
Abstract
Background
The p.Glu109Lys variant (Glu89Lys) is a rare cause of hereditary transthyretin amyloidosis (ATTRv) for which clinical spectrum remains unresolved. We sought to describe the clinical characteristics and outcomes of ATTR Glu89Lys amyloidosis and assess a potential founder effect in Spain.
Methods
Patients with the p.Glu109Lys ATTRv variant from 14 families were recruited at 7 centres. Demographics, complementary tests and clinical course were analysed. Haplotype analysis was performed in 7 unrelated individuals.
Results
Thirty-eight individuals (13 probands, mean age 40.4 ± 13.1 years) were studied. After median follow-up of 5.1 years (IQR 1.7–9.6), 7 patients died and 7 required heart transplantation (median age at transplantation 50.5 years). Onset of cardiac and neurological manifestations occurred at a mean age of 48.4 and 46.8 years, respectively. Median survival from birth was 61.6 years and no individual survived beyond 65 years. Patients treated with disease-modifying therapies exhibited better prognosis (p < 0.001). Haplotype analysis revealed a common origin from an ancestor who lived ∼500 years ago in southeast Spain.
Conclusions
Glu89Lys ATTRv is a TTR variant with a founder effect in Spain. It is associated with near complete penetrance, early onset and mixed cardiac and neurologic phenotype. Patients have poor prognosis, particularly if not treated with disease-modifying therapies.
Introduction
Transthyretin amyloidosis (ATTR) is a systemic disease characterised by abnormal deposition of misfolded transthyretin (TTR) in the extracellular matrix of different tissues, with a marked affinity for peripheral nerves and myocardium. Patients with ATTR can be categorised as wild-type ATTR (ATTRwt), who do not carry pathogenic TTR variants, or hereditary ATTR (ATTRv), who carry monoallelic pathogenic TTR variants [Citation1].
More than 100 pathogenic TTR variants have been described associated with the disease with clear correlations between specific gene variants and phenotypic features [Citation2,Citation3]. Certain variants such as the Val30Met mutation predominantly produce neurological symptoms, whereas other variants such as Val122Ile provoke late-onset cardiomyopathy with mild or no neurological involvement [Citation4,Citation5]. Most variants are, however, associated with mixed clinical manifestations and affect both the heart and the peripheral nerves.
The prevalence of ATTRv amyloidosis is highly variable and depends on the country and race. Some TTR variants are highly prevalent in specific regions due to founder effects, such as the p.Val50Met variant in Portugal, Sweden, Japan and the island of Majorca in Spain [Citation6–8]; p.Glu109Gln in South-West Bulgaria [Citation9]; or p.Thr80Ala in Donegal County, Ireland [Citation10]. This phenomenon has enabled a higher level of awareness of ATTRv amyloidosis in patients from specific areas.
The first case of ATTRv amyloidosis due to the p.Glu109Lys variant was reported in 2000 by Nakamura et al. and, since then, only 11 additional cases have been described worldwide as isolated case reports [Citation11–17]. Two recent studies have suggested that p.Glu109Lys could be a particularly frequent TTR variant in Spain [Citation2,Citation18], but there are no cohorts describing the clinical course and prognosis of patients carrying this particular variant.
Here, we sought to describe the clinical characteristics and natural history of patients with ATTR Glu89Lys amyloidosis and to evaluate its potential founder effect in Spain.
Methods
Population
The major Spanish centres following patients with ATTRv amyloidosis were contacted to participate in this longitudinal cohort study. Inclusion criteria were patients diagnosed with ATTRv amyloidosis who were carriers of the p.Glu109Lys variant (previously reported as p.Glu89Lys) in TTR (Clin Var ID: 1073324). Relatives with this variant were identified through cascade screening and were also included irrespective of their phenotype. Genetic analysis was performed at participating centres or at accredited genetic laboratories using next-generation sequencing (NGS) or Sanger sequencing.
The study complies with the ethical principles of the Helsinki Declaration and was approved by the Hospital Universitario Puerta de Hierro ethics committee. The authors from each participating centre guarantee the integrity of data.
