
Abstract
Background
Hereditary transthyretin amyloidosis (ATTRv amyloidosis) results from pathogenic mutations in the transthyretin (TTR) gene. This analysis aimed to better understand ATTRv amyloidosis development in asymptomatic TTR gene carriers.
Methods
The Transthyretin Amyloidosis Outcomes Survey (THAOS) is an ongoing, global, longitudinal, observational survey of patients with transthyretin amyloidosis, including both inherited and wild-type disease, and asymptomatic TTR gene carriers. Asymptomatic TTR gene carriers were assessed longitudinally to identify those who developed ATTRv amyloidosis after enrolment in THAOS (data cut-off: 1 August 2021).
Results
Of 740 asymptomatic TTR gene carriers, 268 (36.2%) (Val30Met, 212/613 [34.6%]; non-Val30Met, 48/111 [43.2%]) developed ATTRv amyloidosis within a median 2.2 years after enrolment. The most common first symptoms were sensory (49.5%) and autonomic (37.3%) neuropathy in Val30Met patients, and sensory neuropathy (45.8%) and cardiac disorder (22.9%) in non-Val30Met patients. Most patients first presented with a predominantly neurologic phenotype (Val30Met, 77.8%; non-Val30Met, 70.8%).
Conclusions
More than one-third of asymptomatic TTR gene carriers in THAOS developed ATTRv amyloidosis within a median 2 years of enrolment. Val30Met versus non-Val30Met patients had a lower transition rate. Given the importance of early treatment, these findings underscore the need for identification and careful monitoring of at-risk TTR gene carriers to enable prompt treatment.
Trial registration
ClinicalTrials.gov: NCT00628745.
Introduction
Hereditary transthyretin amyloidosis (ATTRv amyloidosis) is a progressive, life-threatening, heterogeneous disease caused by pathogenic mutations in the transthyretin (TTR) gene, resulting in the irreversible deposition of amyloid fibrils in peripheral nerves and vital organs, leading mainly to polyneuropathy and/or cardiomyopathy [Citation1]. More than 140 mutations have been identified in the TTR gene, with the Val30Met (p.Val50Met) mutation being the most common [Citation1,Citation2]. Over 50,000 persons are estimated to be living with ATTRv amyloidosis worldwide, although the disease is likely underdiagnosed [Citation1,Citation3]. The clinical presentation of ATTRv amyloidosis is variable and its symptoms are not specific. Accurate diagnosis can be delayed for years, and ATTRv amyloidosis is frequently confused with other more common disorders, particularly among patients without a family history of previously diagnosed relatives [Citation4–6]. Regardless of the specific TTR mutation or initial symptomatology, one cardinal characteristic of ATTRv amyloidosis is the involvement of multiple systems, with individual symptoms that are not clearly related [Citation5–7]. Initial symptoms can be broadly grouped into peripheral sensory-motor or autonomic neuropathies; carpal tunnel syndrome; and gastrointestinal, cardiovascular, renal, central nervous system and ocular manifestations, and have been well described globally [Citation1,Citation4–6,Citation8–11].
The progression of ATTRv amyloidosis is usually relentless, with death occurring an average of 10–15 years from diagnosis in untreated patients with transthyretin amyloid polyneuropathy, and 2–5 years in untreated patients with transthyretin amyloid cardiomyopathy, depending on multiple factors [Citation1,Citation6,Citation9,Citation12]. The inheritance pattern of ATTRv amyloidosis is autosomal dominant, and disease penetrance is variable [Citation13–17]. While factors contributing to the transition from asymptomatic to symptomatic ATTRv amyloidosis are not fully understood, there is consensus that, as tissue damage associated with the disease is progressive and largely irreversible, at-risk TTR gene carriers should be identified and monitored at least annually to facilitate the earliest possible diagnosis and treatment [Citation5,Citation8,Citation18–22].
