
Abstract
Background
Hereditary transthyretin (ATTRv) amyloidosis is a progressive multisystemic disease of adult-onset that arises from an inherited mutation in the transthyretin gene. Currently available disease severity and progression evaluation tools only cover one single organ or system, impacting data collection uniformity and its use in clinical settings.
Methods
The Jandhyala Method, including a systematic literature review and SMART interviews, was used to observe expert opinion from eight leaders in the treatment of ATTRv across Europe. The aim was to propose a multidisciplinary core dataset (CD) and disease severity scoring (DSS) tools.
Results
The multidisciplinary team of experts identified 140 indicators that form part of the standard diagnostic and monitoring practice (SDMP) and should be collected as the ATTRv CD. Thirty-one (22%) of these indicators informed disease severity and comprised the ATTRv DSS, whilst 25 (18%) were deemed to monitor disease progression.
Conclusions
The resulting CD and DSS have different purposes. The ATTRv CD supports the collection of high-quality data for clinical research, whereas the ATTRv DSS can be rapidly conducted in a clinical setting and aid patient management.
Introduction
Hereditary transthyretin (ATTRv) amyloidosis is a rare, progressive and fatal disease that affects multiple organs [Citation1]. It occurs due to mutations in the transthyretin (TTR) gene (chromosome 18q11.2–12.1) that result either in the destabilisation or the proteolytic cleavage of tetrameric TTR proteins, which contain variant chains [Citation2,Citation3]. As a result, the protein accumulates as amyloid fibrils in multiple tissues and organs, causing progressive dysfunction [Citation4,Citation5]. Consequently, a variety of signs and symptoms arise that are challenging to diagnose and treat.
ATTRv amyloidosis affects more than 50,000 people globally [Citation3]. Diagnosis is often delayed, with an average time to diagnosis of 2.8–4.3 years after the first symptom's appearance [Citation3]. This delay is primarily a result of the varied presentation of symptoms and incomplete family history. Clinically, ATTRv phenotypes are diverse. The disease is mainly characterised by adult-onset polyneuropathy or amyloid cardiomyopathy – although complex mixed phenotypes are common and are often dependent on the type of mutation, age of onset, and geographic origin [Citation6,Citation7]. Consequently, ATTRv amyloidosis can be characterised by various sensory, motor, and autonomic neuropathic manifestations associated with cardiac, gastrointestinal, ocular, and renal complications [Citation7]. The disease is fatal within 2–15 years from the first symptom onset [Citation3]. The average survival from time of diagnosis with ATTRv amyloidosis varies between 2–3 and 10–15 years for cardiac and neuropathic phenotypes, respectively. Patients can continue to have disease progression and reduced survival despite early therapeutic intervention [Citation8,Citation9]. Management of ATTRv amyloidosis with novel anti-amyloid drugs is emerging, and many late-onset cases are now eligible for disease-modifying treatment [Citation3,Citation6].
Monitoring ATTRv amyloidosis
Patients receiving one of these new treatments can be clinically studied in two ways: via interventional ‘trials’ or non-interventional, observational studies. Interventional clinical trials are the gold standard for regulators in determining the efficacy and safety of medicines in a highly selected patient population under arguably artificial conditions. As a result, they are limited in their generalisability to answering the questions of other relevant groups such as prescribers and payors who are interested in patients seen in everyday practice [Citation10,Citation11]. In contrast, observational studies or patient registries are more inclusive, less intrusive, and therefore their findings are more representative of patients in this ‘real-world’ [Citation12,Citation13].
Observing patients in this real-world setting is not without its challenges; rare diseases with complex phenotypes such as ATTRv amyloidosis can be the most problematic. The multisystem nature of the condition brings a further level of complexity; aspects of the disease may be managed by more than one speciality concurrently. The lead responsibility and, therefore, clinical emphasis for the patient across geographies may also vary. It follows that approaches to real-world monitoring of ATTRv patients, embracing or unifying these multidisciplinary perspectives, offers the best opportunity to understand disease severity, its progression and responses to treatments in a holistic and more clinically relevant way. Achieving this objective can involve engaging representatives of all relevant disciplines and observing a consensus on a unified list of indicators pertinent to monitoring the ATTRv patient that could be collected in a patient registry. A recently published novel consensus methodology, The Jandhyala method [Citation14], enables metrics on awareness and consensus amongst participants to be observed without encouraging participants to alter their opinion. This differentiates it from other competing methodologies [Citation15] and has been successful in a multidisciplinary setting.