Data acquisition
Data was retrieved from medical records and anonymized by each centre. The data set included demographics and family history, with special attention to birthplace of subjects as well as to geographical origin of their ancestors, to look for possible common regions of origin between families. Signs and symptoms related to ATTRv amyloidosis were collected at first and last evaluation, focussing on cardiac, neurologic and ophthalmologic features. Phenotypic expression onset of cardiac involvement was defined as first report of left ventricular hypertrophy (maximal wall thickness ≥12 mm) or grade 2 or 3 cardiac uptake on bone scintigraphy; and for neurologic involvement as the appearance of signs or symptoms related to ATTRv amyloidosis and an abnormal neurological test (nerve conduction study or sympathetic skin response) accompanied by likely related signs; and for eye involvement as vitreous opacities or new onset glaucoma, as described [Citation19]. Data from complementary tests such as ECG, laboratory tests, bone scintigraphy, echocardiography and nerve conduction studies, were also included if available. Initiation of specific ATTRv amyloidosis disease-modifying therapies (i.e. tafamidis, patisiran) was also retrieved. Clinical events collected during follow-up included stroke/transient ischaemic accident, pacemaker or implantable cardioverter defibrillator implantation, atrial fibrillation (AF), heart failure (HF) admission, heart transplantation (HT), liver transplantation (LT), or death.
Study endpoints
Penetrance from birth was estimated based on phenotypic expression of cardiac, neurologic and ophthalmologic involvement, and was compared based on sex and phenotype. Survival free from HT was considered as a composite endpoint to describe prognosis, and was analysed according to sex and by treatment with disease-modifying therapies.
Haplotype analysis
Haplotype analysis was performed in one member of each of the 7 families followed at Hospital Universitario Puerta de Hierro Majadahonda, Madrid. DNA extraction from whole blood was carried out by a standard salting-out procedure. The following panel of 11 microsatellite markers (short tandem repeats, STRs) located next to the TTR locus was amplified by polymerase chain reaction (PCR) for haplotype reconstruction: D18S66, D18S49, L1, D18S36, L4, L8, L9, D18S47, D18S1133, D18S57, D18S451. Genetic markers, primer sequences and PCR conditions were selected based on previous haplotype studies for other TTR variants [Citation7,Citation9]. Fragment analysis of each marker was performed by capillary electrophoresis on an ABI Prism 3100 Genetic Analyser (Applied Biosystems, Foster City, CA). The microsatellite data were then analysed by GeneMapper software v.4.0 (Applied Biosystems).
Haplotypes of the p.Glu109Lys TTR carriers were reconstructed on the 11 microsatellites considering a bi-allelic marker at the variant site. For each patient, the haplotype with the highest posterior probability was considered for age estimation. The age of the most recent common ancestor was estimated using the Gamma method, which is based on the high-density single nucleotide polymorphism data surrounding the mutation, as described [Citation20].
Statistical analysis
Continuous variables are expressed as mean ± standard deviation (SD), or as median (interquartile range [IQR]), as appropriate; categorical variables are expressed as counts (percentage of total). Student’s t-test was used to compare continuous variables, with normal distribution assessed by the Shapiro-Wilk test, whereas the non-parametric Wilcoxon rank-sum (Mann-Whitney) test was applied for those not meeting normal distribution. Categorical variables were compared between groups with the parametric Chi-square test or non-parametric Fisher’s exact test. Matched tests were applied to analyse changes in clinical status of patients receiving disease-modifying therapies. Survival analysis with Kaplan-Meier curves was used to describe penetrance and composite survival free from death and HT. Cox proportional hazard regression was performed to analyse the impact of sex and disease-modifying therapies on prognosis. STATA software version 15.1 (StataCorp, College Station, TX) was used for statistical analysis. A two-tailed p-value <0.05 was considered statistically significant.