The Transthyretin Amyloidosis Outcomes Survey (THAOS) is an ongoing, global, longitudinal, observational survey of patients with transthyretin amyloidosis (ATTR amyloidosis), including both inherited and wild-type disease, and asymptomatic gene carriers with TTR mutations [Citation23]. A major strength of THAOS has been the inclusion of at-risk TTR gene carriers, providing the opportunity to monitor and describe the natural history and development of ATTRv amyloidosis. This THAOS analysis examined a large, real-world, asymptomatic TTR gene carrier population longitudinally to determine their risk for transition to ATTRv amyloidosis. Demographic and clinical profiles of those at-risk TTR gene carriers who did and did not manifest ATTRv amyloidosis were also assessed to better understand possible prognosticators of disease progression.
Materials and methods
The design of THAOS (ClinicalTrials.gov: NCT00628745) has been published previously [Citation23]. All THAOS sites received ethical or institutional review board approval prior to patient enrolment and followed the International Council for Harmonisation Good Pharmacoepidemiology Practice guidelines, and the principles of the Declaration of Helsinki. Each patient provided written informed consent.
The analysis population consisted of asymptomatic TTR gene carriers with at least one follow-up visit (data cut-off date: 1 August 2021). Asymptomatic TTR gene carriers were defined as those with no definitely ATTR amyloidosis-related symptoms at enrolment. Symptoms were categorised by level of relatedness to ATTR amyloidosis based on the investigator’s designation on the clinical report form. Investigators could select one of three options (‘yes,’ ‘no’ or ‘possibly’) for each symptom to indicate its relatedness. In this analysis, symptoms marked ‘yes’ were considered definitely ATTR amyloidosis-related, those marked ‘no’ were considered not ATTR amyloidosis-related, and those marked ‘possibly’ were considered possibly ATTR amyloidosis-related.
Patients with wild-type ATTR amyloidosis or those participating in a clinical trial were excluded from the analysis.
Demographics and clinical characteristics, including genotype, were collected at enrolment. Change in symptomatic status and first presenting ATTR amyloidosis symptoms (see Supplemental Methods) were collected over time post enrolment. Patients who developed ATTRv amyloidosis were also examined by genotype subgroup (Val30Met or non-Val30Met), and first presenting symptoms were examined by phenotype (see Supplemental Methods).
A subset of asymptomatic TTR gene carriers designated as possibly symptomatic at enrolment was also assessed. Possibly symptomatic patients were defined as those who had no definitely ATTR amyloidosis-related symptoms and at least one possibly ATTR amyloidosis-related symptom at enrolment.
Statistical analysis
The primary analysis described the population of asymptomatic TTR gene carriers at enrolment and assessed this population longitudinally to determine who remained asymptomatic at all post-enrolment visits and who developed ATTRv amyloidosis (i.e. had at least one definitely ATTR amyloidosis-related symptom) at any post-enrolment visit.
A secondary analysis described the population of possibly symptomatic patients at enrolment and determined the following: (1) those who remained possibly symptomatic (i.e. continued to have no symptoms definitely ATTR amyloidosis-related, but at least one symptom possibly ATTR amyloidosis-related post enrolment); (2) those who became asymptomatic (i.e. reported no symptoms definitely or possibly ATTR amyloidosis-related post enrolment); and (3) those who developed ATTRv amyloidosis (i.e. reported at least one definitely ATTR amyloidosis-related symptom post enrolment).
Demographic and clinical characteristics at enrolment of each population and genotype subgroup, and first presenting symptoms and phenotype in patients who developed ATTRv amyloidosis, were summarised descriptively.
Results
Demographic characteristics of asymptomatic TTR gene carriers at enrolment
This analysis included 740 asymptomatic TTR gene carriers from 15 countries, the majority from Portugal (). Of these 740 asymptomatic TTR gene carriers, 62.7% were female, and the predominant genotype was Val30Met (84.7%) ( and ). Additional genotypes are listed in . Mean follow-up duration was 6.6 years.
Figure 1. Geographic distribution of asymptomatic TTR gene carriers at enrolment. Countries contributing five or fewer patients not shown: Bulgaria (n = 2), Japan (n = 3), Mexico (n = 5), the Netherlands (n = 2) and South Korea (n = 2).