ATTRv core dataset
A generally accepted limitation of a patient registry compared to a clinical trial is the expectation of missing data from that requested. As no examinations and investigations can be either mandated by the protocol or fall outside routine practice, which is in contrast to a clinical trial, the dataset's quality is understandably lower and more difficult to interpret. It follows that, when designing a core dataset for a patient registry [Citation10], Standard Diagnostic and Monitoring Practice (SDMP) across all participating geographies should be taken into account. Furthermore, the amount of data requested should be acceptable to the investigators and easily achievable in a clinical setting whilst still reaching the scientific objective threshold. The data format must also be standardised to ensure that the contributing datasets can be reliably combined into a single, uniform aggregated dataset. Meeting these criteria will guarantee the collection of a unified dataset of optimal quality to form a successful ATTRv CD [Citation11]. Currently, no ATTRv-specific CD exists, and existing ATTRv disease assessments typically utilise clinical tools such as the Familial Amyloid Polyneuropathy (FAP) staging system and the Polyneuropathy Disability (PND) score [Citation6]. Clinical scales such as the Composite Autonomic Symptom Scale-31 (COMPASS-31) questionnaire, the Compound Autonomic Dysfunction Test (CADT) questionnaire and the Neuropathy Impairment Score (NIS) are also used [Citation6]. Though helpful, these clinical scales only provide a generic indicator of overall disease status. They are not sufficiently sensitive enough to track disease progression in patients with ATTRv amyloidosis in the short-term. They only address the polyneuropathy aspect of the disease [Citation6].
Furthermore, they may not form part of SDMP across all treatment centres. Consequently, an ATTRv CD to be used across multiple centres and countries needs to be created by following SDMP. This will enable current limitations to be addressed and clear the path for high-quality observational research to be conducted to answer stakeholders’ remaining questions – specifically those that are not answerable by the conduct of clinical trials.
ATTRv disease severity score
Endpoints in clinical trials are constructs that measure direct clinical benefits such as more prolonged survival, improved function, and symptomatic improvement [Citation13]. However, the selection of non-relevant, or exclusion of relevant, endpoints may not detect or accurately assess the full severity of the disease in a patient. The development of disease-specific severity scores addresses this measurement bias by measuring the full range of disease phenotype indicators. Therefore, and as an alternative to single measures, an ATTRv DSS limited to a complete list of relevant items contained within the ATTRv CD can be expected to facilitate accurate patient identification, assessment and monitoring. Such a scoring system can assess and quantify the severity of a disease and the response to treatment [Citation12].
Through a dedicated assessment of all indicators contained in the CD, a holistic view of the burden of disease in patients with ATTRv amyloidosis can be achieved with an associated score at a particular point in time. Such scores, if performed serially, can enable relevant items to inform disease severity and progression and describe the contributing changes in the values of indicators, domains, and overall scores. Significant differential changes in sub-domain values can be detected between individuals with similar overall scores ( and ).
Figure 1. ATTRv disease phenotype aligned to best fit system organ classification..