Results
Study participants
Information was collected from 38 subjects with the p.Glu109Lys TTR variant from 14 non-related families followed at 7 Spanish centres between 2003 and 2021, with the exception of one single patient evaluated in 1994. All patients were heterozygous. Patients included by each participating centre are listed in Table S1 in the Supplemental Material. The main characteristics of patients at initial evaluation of probands (n = 13) and relatives (n = 25) are listed in . Overall, mean age at initial evaluation was 40.4 ± 13.1 years and 39.5% were female. Among probands, 9 patients (69%) were initially evaluated because of cardiac symptoms, and 4 (31%) had neurologic symptoms that prompted evaluation. The flowchart of patients according to phenotypic expression at initial evaluation and at last follow-up is described in . In total, 8 (61.5%) probands had a mixed cardiac and neurologic phenotype at initial evaluation, 4 (31%) had isolated cardiomyopathy, and 1 (8%) had mixed cardiac and neurologic symptoms with concomitant ophthalmologic involvement. Of the 25 relatives evaluated, 17 (68%) had no organ involvement at initial evaluation, and 8 (32%) already had signs and symptoms of the disease (4 isolated neuropathy, 3 isolated cardiac disease and 1 mixed cardiac and neurologic phenotype). Relatives with phenotypic expression at first evaluation were significantly older than unaffected peers (45.7 ± 5.8 vs 30.3 ± 12.2; p = 0.003). Carriers without ATTRv amyloidosis signs or symptoms at initial evaluation (n = 17) were followed for a median of 6.5 years (IQR 1.71–8.62). During this period, 6 subjects (35%) developed phenotypic expression (4 mixed cardiac and neurologic involvement and 2 isolated neuropathy). Mean age at phenotypic expression among healthy carriers at initial evaluation who developed clinical manifestations was 41.2 ± 9.8 years compared with 48.5 ± 5.7 years in probands (p = 0.054). Overall, 13 patients (48%) were diagnosed based on histological confirmation of TTR deposit (9 cases [33%] through endomyocardial biopsy and 4 cases [15%] through extracardiac biopsy. Non-invasive diagnosis was established in 14 patients (52%) based on cardiac uptake on bone scintigraphy in the absence of monoclonal gammopathy (N = 9) or based on appearance of neurological signs or symptoms related to ATTRv amyloidosis and abnormal neurological test (N = 5).
Figure 1. Patient flowchart according to phenotypic expression. CAR: Cardiac; NEUR: Neurologic; OPHT: Ophthalmologic.
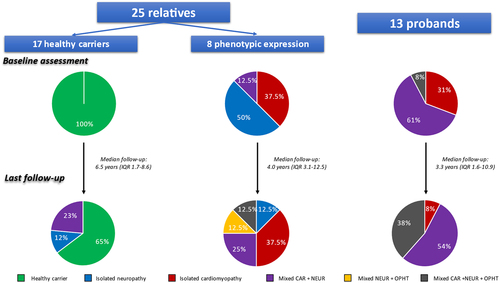
Table 1. Baseline clinical characteristics of p.Glu109Lys variant carriers (N = 38).
Patients with cardiomyopathy at initial evaluation (13 probands and 4 relatives) had a median NT-ProBNP level of 911 pg/mL (IQR 218–1400). All patients with baseline technetium-labeled bone scintigraphy (N = 11) had intense cardiac uptake (Perugini grade 3). One patient (6%) had prior history of pacemaker implantation because of third-degree atrioventricular block, and 3 patients (18%) had AF. Mean myocardial wall thickness was 16.4 ± 4.2 mm and mean left ventricular ejection fraction (LVEF) was 53.4 ± 16.8%.
Neurological involvement was present in 14 individuals (37%; 9 probands and 5 relatives). Superficial (vibratory and proprioceptive) sensory loss was the most frequent manifestation, affecting 10 (77%) of the 14 patients with neuropathy, followed by orthostatic hypotension (6, 43%), gastrointestinal disturbances (constipation/diarrhea) (4, 29%), motor dysfunction (3, 23%) and deep (thermal and pain) sensory loss (2, 15%). Only 1 patient (proband) had ophthalmologic involvement at initial evaluation, whereas carpal tunnel syndrome was present in 9 patients (24%).
Penetrance
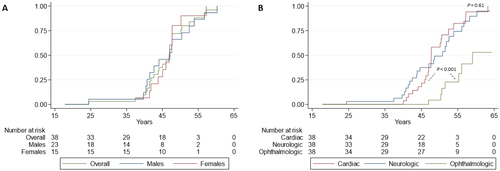
Overall penetrance was 10, 72 and 96% at 40, 50 and 60 years, respectively. Overall penetrance from birth and according to sex is shown in , with no differences between males and females (hazard ratio [HR], 1.13; 95% confidence interval [CI], 0.51–2.49; p = 0.77). Penetrance according to phenotype expression is shown in . Mean age of cardiac and neurologic involvement was 48.4 ± 5.6 and 46.8 ± 8.2 years, respectively (p = 0.61). Seven patients (18%) developed ophthalmologic involvement during the follow-up with a mean age at onset of 52.8 ± 4.1 years, suggesting incomplete penetrance with a significant delayed presentation as compared with cardiac and nerve involvement (p < 0.001).