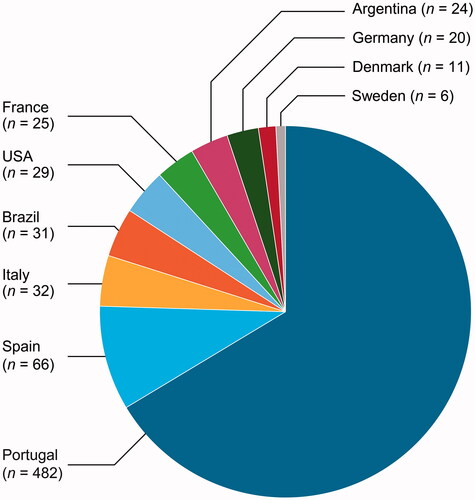
Table 1. Genotype distribution in asymptomatic TTR gene carriers.
Table 2. Demographic characteristics of asymptomatic TTR gene carriers at enrolment in THAOS.
Demographic and clinical characteristics of TTR gene carriers who remained asymptomatic versus those who developed ATTRv amyloidosis
More than one-third of asymptomatic TTR gene carriers (n = 268 [36.2%]) developed ATTRv amyloidosis post enrolment, with a median duration from enrolment to symptom onset of 2.2 years (). Patients were diagnosed based on tissue biopsy (n = 161) or family history (i.e. diagnosis of ATTR amyloidosis in a family member by genotyping, biopsy, or both; n = 82); 25 patients had a missing or unknown diagnostic method.
Of the 268 patients who developed ATTRv amyloidosis, 212 carried the Val30Met mutation and 48 carried a non-Val30Met mutation (). Val30Met patients had a lower rate of transition from asymptomatic to symptomatic (34.6% vs. 43.2%) and lower mean age at symptom onset (40.5 vs. 48.0 years) compared with non-Val30Met patients ().
Overall, those who remained asymptomatic were more likely to be female and to have had a shorter mean follow-up duration than those who developed ATTRv amyloidosis (). Clinical characteristics were broadly similar between those who remained asymptomatic and those who developed ATTRv amyloidosis ().
Table 3. Clinical characteristics of asymptomatic TTR gene carriers at enrolment in THAOS.
First presenting symptoms in asymptomatic TTR gene carriers who developed ATTRv amyloidosis
Val30Met patients most often presented with symptoms involving the neurologic and gastrointestinal systems first (). The most common first presenting individual symptoms were neuropathic pain/paraesthesia, tingling and unintentional weight loss (), and the most common first presenting symptoms by category were sensory neuropathy and autonomic neuropathy (). Predominantly neurologic was the most common first presenting phenotype ().
Figure 2. First presenting symptom categories by genotype in asymptomatic patients who developed ATTRv amyloidosis post enrolment. Includes patients who were asymptomatic at enrolment and reported at least one definitely ATTR amyloidosis-related symptom post enrolment. First symptoms recorded as definitely ATTR amyloidosis-related by the investigator are shown by category.
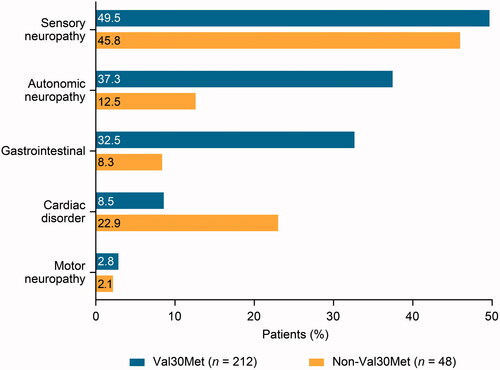
Figure 3. First presenting phenotype overall and by genotype in patients who developed ATTRv amyloidosis post enrolment. Includes patients who were asymptomatic at enrolment and reported at least one definitely ATTR amyloidosis-related symptom post enrolment.