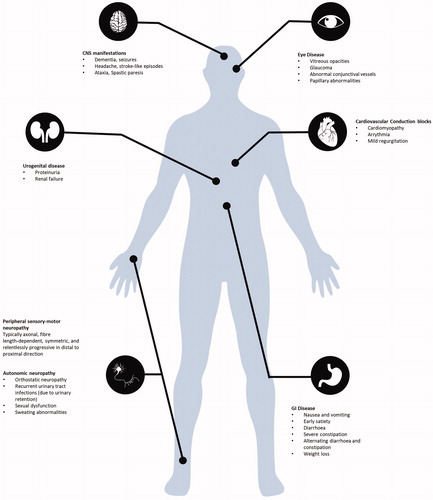
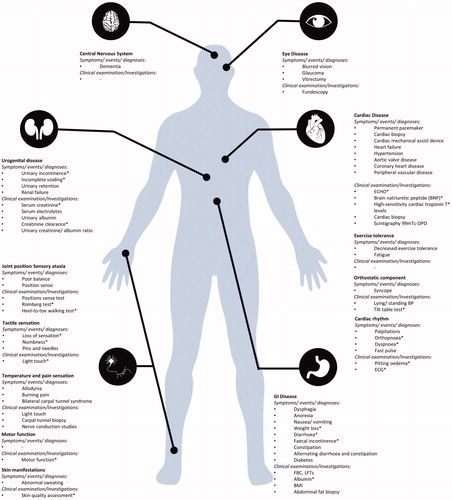
Figure 2. ATTRv core dataset indicators aligned to best fit system organ classification. *Indicators informing disease severity and featured in ATTRv disease severity score.
Methods
The Jandhyala Method [Citation14,Citation15] was utilised, including a systemic literature review to generate a list of proposed items to be included in the CDS. Following this, SMART interviews were conducted with each expert to assess, in turn, whether each item: formed part of standard diagnosis and monitoring practice in the expert’s centre and if, in their opinion, it informed disease severity and could be used to monitor disease progression. Finally, a correction was applied to the responses to balance them equally across the disciplines of the experts. Full details of the method can be found in the Supplementary material.
Results
Jandhyala method
Systematic literature review
A total of 14,989 publications were identified through PubMed and proceeded to screening. In addition, eight existing registries found via internet searches, as well as the Cochrane Register of Controlled Trials, were used to identify a further 875 publications on clinical trials. In total, using the PRISMA Literature Review Protocol, 15,864 studies were identified from all sources, and, following the removal of duplicate items (N = 590), 15,274 studies progressed to title and abstract screening. Of these, 1492 studies progressed to full-text review. After excluding 1190 articles, data were extracted from the remaining 302 articles ().
Figure 3. PRISMA flow diagram illustrating the process of obtaining the ATTRv core data set from the systematic literature review.
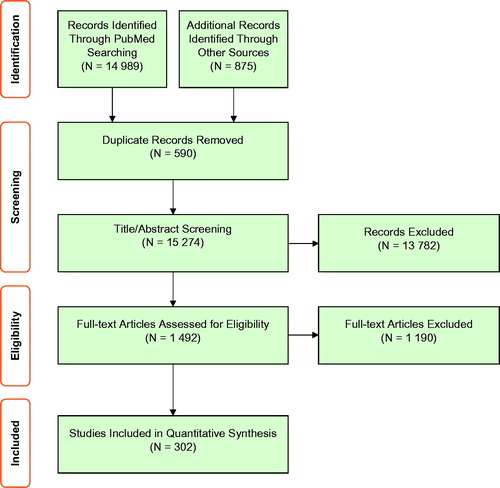
A total of 982 items were identified from the data extracted from these studies. Data synthesis was conducted following the removal of duplicate items, combination code merging, and bi-directional frequency counting with the Awareness Round (1) expert responses.
Awareness and consensus round surveys
Eight experts in the management of ATTRv amyloidosis from across Europe agreed to participate in this study. Two experts were from Portugal, and two were from France, while the United Kingdom, Italy, Spain, and Sweden all had one expert each. Their specialities were as follows: three neurologists, two cardiologists, and one nephrologist, gastroenterologist, and internal medicine, respectively.
All eight European ATTRv experts completed the Awareness Round (1) survey. One hundred and fifteen items were coded from their free-text responses. When combined with unique items extracted from the systematic literature review, a total of 698 items were agreed for inclusion in the Consensus Round (2) survey ().
Figure 4. Flow diagram illustrating the attrition of the clinical items and indicators throughout the project timeline. *Items from SMART interviews that were considered by >50% of the experts to be part of the Standard Diagnostic and/or Monitoring Practice (SDMP) were included as Indicators at the Core Dataset stage.