Figure 2. Penetrance of p.Glu109Lys ATTR variant according to sex (A) and phenotype (B).
Clinical events and follow-up
Overall median follow-up time from first evaluation was 5.1 years (IQR 1.7–9.6). No patient was lost to follow-up, although medical records of 1 patient (deceased in 1997) were incomplete regarding clinical events. Clinical events are summarised in . Three patients (8%) were admitted because of HF during follow-up and, additionally, 3 patients (14%) progressed from New York Heart Association (NYHA) I to II/III. Four patients (12%) developed new-onset AF, 2 (5%) had a stroke, and 3 (9%) had a new pacemaker implanted because of symptomatic slow AF, alternating bundle-branch block and syncope with prolonged H-V interval. PND progress during follow-up was observed in 10 patients (28%). At last follow-up, 6 patients (17%) had a PND II or greater.
Table 2. Clinical events.
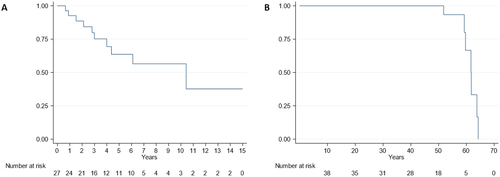
Seven patients (18%) received a HT during follow-up at a median age of 50.5 years (IQR 45.8–53.2) and 7 (18%) died during follow-up due to: sudden death (3), advanced HF (1), neurological progression (1), cancer (1) and pneumonia (1). shows survival free from HT from diagnosis and shows overall mortality from birth. Median survival free from HT was 10.4 years (standard error [SE] 0.82) from diagnosis. Overall median life expectancy from birth was 61.6 years (SE 0.96). Of note, no patient survived beyond 65 years.
Figure 3. Survival in ATTR Glu89Lys amyloidosis. (A) Survival free of heart transplantation from disease onset in affected individuals (n = 27). (B) Survival free of death from birth in the overall cohort (n = 38).
Treatment
Phenotypic expression, disease-modifying therapies received and outcomes in the 27 patients with ATTRv amyloidosis manifestations at baseline or during follow-up are summarised in Figure S1 in the Supplemental Material. Thirteen patients (48%) with neuropathy received tafamidis, of which 5 patients (38.5%) switched to patisiran due to neurological progression after a median of 13.6 months (IQR 11.0–13.6) under treatment. One patient withdrew from tafamidis after 22.3 months of treatment due to personal reasons without adverse events or neurological progression. Seven patients (26%) underwent LT after a median of 4.9 years (IQR 0.9–8.0) from diagnosis. All patients who underwent LT also had a HT, in 2 cases simultaneously with LT and in 5 cases before LT with a median of 23.2 months (IQR 20.3–25.3). Two patients (7%) received off-label diflunisal and 5 patients (18.5%) did not receive any disease-modifying treatment. Two patients with isolated cardiac involvement and one with isolated mild autonomic manifestations (sweat dysfunction) were not receiving any disease-modifying therapy at last follow-up.
Baseline characteristics of patients who maintained treatment with tafamidis and those who switched to patisiran due to neurological progression are shown in Table S2 in Supplemental Material. Although no statistically significant differences were found between groups, those who were switched to patisiran tended to be older when tafamidis was initiated and had lower LVEF, these patients also had a significant progression of NTProBNP levels at 1 year since start of tafamidis (611 vs 1233.4 pg/mL; p = 0.04) (Figure S2 supplementary material).
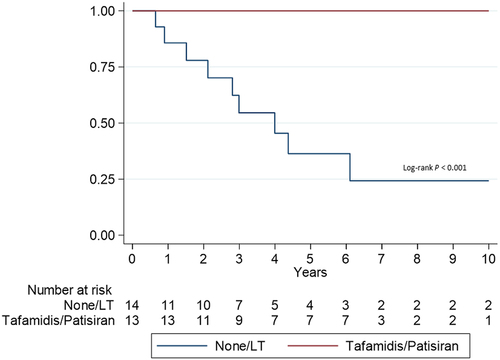
We compared survival free from HT from ATTRv amyloidosis diagnosis in patients treated with tafamidis/patisiran with those not treated with these drugs (). Patients treated with tafamidis/patisiran had better prognosis (p < 0.001) despite similar clinical characteristics between treated and untreated patients (except for LVEF, which was lower in patients not receiving tafamidis/patisiran) (Table S3 in Supplemental Material).