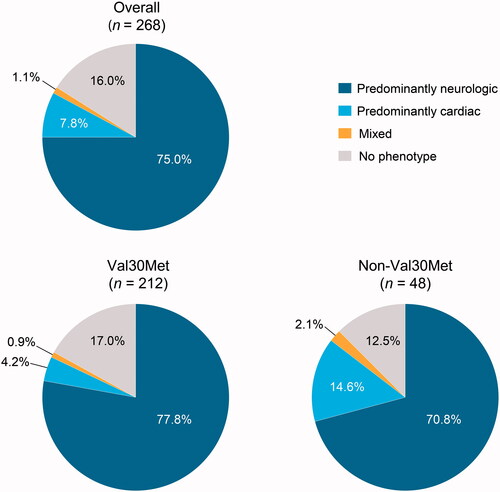
Table 4. First presenting symptomsa by body system in asymptomatic TTR gene carriers who developed ATTRv amyloidosis post enrolment.b
Table 5. First presenting symptomsa in asymptomatic TTR gene carriers who developed ATTRv amyloidosis post enrolment.b
In non-Val30Met patients, first presenting symptoms by body system were most commonly neurologic and cardiovascular (). The most common first presenting individual symptoms were neuropathic pain/paraesthesia, tingling, and carpal tunnel syndrome (), and the most common first presenting symptom categories were sensory neuropathy and cardiac disorder (). Predominantly neurologic was the most common first presenting phenotype ().
Subset analysis of possibly symptomatic patients at enrolment
There were 282 possibly symptomatic patients at enrolment of whom 64.2% were female (). Post enrolment, 44.3% remained possibly symptomatic, 22.0% became asymptomatic, and 33.7% developed ATTRv amyloidosis.
Table 6. Demographic characteristics of possibly symptomatic patients at enrolment in THAOS.
Demographic and clinical characteristics for those who remained possibly symptomatic, those who became asymptomatic, and those who developed ATTRv amyloidosis were broadly similar ( and ). Mean duration of follow-up was longer in those who developed ATTRv amyloidosis than those who remained possibly symptomatic or became asymptomatic ().
Table 7. Clinical characteristics of possibly symptomatic patients at enrolment in THAOS.
Discussion
In this large, longitudinal analysis from THAOS, over 35% of asymptomatic TTR gene carriers developed ATTRv amyloidosis within a median of 2.2 years. Val30Met patients had a lower rate of transition from asymptomatic to symptomatic (34.6% vs. 43.2%) and lower mean age at symptom onset (40.5 vs. 48.0 years) than non-Val30Met patients. Furthermore, of those patients who were classified as possibly symptomatic at enrolment, 34% were subsequently confirmed to have symptoms consistent with ATTRv amyloidosis, while 22% reverted to asymptomatic status.
These findings highlight the need for frequent and ongoing monitoring of asymptomatic TTR gene carriers to ensure proper clinical management of these patients. ATTR amyloidosis is an often rapidly progressive disease and can ultimately lead to organ failure and death [Citation1]. Historically, treatment of ATTR amyloidosis has been limited to symptom management and liver and/or heart transplantation, which carry substantial risk to the patient [Citation24,Citation25]. Treatments that suppress expression or stabilise TTR protein to slow or halt disease progression have recently become available and are more effective in the earlier stages of the disease [Citation25,Citation26]. With the current availability of these disease-modifying agents, early identification of patients with ATTR amyloidosis has become increasingly important.
These findings also support the adherence to consensus guidelines on the management and follow-up of at-risk asymptomatic TTR gene carriers, including at least annual monitoring [Citation18,Citation21,Citation22]. The first presenting symptoms in this analysis included autonomic dysfunction, cardiovascular disease, gastrointestinal problems, carpal tunnel syndrome, renal impairment and ocular involvement, and are consistent with the earliest ‘red flag’ symptoms of this spectrum disease, previously described [Citation5,Citation6]. Approximately half of Val30Met patients first presented with sensory neuropathy, and a substantial proportion first presented with autonomic neuropathy and gastrointestinal symptoms. Sensory neuropathy was also among the first presenting symptoms in approximately half of non-Val30Met patients, while cardiac disorder was seen in one-quarter of patients. First presenting motor neuropathy was rare in both groups.