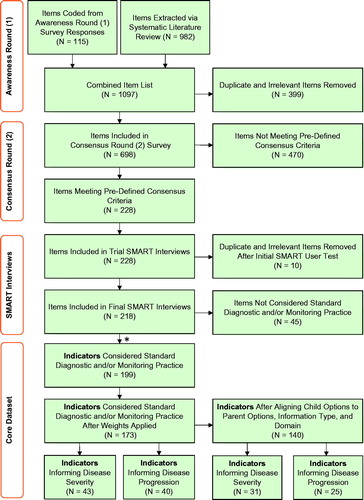
Seven out of eight experts completed the Consensus Round (2) survey. Two hundred and twenty-eight out of 698 (33%) met the consensus threshold and were included for appraisal in the SMART interviews ().
SMART interviews
All eight European ATTRv experts completed the SMART interviews. However, one expert was time-constrained in completing a proportion of the subject matter.
A pilot interview with the senior author was conducted to ensure that the interview structure, materials, and items were optimal. Any items identified as duplicated, misclassified, or irrelevant were excluded (N = 10) from the full SMART interview exercise (N = 218).
One hundred and ninety-nine items were agreed to be SDMP and formed the CD indicators by over 50% of the experts. Ninety (45%) of these CD indicators were agreed to inform disease severity, and 77 (39%) indicators were agreed to inform disease progression by over 50% of the experts. All the disease progression-informing indicators were included in the disease severity subset. Once the weights across all the stakeholder disciplines were balanced and equalised to eliminate bias, 173 indicators were retained in the CD. Forty-three (25%) of these indicators informed disease severity, and 40 (23%) of them informed disease progression. These same indicators were further aligned by information and domain type, which resulted in a final list of 140 indicators for the CD. Thirty-one (22%) of these indicators informed disease severity, and 25 (18%) informed disease progression.
Core dataset, disease severity score and disease progression
An ATTRv CD structure was developed to house all 140 indicators (Supplementary Table 1) using category headings which were selected to best reflect both the nature of the indicator and their position in the sequence of a routine consultation, namely: Demographics, Symptoms, Events/diagnosis, Clinical examinations, and Investigations ().
Table 1. ATTRv core dataset baseline demographics.
The CD is reduced further to create the DSS, which has 31 indicators across the same category headings as the CD. In the clinic setting, the collection of the DSS indicators can be achieved independently without committing to the full core dataset. Clinicians need only ask 10 questions (9 symptoms and 1 event/diagnosis) and conduct 6 clinical bedside examinations and 5 investigations to assess all 31 indicators contained within the DSS (). Alternatively, the DSS can be automatically generated from the CD through a simple calculation programmed into its electronic data capture tool.
Table 2. ATTRv core dataset and disease severity score indicators.
The DSS indicators were arranged into domains with each assigned a weighting taken from the average of those provided by the experts (Supplementary Table 2): Joint position (proprioception) (3.62); Tactile sensation (6.87); Gastrointestinal disease (9.87); Urogenital disease (5.75); Skin manifestations (1.63); Cardiovascular manifestations (21.50); Cardiac disease (21.50); and motor (14.37). Values are assigned for each category of indicator severity provided. The most significant cardiovascular indicators were symptoms of orthopnoea (1.26) and dyspnoea (1.21), while the event/diagnosis of heart failure (1.53) was considered the most significant cardiac disease indicator. The maximum severity score achievable is 38.93 (Supplementary Tables 1 and 2).
All indicators, agreed by the experts, informing disease progression formed a subset of those retained in the disease severity score (25/31) (Supplementary Table 2).
Standardised assessments of ATTRv amyloidosis
Standardised assessments contained a degree of overlap across indicators within the group and indicators included in the other sections of the CD and DSS. These were retained for the CD (N = 9) but were excluded from the disease severity and disease progression scoring. This was primarily because completion of these assessments would introduce an irreconcilable degree of double counting for some indicators. Furthermore, the time taken to complete some questionnaires would be incompatible with the clinic setting, limit the adequate completion of the disease severity score, and impact both its utility and adoption.
Discussion
The Jandhyala Method was successfully implemented here to observe a consensus of expert opinion on the indicators relevant to monitoring ATTRv patients that should form a Core Dataset and Disease Severity Score. Both instruments were duly developed to utilise raw data from questioning in clinic, clinical examination findings, and investigation results.