Figure 4. Survival free of heart transplantation according to treatment. LT: Liver transplantation.
Geographical distribution
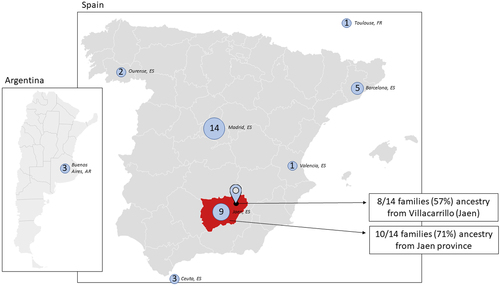
The place of birth of all patients included is shown in , with Madrid as the region of origin of most individuals (n = 14, 37%) followed by Jaen (n = 9, 24%) and Barcelona (n = 5, 13%). When examining the geographical origin of the 14 families evaluated, 12 (86%) were from Andalusia, in southern Spain, with a clear cluster located in the province of Jaen (10/14 families; 71%). Notably, when we evaluated more precisely the town of origin of the families we found that for 8 families (57%) the exact origin was Villacarrillo, a town of 10,673 inhabitants in the north of the province of Jaen.
Figure 5. Geographical origin of patients. Numbers displayed represent the province of birth of patients included in the cohort. Jaen province, where most families shared common ancestry, is highlighted in red and the town of Villacarrillo highlighted. AR: Argentina; ES: Spain; FR: France.
Haplotype analysis
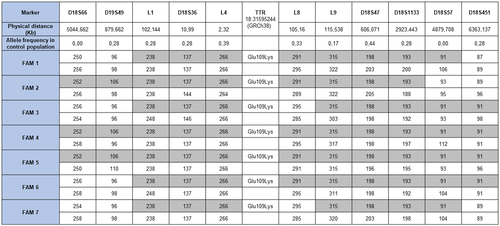
Due to the shared origin in a significant number of families, a founder effect of this variant that likely arose in a common ancestor was suspected. We performed a haplotype analysis to test this hypothesis. Using the copy number of 11 STRs covering a total of 11.4 megabases (Mb) surrounding the pGlu109Lys TTR variant, we generated the haplotype from 7 unrelated carriers and found that they shared a DNA segment of 5 Mb (). Assuming a correlated genealogy, the most recent common ancestor of the 7 families was estimated to be 19.7 generations in the past (95% confidence interval [CI], 6.2–69.7). Considering the length of a generation to be 25 years, we estimated that the 7 families originated from a common ancestor who lived approximately 500 years ago.
Figure 6. Haplotype analysis. kb: kilobases; FAM: Family.
Discussion
The present study represents the largest cohort of patients with ATTR Glu89Lys amyloidosis described to date. We found that the disease is characterised by complete penetrance by the age of 60, early onset and mixed cardiac and neurologic phenotype in the majority of cases, with some patients also experiencing ophthalmologic complications. Moreover, we found that patients with p.Glu109Lys variant have a grim prognosis with poor survival beyond the seventh decade of life. Lastly, we show that the ATTR p.Glu109Lys variant has a founder effect in southern Spain and arose from a common ancestor who lived in the town of Villacarrillo (Jaen, Spain) around 500 years ago.
Villacarrillo is a medium-sized town located in the rural province of Jaen in southeast Spain, famous for its intensive olive oil production. This region witnessed a mass exodus in the 1950s and 1960s to other more industrialised Spanish regions (Madrid, Barcelona and Valencia), but also to other industrialised countries [Citation21]. It is estimated that more than 25% of the population of Jaen migrated during the 1960s and the population of Villacarrillo shrank from 20,056 inhabitants in 1950 to only 10,673 in 2020 [Citation22]. This might explain why the first cases of ATTR Glu89Lys amyloidosis were described in other industrialised countries, and why only a minority of the patients included in our cohort were born in the Jaen province.