In this analysis, a larger proportion of asymptomatic TTR gene carriers at enrolment were female. Furthermore, female patients were less likely to develop ATTRv amyloidosis post enrolment compared with males. This finding may reflect a later average age of onset in female patients, which has been previously reported [Citation15,Citation27,Citation28]. As such, it is possible that more female patients would transition from asymptomatic to symptomatic ATTRv amyloidosis with even longer follow-up.
Study strengths and limitations
Strengths of this analysis include the large sample size, geographic heterogeneity, and the duration of follow-up. Over 700 asymptomatic TTR gene carriers from 15 different countries worldwide were included, and this analysis was represented by an average overall follow-up time of 6.6 years. A limitation of this analysis was the longer mean duration of follow-up in asymptomatic TTR gene carriers who developed ATTRv amyloidosis (7.6 years) compared with those who remained asymptomatic (6.1 years). This may represent a form of length bias [Citation29], with the longer follow-up allowing more time for patients to develop ATTRv amyloidosis. Comparisons of baseline characteristics between the two groups should, therefore, be interpreted with caution. Additionally, a majority of patients in this analysis were from a single country, Portugal, which might have influenced our results. Lastly, neuropathy was diagnosed by symptoms and not by examination, the evaluation of neuropathic pain and paraesthesia together did not allow for differentiation between small and large fibre involvement, and other risk factors for neuropathy aside from ATTR amyloidosis were not systematically assessed.
Conclusions
This natural history analysis revealed that over 35% of at-risk asymptomatic TTR gene carriers developed ATTRv amyloidosis within 2 years of enrolment into THAOS. Those with non-Val30Met mutations developed ATTRv amyloidosis at a higher rate than those with the Val30Met mutation. These findings underscore the need to both identify and monitor at-risk TTR gene carriers for the earliest signs and symptoms of ATTRv amyloidosis to enable intervention with approved ATTR amyloidosis disease-modifying treatments as soon as possible [Citation18,Citation21,Citation30].
| Abbreviations | ||
| ATTR amyloidosis | = | transthyretin amyloidosis |
| ATTRv amyloidosis | = | hereditary transthyretin amyloidosis |
| BMI | = | body mass index |
| mBMI | = | modified body mass index |
| Norfolk QoL | = | Norfolk Quality of Life Questionnaire |
| Q | = | quartile |
| SD | = | standard deviation |
| THAOS | = | Transthyretin Amyloidosis Outcomes Survey |
| TTR/TTR | = | transthyretin (gene/protein) |
| VAS | = | visual analog scale |
Acknowledgements
We thank Dr Jan Kiszko and Dr Moh-Lim Ong for their contributions to earlier versions of this work. We thank all THAOS patients and investigators for their important contributions to this study.
Disclosure statement
T. Coelho reports having served as a medical advisor for Pfizer, honoraria for presentations, and funding for scientific meeting expenses (travel, accommodation, and registration) from Pfizer. I. Conceição reports research funding, consulting fees, and travel support for advisory boards and meetings from Pfizer. M. Waddington-Cruz reports research funding, consulting fees, and travel support for advisory boards and meetings from FoldRx Pharmaceuticals and Pfizer. D. Keohane, M.B. Sultan, D. Chapman, and L. Amass are full-time employees of Pfizer and hold stock and/or stock options with Pfizer.
Data availability statement
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual anonymized participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Additional information
Funding
References
- Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31.
- Rowczenio D, Wechalekar A. Mutations in hereditary amyloidosis [Internet]. 2015. [cited 2021 Oct 11]. Available from: http://amyloidosismutations.com/mut-attr.php.
- Hawkins PN, Ando Y, Dispenzeri A, et al. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625–638.
- Ruberg FL, Grogan M, Hanna M, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73(22):2872–2891.
- Conceição I, Gonzalez-Duarte A, Obici L, et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5–9.
- Witteles RM, Bokhari S, Damy T, et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail. 2019;7(8):709–716.