Multidisciplinary insights
The background disciplines of the recruited experts reflected the truly multidisciplinary care being provided to ATTRv patient across Europe with contributions from the fields of neurology, cardiology, gastroenterology, nephrology, and internal medicine. This confirms the disease's multifactorial nature and the breadth of variation in the disease phenotype to require their specialisms’ involvement. In a care provider network such as this, where the local clinical emphasis may be focussed in certain areas to a greater degree than others, the importance of a unifying CD and DSS tailored for patients with ATTRv amyloidosis, providing representation of all relevant aspects of the disease to all specialists involved can be appreciated. They also offer a realistic opportunity to combine small, disparate populations of ATTRv patients into a single homogenous one available for more highly powered research.
In this research, a preponderance of neurologists and cardiologists meant that a balancing of opinion by speciality was justified to ensure the less well-represented disciplines’ suggestions were recognised in the CD. The exercise had a welcome effect of reducing the total indicators in the CD, DSS and DP from 199 to 140, 90/199 to 31/140 and 77/199 to 25/140, respectively.
The profile of the experts enabled meaningful interdisciplinary discussions to take place on the structure of the CD with domain headings and item categories requiring some revision. Initially, a disease aetiology approach was used. For instance, neuropathy was divided into small fibre disease and large fibre disease. This proved problematic, as some indicators could not be attributed to a single system or organ class. For example, experts felt that it was not appropriate to align orthostatic hypotension with only one system or organ (either neurological findings or cardiac findings). This was because orthostatic hypotension could be caused by pathology from either of the two. Consequently, specific improvements to the CD were recommended and adopted through discussion with the experts. One such change included incorporating new, more inclusive domain headings, such as ‘cardiac manifestations’ and ‘skin manifestations’.
The following deals with categorising indicators into symptoms, events/diagnosis, clinical examinations, and investigations. Initial concerns relating to the reliability, or reproducibility, of soliciting/eliciting symptoms and signs of ATTRv amyloidosis were raised. Through the CD's development and the feedback obtained during the SMART interviews, the experts indicated that specialist clinicians might feel that some of the indicators lie ‘outside’ of their speciality. This would negatively impact their scoring. To address this, the development of a user manual was recommended to outline key definitions and remind the user on how to perform clinical examinations such as the Romberg test or assessing reflexes if required. Development of such a user manual is planned.
Disease phenotype complexity
An objective indication of the true breadth of the disease phenotype and its distribution across several organ systems was demonstrated by the inclusion of 199 items (later refined to 140 indicators) meeting the consensus threshold for inclusion in the ATTRv CD. Consequently, the importance of utilising a standardised tool such as the CD and DSS to assess disease severity and disease progression can be seen, particularly regarding clinicians and researchers becoming more sensitive to changes in the clinical manifestations managed by other disciplines.
Understandably, the CD with its 140 indicators, though indicative of the level of detail required to monitor ATTRv patients, its implementation in a busy clinic setting was thought to be problematic. The availability of the abbreviated set of indicators in the DSS () was seen as a better approach in this context, where it could be completed using a paper or electronic version as an aide-memoire. This would enable efficient calculation and tracking in the patient notes. The CD could reasonably be reserved for formal research (Supplementary Table 1), such as patient registries and clinical trials.
Standardised assessments
Several standardised assessments were initially identified through the process outlined above in both the CD and DSS. A closer review of these tools revealed a significant overlap with indicators already included elsewhere in the CD. An inability to reconcile the inevitable double counting of information, particularly in the DSS, and the prohibitive time required to administer these tools in the clinic setting contributed to their exclusion from the final DSS. However, they remain represented in the CD for research purposes. Consequently, the DSS now lacks a patient-reported outcome measure. However, the inclusion of one specifically developed for the clinical setting at a later date would be desirable.
Adoption of core dataset as a disease severity and progression tool
To the authors’ knowledge, this study is the first of its kind to develop an ATTRv CD that can be used as a disease severity and disease progression tool. Though a clear need for it has been expressed, it faces the expected phases of adoption seen by any new approach or intervention.