Recognising the endemic nature of the ATTR p.Glu109Lys variant in Spain and its founder effect in Villacarrillo has important clinical implications. Firstly, geographical delimitation of the founder mutation to a medium-sized town should translate into a higher level of awareness of suspicion of ATTRv amyloidosis in patients with unexplained neuropathy or cardiomyopathy from that area. Interestingly, only 1 patient from the study currently lives in Villacarrillo and when this study was completed, the patient was the only individual diagnosed with ATTRv amyloidosis in the town. It is very likely that additional individuals will be diagnosed with ATTRv amyloidosis as a consequence of our study. A lower suspicion threshold would be desirable in individuals with documented ancestry from Villacarrillo or the Jaen province currently living in other countries or regions. Identification of new additional ATTRv amyloidosis populations following migration routes has been reported for other ATTRv founder variants–for example, the Portuguese p.Val50Met variant in Brazil [Citation7]. As the p.Glu109Lys variant arose in Villacarrillo before the Spanish migration to America, it would not be unexpected that a similar pattern to that occurring with Portuguese and Brazil will emerge with this variant and other South-American countries. Indeed, one of the families included in this study and haplotyped came from Argentina.
This study provides relevant information to describe the natural history of ATTRv amyloidosis caused by the p.Glu109Lys variant. This entity is associated with a mixed cardiac and neurologic phenotype with complete penetrance by the age of 60 years and with mean age of phenotypic expression before the age of 50. A trend towards a younger age of phenotypic expression was observed in healthy carriers as compared with probands. This could be caused by genetic anticipation, which has been widely described in families with ATTR Val30Met amyloidosis in different endemic countries [Citation23–25], or, more likely, as a result of earlier and closer evaluation following identification of healthy carrier status. Nevertheless, based on our findings we can determine that the mean age of first phenotypic expression in ATTR Glu89Lys amyloidosis is ∼40 years, which is later than the predominantly neurologic Val30Met Portuguese variant but earlier than other founder variants with mixed phenotype like p.Glu109Gln or p.Thr80Ala [Citation26].
We also observed that cardiac and neurological involvement follow a parallel course over the years, while ophthalmologic involvement is delayed by several years and shows incomplete penetrance. In contrast to previous reports in small kindreds, superficial sensory loss is the most common neurological manifestation rather than deep sensory loss [Citation14].
ATTRv Glu89Lys amyloidosis is associated with poor prognosis, as median life expectancy in our cohort was only 62 years and none of the studied individuals survived beyond 65 years of age despite the fact that LT and HT were performed in many cases. Nevertheless, the significant increase in survival in our cohort with disease-modifying therapies offers a glimpse of hope, although the lead-time bias might explain in part this finding. Even with this limitation, we certainly hope that the recently available disease-specific therapies will change the natural history of this devastating disease. On the other hand, it is likely that the extension of survival in p.Glu109Lys carriers will result in increased ophthalmologic disease, as eye involvement is not prevented with current ATTR therapies and seems to affect a significant number of individuals with this variant. Clinicians following patients with the p.Glu109Lys TTR variant should be aware of this particularity and ensure an appropriate ophthalmological follow-up in these patients.
In contrast to current recommendations that propose to begin clinical follow-up 10 years ahead of predicted age of disease onset in healthy ATTRv carriers [Citation1,Citation19], we would suggest that clinical follow-up in p.Glu109Lys carriers should be started in early adulthood (20–25 years) based on the possible genetic anticipation effect seen and the outlier cases with very early onset found in our cohort. Moreover, due to the observed complete penetrance and poor prognosis, we would recommend early and exhaustive genetic counselling among relatives of affected patients that also includes reproductive counselling in relatives identified as genetic carriers.
Lastly, we observed that almost 40% of patients with Glu89Lys ATTRv amyloidosis and polyneuropathy required to be switched from tafamidis to patisiran due to neurological progression after 1 year of treatment, suggesting that a significant proportion of patients might be non-responders to TTR stabilisers. Predictors of response to tafamidis in patients with ATTR Val30Met amyloidosis polyneuropathy have been previously analysed by Monteiro et al. who found male sex, advanced disease stage and native TTR basal levels as predictors of worse response [Citation27], Although our limited sample precluded statistically sound analysis, there was a trend for older age and lower LVEF in patients who switched from tafamidis to patisiran compared with those who remained on tafamidis, suggesting that ATTRv Glu89Lys amyloidosis patients with advanced disease might respond worse to stabilisers.
