapy with diuretics and optimisation of volume status and heart rhythm.
Inotersen has shown a well-tolerated safety profile in this cohort of elderly patients with significantly impaired cardiac reserve. Lowering of platelet counts has been a consistent finding but no significant bleeding has been related to drug administration even in patients taking antithrombotic drugs. No signs of glomerulonephritis have been noted in any subject.
Close monitoring of cardiac parameters has shown efficacy of inhibition of TTR production as measured by both structural and functional status of disease. As of one year of the study the 15 subjects analysed mainly showed lack of progression of disease [Citation7]. Now data from subjects at both 2 and 3 years of treatment indicate not only lack of disease progression, but in some subjects, a decrease in cardiac amyloid burden (). This finding suggests that ATTR cardiac amyloid deposits may represent a chemical state where decreasing the substrate for fibril formation may reverse an equilibrium and result in reabsorption of the deposited amyloid. This has been documented for immunoglobulin light chain (AL) amyloidosis when the offending plasma cell dyscrasia is effectively treated and AA (secondary, reactive) amyloidosis when plasma serum amyloid A levels have normalised. The present study suggests that a similar effect can be attained by restricting the ATTR fibril precursor protein. Amyloid, while defined by the main protein constituent, is really a complex substance with varying proportions of a number of biologic entities including glycosaminoglycans and serum amyloid P (SAP). Disruption of this state by restricting availability of the main protein constituent may lead to dissolution.
In conclusion: Analysis of patients treated with inotersen for 2 and 3 years is consistent with not only lack of progression of disease but in some cases reduction of cardiac amyloid burden. While most notable in patients with hereditary ATTR amyloidosis, lack of progression of cardiomyopathy in patients with wild-type ATTR disease is encouraging. The latter is an age related phenomenon and medical treatment to inhibit TTR production may significantly impact life expectancy.
| Abbreviations | ||
| 6-MWT | = | 6-minute walk test |
| ASO | = | antisense oligonucleotide |
| ATTR | = | transthyretin amyloidosis |
| BNP | = | beta type natriuretic peptide |
| CHF | = | congestive heart failure |
| cvi | = | circle cardiovascular imaging |
| GFR | = | glomerular filtration rate |
| IVS | = | interventricular septum |
| LV | = | left ventricle |
| LVEF | = | left ventricular ejection fraction |
| LVPW | = | left ventricular posterior wall |
| MRI | = | magnetic resonance imaging |
| mRNA | = | messenger RNA |
| SAP | = | serum amyloid P component |
| NYHA | = | New York Heart Association class |
| TSH | = | thyroid stimulating hormone |
| TTR | = | transthyretin |
| T4 | = | thyroxine |
Acknowledgments
The authors thank Elizabeth J. Ackermann amd Brett P. Monia for assistance with protocol development and data analysis for this study and Ionis Pharmaceuticals (Carlsbad, California) and Akcea Therapeutics (Boston, MA), for providing inotersen (Tegsedi™).
Disclosure statement
Dr. Benson has received consulting fees from Ionis Pharmaceuticals and Akcea Therapeutics. Dr. Dasgupta has received consulting fees from Ionis Pharmaceuticals, and Pfizer. Dr. Dasgupta is on the Speaker’s Bureau for Akcea Therapeutics and Pfizer.
Additional information
Funding
References
- Dungu JN, Anderson LJ, Whelan CJ, et al. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546–1554.
- Lobato L, Rocha A. Transthyretin amyloidosis and the kidney. Clin J Am Soc Nephrol. 2012;7(8):1337–1346.
- Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286–1300.
- Grogan M, Scott CG, Kyle RA, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol. 2016;68(10):1014–1020.
- Benson MD, Kluve-Beckerman B, Zeldenrust SR, et al. Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve. 2006;33(5):609–618.
- Ackermann E, Guo S, Benson MD, et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-Generation antisense oligonucleotides. Amyloid. 2016;23(3):148–157.
- Benson MD, Dasgupta NR, Rissing SM, et al. Safety and efficacy of a TTR specific antisense oligonucleotide in patients with transthyretin amyloid cardiomyopathy. Amyloid. 2017;24:219–225.
- Benson MD, Dasgupta NR. Editorial comment – Amyloid cardiomyopathy. J Am Coll Cardiol. 2016;68(1):25–28.
- Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203–1212.
- Lang RM, Badano LP, Mor-Avi V, et al. Recommendations for cardiac chamber quantification by echochardiography in adults: an update for the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2015;28(1):1–39.
- Geary RS, Wancewicz E, Matson J, et al. Effect of dose and plasma concentration on liver uptake and pharmacologic activity of a 20-methoxyethyl modified chimeric antisense oligonucleotide targeting PTEN. Biochem Pharmacol. 2009;78(3):284–291.
- Waddington-Cruz M, Benson MD. A review of tafamidis for the treatment of transthyretin-related amyloidosis. Neurol Ther. 2015;4:61–79.
- Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a clinical trial. JAMA. 2013;310(24):2658–2667.
- Coehlo T, Maia LF, Martins da Silva A, et al. Long-term effects of tafamidis for the treatment of transthyretin amyloid polyneuropathy. J Neurol. 2013;260:2802–2814.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016.
- Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidoitic polyneuropathy (FAP-met30). Clin Genet. 2008;40(3):242–246.
- Yamashita T, Ando Y, Okamoto S, et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology. 2012;78(9):637–643.
- Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277–282.
- Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31.
- Adams D, Gonzalez-Duarte A, Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21.
- Solomon SD, Adams D, Kristen A, et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation. 2019;139(4):431–443.
- Ruberg FL, Maurer MS, Judge DP, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J. 2012;164(2):222–228.