The efforts taken to ensure the indicators included are those which are customarily conducted and collected go some way to increasing the chances of a smooth adoption. However, the DSS has not yet been validated. This will be an essential next step towards utilising the DSS as a more interactive tool for supporting the calculation in the clinic setting. Its speedy conclusion will further increase confidence in the tools’ utility and remove this potential barrier to adoption, thus, serving as a significant subject matter for further discussion.
The result is intended to be a tool which can homogenise the observations of ATTRv patients around the world, enabling meaningful answers to research questions to be achieved using a larger patient population, potentially in the form of a patient registry.
The phenotype of ATTRv amyloidosis affected individuals remains broad, as does the continued need to maintain a multidisciplinary approach to assessment and treatment. This study, along with the tools that have been created, is intended to serve as a genuine contribution towards quantifying ATTRv disease severity and progression. It also represents a ‘starting point’ for further development of a unified understanding of these patients.
Limitations
The methodology, being both rigorous and lengthy, presented over 600 items to the experts to assess whether they should be retained. Such an exercise may well have led to a concentration bias by the experts. However, results were assessed only as combined responses. Experts were asked to highlight any areas where relevant information may be missing at the SMART interview stage, thus, mitigating the risk of any items being lost due to lapses in concentration.
Additionally, the study recruited eight experts from six European countries. This encourages legitimate questions regarding the generalisability of the work to those territories not represented by an expert. However, the tertiary nature of the specialities managing individuals with ATTRv and implementing the systematic literature review, which reviewed publications from all territories, may provide some reassurance on this point.
This study identified a list of 140 indicators that the multidisciplinary group of European experts agreed as falling within SDMP and should be collected as part of an ATTRv CD. A subset of these indicators has been arranged into domains, weighted and scored to achieve an ATTRv DSS with 31 indicators, 25 of which are deemed to monitor disease progression. The ATTRv CD is amenable for clinical research, whereas the ATTRv DSS is appropriate for administration in a clinic setting. The DSS, involving only 10 questions on symptoms and events, six clinical examinations, and 5 investigations, can be completed rapidly in the clinic or at the bedside. The intention is that, by providing this unified framework, within which multiple populations of ATTRv patients spread across many geographies and disciplines can be effectively combined and observed, using the same indicators, a much larger, uniform real-world population will become available for research. When implemented in a patient registry, this ATTRv CD may well address the longstanding issues associated with missing data in observational studies and ultimately result in higher quality research. Once validated and adopted, the ATTRv DSS may improve the overall management of patients with ATTRv by detecting changes in disease severity and subsequent disease progression, thus informing individualised changes to patient management plans and improving patient outcomes.
| Abbreviations | ||
| ATTRv | = | hereditary transthyretin amyloid protein |
| CADT | = | compound autonomic dysfunction test |
| CD | = | core dataset |
| COMPASS-31 | = | Composite Autonomic Symptom Scale-31 |
| DSS | = | Disease Severity Scoring |
| FAP | = | familial amyloid polyneuropathy |
| NIS | = | Neuropathy Impairment Score |
| PND | = | polyneuropathy disability |
| PRISMA | = | Preferred Reporting Items for Systematic Reviews and Meta-Analyses |
| SDMP | = | Standard Diagnostic and Monitoring Practice |
| SMART | = | Specific Measurable Achievable Relevant Time-bound |
| TTR | = | transthyretin |
Supplemental Material
Download Zip (266.9 KB)Acknowledgements
The authors thank Professor Violaine Planté-Bordeneuve (Department of Neurology, Henri Mondor University Hospital, Créteil Cedex, France) and Dr Laura Obici (Amyloidosis Research and Treatment Centre, IRCCS Fondazione Policlinico San Matteo, Pavia, Italy) for their contribution to this study.