Limitations
Some limitations should be considered regarding this study. Firstly, it was a retrospective longitudinal cohort study in which some patients did not have available current therapies. Although studies like this help to determine what to expect regarding clinical manifestations, disease and natural history of carriers of the p.Glu109Lys TTR variant, the results here are likely to change in upcoming years as a result of therapeutic advances in the field. Conversely, phenotype and severity might have been influenced by disease-modifying therapies received by some patients during follow-up. Furthermore, even though this cohort is the largest cohort of Glu109Lys carriers reported to date, given the rarity of this disease, the low number of subjects included limits the possibility of identifying prognostic factors.
Conclusions
p.Glu109Lys is a pathogenic TTR variant with a founder effect in southern Spain that originated in the town of Villacarrillo around 500 years ago. ATTRv amyloidosis caused by this variant is characterised by complete penetrance by the age of 60, early onset and mixed cardiac and neurologic phenotype, as well as ophthalmologic involvement in a significant number of cases. Overall, patients have poor prognosis, particularly if not treated with disease-modifying therapies.
| Abbreviations | ||
| AF | = | Atrial fibrillation |
| ATTR | = | Transthyretin amyloidosis |
| ATTRv | = | Hereditary transthyretin amyloidosis |
| ATTRwt | = | Wild-type transthyretin amyloidosis |
| CI | = | Confidence interval |
| HF | = | Heart failure |
| HR | = | Hazard ratio |
| HT | = | Heart transplantation |
| IQR | = | Interquartile range |
| LVEF | = | Left ventricular ejection fraction |
| LT | = | Liver transplantation |
| Mb | = | Megabases |
| NGS | = | Next-generation sequencing |
| NIS | = | Neurologic impairment score |
| NYHA | = | New York Heart Association |
| PCR | = | Polymerase chain reaction |
| SD | = | Standard deviation |
| SE | = | Standard error |
| STR | = | Short tandem repeats |
| TTR | = | Transthyretin |
Supplemental Material
Download MS Word (232.7 KB)Acknowledgement
The authors thank the DNA sequencing and Molecular Biology Unit from Instituto de Investigación Puerta de Hierro - Segovia de Arana (Madrid, Spain) for their support in this project.
Disclosure statement
Fernando de Frutos reports speaking fees from Pfizer. Esther Gonzalez-Lopez reports speaking fees from Pfizer and Alnylam, consulting fees from Pfizer and Proclara, and research/educational support to her institution from Pfizer, Bridgebio and Alnylam. Lucia Galan received honoraria as an advisor and speaker from Alnylam, Akcea, Genzyme, Grunenthal, Sobi and Pfizer. José González-Costello reports speaking fees from Pfizer, Alnylam, AstraZeneca, Boehringer-Ingelheim, Abbott and Orion, and consulting fees from Pfizer, Alnylam, AstraZeneca, Abbott, Novartis and Boehringer-Ingelheim. Maria Angeles Espinosa Castro reports speaking fees from Alnylam. Teresa Sevilla reports consulting fees from Pfizer, Alnylam and Akcea. Pablo Garcia-Pavia reports speaking fees from Pfizer, Bridgebio, and Alnylam, consulting fees from Pfizer, Bridgebio, Neuroinmmune, Alnylam, AstraZeneca, NovoNordisk and Alexion, and research/educational support to his institution from Pfizer, Bridgebio and Alnylam. The remaining authors declare that they have no conflicts of interest relevant to the content of this manuscript.
Additional information
Funding
References
- Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2021;42(16):1554–1568.
- Damy T, Kristen AV, Suhr OB, et al. Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the transthyretin amyloidosis outcomes survey (THAOS). Eur Heart J. 2019;40(21):1707–1715.
- Maurer MS, Hanna M, Grogan M, et al. Genotype and phenotype of transthyretin cardiac amyloidosis. J Am Coll Cardiol. 2016;68(2):161–172.
- Reinés JB, Vera TR, Martín MU, et al. Epidemiology of transthyretin-associated familial amyloid polyneuropathy in the Majorcan area: son Llàtzer Hospital descriptive study. Orphanet J Rare Dis. 2014;9(1):29– 6.
- Quarta CC, Buxbaum JN, Shah AM, et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372(1):21–29.