- Gertz MA. Hereditary ATTR amyloidosis: burden of illness and diagnostic challenges. Am J Manag Care. 2017;23(7 Suppl):S107–S112.
- Adams D, Slama M. Hereditary transthyretin amyloidosis: current treatment. Curr Opin Neurol. 2020;33(5):553–561.
- Koike H, Misu K, Ikeda S, et al. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset form. Arch Neurol. 2002;59(11):1771–1776.
- Planté-Bordeneuve V, Lalu T, Misrahi M, et al. Genotypic-phenotypic variations in a series of 65 patients with familial amyloid polyneuropathy. Neurology. 1998;51(3):708–714.
- González-Duarte A, Lem-Carrillo M, Cárdenas-Soto K. Description of transthyretin S50A, S52P and G47A mutations in familial amyloidosis polyneuropathy. Amyloid. 2013;20(4):221–225.
- Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75(3):408–427.
- Hellman U, Alarcon F, Lundgren H-E, et al. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid. 2008;15(3):181–186.
- Planté-Bordeneuve V, Carayol J, Ferreira A, et al. Genetic study of transthyretin amyloid neuropathies: carrier risks among French and Portuguese families. J Med Genet. 2003;40(11):e120.
- Saporta MA, Zaros C, Cruz MW, et al. Penetrance estimation of TTR familial amyloid polyneuropathy (type I) in Brazilian families. Eur J Neurol. 2009;16(3):337–341.
- Quarta CC, Buxbaum JN, Shah AM, et al. The amyloidogenic V122I transthyretin variant in elderly Black Americans. N Engl J Med. 2015;372(1):21–29.
- Rowczenio D, Quarta CC, Fontana M, et al. Analysis of the TTR gene in the investigation of amyloidosis: a 25-year single UK center experience. Hum Mutat. 2019;40(1):90–96.
- Conceição I, Damy T, Romero M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid. 2019;26(1):3–9.
- Planté-Bordeneuve V. Update in the diagnosis and management of transthyretin familial amyloid polyneuropathy. J Neurol. 2014;261(6):1227–1233.
- Ueda M, Ando Y. Recent advances in transthyretin amyloidosis therapy. Transl Neurodegener. 2014;3:19.
- Ueda M, Sekijima Y, Koike H, et al. Monitoring of asymptomatic family members at risk of hereditary transthyretin amyloidosis for early intervention with disease-modifying therapies. J Neurol Sci. 2020;414:116813.
- Schmidt HH, Barroso F, González-Duarte A, et al. Management of asymptomatic gene carriers of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2016;54(3):353–360.
- Planté-Bordeneuve V, Suhr OB, Maurer MS, et al. The Transthyretin Amyloidosis Outcomes Survey (THAOS) registry: design and methodology. Curr Med Res Opin. 2013;29(1):77–84.
- Burton A, Castano A, Bruno M, et al. Drug discovery and development in rare diseases: taking a closer look at the tafamidis story. Drug Des Devel Ther. 2021;15:1225–1243.
- Muller ML, Butler J, Heidecker B. Emerging therapies in transthyretin amyloidosis - a new wave of hope after years of stagnancy? Eur J Heart Fail. 2020;22(1):39–53.
- Waddington Cruz M, Amass L, Keohane D, et al. Early intervention with tafamidis provides long-term (5.5-year) delay of neurologic progression in transthyretin hereditary amyloid polyneuropathy. Amyloid. 2016;23(3):178–183.
- Parman Y, Adams D, Obici L, et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol. 2016;29(Suppl 1):S3–S13.
- Caponetti AG, Rapezzi C, Gagliardi C, et al. Sex-related risk of cardiac involvement in hereditary transthyretin amyloidosis: insights from THAOS. JACC Heart Fail. 2021;9(10):736–746.
- Morrison AS. The effects of early treatment, lead time and length bias on the mortality experienced by cases detected by screening. Int J Epidemiol. 1982;11(3):261–267.
- Obici L, Kuks JB, Buades J, et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016;29(Suppl 1):S27–S35.