Disclosure statement
JW has received grants for lectures and advisory board meetings from Pfizer Inc., Alnylam Pharmaceuticals, and Akcea Therapeutics. PG-P reports speaking fees from Pfizer Inc, Eidos Therapeutics, Alnylam Pharmaceuticals and Akcea Therapeutics. Consulting fees from Pfizer Inc, Eidos Therapeutics, Neuroimmune, Alnylam Pharmaceuticals and Akcea Therapeutics. Research support to his institution from Pfizer Inc, Eidos Therapeutics and Alnylam Pharmaceuticals. JG is an advisory board member for Akcea Therapeutics, Alnylam Pharmaceuticals and Eidos Therapeutics. TD has received research grants and consulting fees from Akcea Therapeutics, Alnylam Pharmaceuticals, Pfizer Inc, GlaxoSmithKline Plc, Prothena and Neuroimmune. IC acknowledges financial support as primary investigator of clinical studies from FoldRx Pharmaceuticals/Pfizer Inc., Alnylam Pharmaceuticals and Ionis Pharmaceuticals and has received research support from Pfizer Inc, and serves on the THAOS scientific advisory board, financially supported from Pfizer Inc. Dr Isabel Conceição also participates in medical advisory boards promoted by Alnylam Pharmaceuticals and Pfizer Inc. TC received financial support from Alnylam Pharmaceuticals, Ionis and Pfizer Inc. to attend scientific meetings and personal fees from Alnylam Pharmaceuticals and Pfizer Inc. to provide scientific lectures. Dr Coelho has also received support from FoldRx Pharmaceuticals/Pfizer Inc., as a clinical investigator and for scientific meeting expenses. JS is employed by Akcea Therapeutics, a manufacturer of a treatment option for ATTRv amyloidosis.
Data availability statement
Data are available upon reasonable request from the authors.
Additional information
Funding
References
- Reddy S, Chang E, Tarbox M, et al. The clinical and economic burden of newly diagnosed hereditary transthyretin (ATTRv) amyloidosis: a retrospective analysis of claims data. Neurol Ther. 2020;9(2):473–482.
- Suhr OB, Lundgren E, Westermark P. One mutation, two distinct disease variants: unravelling the impact of transthyretin amyloid fibril composition. J Intern Med. 2017;281(4):337–347.
- Gonzalez-Duarte A. Autonomic involvement in hereditary transthyretin amyloidosis (hATTR amyloidosis). Clin Auton Res. 2019;29(2):245–251.
- Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8(1):1–18.
- Adams D, Koike H, Slama M, et al. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15(7):387–404.
- Luigetti M, Romano A, Di Paolantonio A, et al. Diagnosis and treatment of hereditary transthyretin amyloidosis (hATTR) polyneuropathy: current perspectives on improving patient care. Ther Clin Risk Manag. 2020;16:109.
- Koike H, Fukami Y, Nishi R, et al. Clinicopathological spectrum and recent advances in the treatment of hereditary transthyretin amyloidosis. Neurol Clin Neurosci. 2019;7(4):166–173.
- Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep. 2014;11(1):50–57.
- Moshe-Lilie O, Dimitrova D, Heitner SB, et al. TTR knockdown therapy in patients with hATTR amyloidosis who have disease progression despite liver transplant. Neurology. 2019;92(15 Suppl):P5.4–013.
- Trotter JP. Patient registries: a new gold standard for “real world” research. Ochsner J. 2002;4(4):211–214.
- von Martial S, Brix TJ, Klotz L, et al. EMR-integrated minimal core dataset for routine health care and multiple research settings: a case study for neuroinflammatory demyelinating diseases. PLoS One. 2019;14(10):e0223886.
- Daniel BS, Hertl M, Werth VP, et al. Severity score indexes for blistering diseases. Clin Dermatol. 2012;30(1):108–113.
- McKee AE, Farrell AT, Pazdur R, et al. The role of the US Food and Drug Administration review process: clinical trial endpoints in oncology. Oncologist. 2010;15(S1):13–18.
- Jandhyala R. A novel method for observing proportional group awareness and consensus of items arising from list-generating questioning. Curr Med Res. 2020;36(5):883–893.
- Jandhyala R. Delphi, non-RAND modified Delphi, RAND/UCLA appropriateness method and a novel group awareness and consensus methodology for consensus measurement: a systematic literature review. Curr Med Res. 2020;36(11):1873–1887.