- Ohmori H, Ando Y, Makita Y, et al. Common origin of the Val30Met mutation responsible for the amyloidogenic transthyretin type of familial amyloidotic polyneuropathy. J Med Genet. 2004;41(4):1–5.
- Zaros C, Genin E, Hellman U, et al. On the origin of the transthyretin val30met familial amyloid polyneuropathy. Ann Hum Genet. 2008;72(Pt 4):478–484.
- Olsson M, Jonasson J, Cederquist K, et al. Frequency of the transthyretin Val30Met mutation in the Northern Swedish population. Amyloid. 2014;21(1):18–20.
- Kirov A, Sarafov S, Pavlova Z, et al. Founder effect of the Glu89Gln TTR mutation in the Bulgarian population. Amyloid. 2019;26(4):181–185.
- Reilly MM, Staunton H, Harding AE. Familial amyloid polyneuropathy (TTR ala 60) in North West Ireland: a clinical, genetic, and epidemiological study. J Neurol Neurosurg Psychiatry. 1995;59(1):45–49.
- Nakamura M, Asl KH, Benson MD. A novel variant of transthyretin (Glu89Lys) associated with familial amyloidotic polyneuropathy. Amyloid. 2000;7(1):46–50.
- Niederhauser J, Lobrinus JA, Ochsner F, et al. Successful heart and liver transplantation in a swiss patient with Glu89Lys transthyretin amyloidosis. Transplantation. 2011;91(6):e40–e42.
- Sandhu R, Westcott M, Pavesio C, et al. Retinal microangiopathy as an initial manifestation of familial amyloid cardiomyopathy associated with transthyretin E89K mutation. Retin Cases Brief Rep. 2013;7(3):271–275.
- Cruz S, Cortes-Vicente E, Illa I, et al. A novel variant of transthyretin (Glu89Lys) associated with familial amyloidotic polyneuropathy. Amyloid. 2017;7(6):46–50.
- Bourque PR, McCurdy AR, Mielniczuk LM, et al. Cardiac amyloidosis phenotype associated with a Glu89Lys transthyretin mutation. Can J Cardiol. 2017;33(6):830.e5–830-e7.
- Reynolds MM, Veverka KK, Gertz MA, et al. Ocular manifestations of familial transthyretin amyloidosis. Am J Ophthalmol. 2017;183:156–162.
- Gawor M, Holcman K, Franaszczyk M, et al. Spectrum of transthyretin gene mutations and clinical characteristics of polish patients with cardiac transthyretin amyloidosis. Cardiol J. 2020. Online published.
- Álvarez Rubio J, Manovel Sánchez AJ, González-Costello J, et al. Characterization of hereditary transthyretin cardiac amyloidosis in Spain. Rev Esp Cardiol (English Edition). 2022;75(6):488–495.
- Conceição I, Damy T, Romero M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid. 2019;26(1):3–9.
- Gandolfo LC, Bahlo M, Speed TP. Dating rare mutations from small samples with dense marker data. Genetics. 2014;197(4):1315–1327.
- Recaño VJ. La emigración andaluza en España. Bol Econ Andal. 1998;24:119–143.
- Instituto de Estadística y Cartografía de Andalucía. (Updated on 15/03/2021). https://www.juntadeandalucia.es/institutodeestadisticaycartografia/badea/operaciones/consulta/anual/6777?CodOper=b3_128&codConsulta=6777
- Lemos C, Coelho T, Alves-Ferreira M, et al. Overcoming artefact: anticipation in 284 portuguese kindreds with familial amyloid polyneuropathy (FAP) ATTRV30M. J Neurol Neurosurg Psychiatry. 2014;85(3):326–330.
- Cisneros-Barroso E, González-Moreno J, Rodríguez A, et al. Anticipation on age at onset in kindreds with hereditary ATTRV30M amyloidosis from the Majorcan cluster. Amyloid. 2020;27(4):254–258.
- Gorram F, Olsson M, Alarcon F, et al. New data on the genetic profile and penetrance of hereditary Val30Met transthyretin amyloidosis in Sweden. Amyloid. 2021;28(2):84–90.
- Sattianayagam PT, Hahn AF, Whelan CJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J. 2012;33(9):1120–1127.
- Monteiro C, Mesgazardeh JS, Anselmo J, et al. Predictive model of response to tafamidis in hereditary ATTR polyneuropathy. JCI Insight. 2019;4(12):e126526.