
ABSTRACT
Objectives: Arsenic is a ubiquitous element that is widely distributed in the environment to which man and animals are exposed. Cardiovascular disease is one of the aftermaths of chronic arsenic exposure-related morbidity and mortality. This study sought to investigate the possibility of reversal from arsenic-induced cardio-renal toxicity following exposure and subsequent withdrawal. The study also seeks to understand the mechanism of action of this reversal.
Methods: Rats were orally exposed to sodium arsenite at 10, 20 and 40 mg/kg daily for 4 weeks followed by 4 weeks of withdrawal.
Results: Exposure to arsenic caused a significant increase in malondialdehyde, H2O2 generation but decrease total thiol and reduced glutathione levels in both cardiac and renal tissues. Furthermore, increases in superoxide dismutase, glutathione-S-transferase and catalase with significant increases in glutathione peroxidase activities were observed in the cardiac tissues. On the contrary, a significant reduction in the renal antioxidant enzyme activity was recorded following exposure. Also, antioxidant defense system did not return to apparent values after arsenic withdrawal. Immunohistochemistry revealed a reduction in the expression of the pro-survival protein–protein kinase B (Akt/PKB) following exposure to arsenic and this was not reversed by withdrawal
Discussion: Exposure to arsenic caused cardio-renal toxicity via induction of oxidative stress and down-regulation of Akt/PKB expressions.
Introduction
Arsenic is a toxic heavy metal and its toxicity is of global health concern. Toxic levels of arsenic in the blood interfere with many metabolic processes. Toxic arsenic induces oxidative stress through the generation of reactive oxygen species (ROS), leading to multi-system organ failure, chronic diseases and death [Citation1]. Arsenic is ubiquitous and exists in multiple forms such as organic and inorganic compounds widely distributed in the environment to which man and animals are exposed. Arsenic exposure occurs mainly through contaminated food and water sources leading to human and ground water exposures [Citation2]. Arsenic toxicity is largely dependent on its form; trivalent or pentavalent with trivalent salt more potent to cause cancers, and other pathologies.
Oxides of arsenic such as sodium arsenite are components of some underground rocks, thus, present in some water sources as contaminants [Citation3]. Poisoning can also be linked to occupations such as mining and smelting of ore as well as higher concentrations have been found in water containing leached arsenics from rocks in an area endemic with high deposit of arsenic in the environment [Citation3]. Sodium arsenite is primarily used as a pesticide and has also been utilized as antiseptic, hide preservative, and in dye and soap production. Its persistent ingestion has been associated with various cardiovascular, neurologic and carcinogenic effects such as ischemic heart disease, and peripheral neuropathy [Citation4]. Once absorbed, arsenic accumulates in the liver, kidneys, heart and lungs while lower levels are found in muscles and neuronal tissues [Citation5].
Various reports indicate that the generation of ROS, oxidative stress and cellular damage through lipid peroxidation is the major pathway for the pathogenesis of sodium arsenite [Citation6,Citation7]. It has been reported that intracellular ROS levels produced is dependent on the concentration of sodium arsenite which further determine the type and severity of biological/pathological effects [Citation8]. However, sodium arsenite effect has been associated with hormetic properties in which low dose stimulation and high dose inhibition is expected. Chronic exposure to arsenic is expected to lead to accumulation in tissues and cause cellular dysfunction, however, human activities such as relocation or removal of the source of arsenic might reduce or eliminate its toxic effects. This study therefore sought to investigate the probable reversal of arsenic-induced cardio-renal toxicity following exposure and subsequent withdrawal from exposed subjects and its molecular mechanism of action. We therefore test the hypothesis that exposure of animals to arsenic acid as well as subsequent withdrawal does not have public health importance.
Materials and methods
Experimental animals and design
Eighty adult Wistar rats (120–200 g) were obtained from the Experimental Animal Unit of the Faculty of Veterinary Medicine, University of Ibadan and housed under controlled light cycle (12 hour light/12 hour dark). They were randomly divided into 4 groups of 20 animals per group and maintained on a standard commercial pelletized rat feed and water was provided ad libitum.
Group A served as control and received water alone while groups B, C and D received 10, 20 and 40 mg/kg of sodium arsenite, respectively, daily for 4 weeks. All treatments were administered orally. This was followed by withdrawal of treatment and animals thereafter placed on clean tap water for another 4 weeks. After withdrawal, 40 rats were sacrificed by cervical dislocation. The remaining 40 rats that were placed on clean water tap for 4 weeks were also sacrificed at the end of the experiment. The whole experiment lasted for 8 weeks.
The animals were cared for according to the criteria outlined in the Guide for the Care and the Use of Laboratory Animals prepared by the National Academy of Science and published by the National Institute of Health. During the experiment, ethics regulations were followed in accordance with national and institutional guidelines for the protection of the welfare of animals [Citation9].
Blood collection and post-mitochondrial fraction preparation
About 3 mL of blood was collected from the retro-orbital venous plexus of the animals into heparinized vials before sacrifice by cervical dislocation. The blood was centrifuged at 4000 rev min−1 for 15 minutes to obtain the plasma.
The hearts and kidneys were harvested, rinsed and homogenized in potassium buffer (0.1 M, pH 7.4) and the resulting homogenate was centrifuged at 10,000 rev min−1 (4°C) for 10 minutes to obtain the post-mitochondrial fractions.
Biochemical assays
Protein concentration of the post-mitochondrial fractions of the hearts and kidneys was determined by Biuret method as described by Gornal et al. [Citation10]. Reduced glutathione (GSH) was estimated at 412 nm according to the method of Beutler et al. [Citation11]. Glutathione peroxidase (GPx) activity was determined as described by Rotruck et al. [Citation12]. Glutathione-S-transferase (GST) was measured by the method of Habig et al. [Citation13] using 1-chloro-2, 4-dinitrobenzene as substrate. The catalase (CAT) activity was determined according to the method of Sinha [Citation14]. Superoxide dismutase (SOD) was determined by measuring the inhibition of auto-oxidation of epinephrine at pH 10.2 as described by Misra and Fridovich [Citation15] with slight modification as described by Oyagbemi et al. [Citation16]. The MDA level measured as described by Varshney and Kale [Citation17]. Lipid peroxidation in µmol MDA formed/mg protein was computed with a molar extinction coefficient of 1.56 × 105 M−1 cm−1. Hydrogen peroxide (H2O2) generation was estimated as described by Wolff [Citation18]. The cardiac and renal nitric oxide (NO) content was quantified as previously described by Olaleye et al. [Citation19]. All biochemical assays were carried out on the post-mitochondrial fractions of cardiac and renal tissues. Plasma creatinine (CR2337) and blood urea nitrogen (UR2821) levels were assayed using Randox kits (Randox Laboratories Ltd, Crumlin, UK) according to manufacturer’s manual.
Histopathology
Small slices of hearts and kidneys were collected in 10% buffered formalin for proper fixation. These tissues were processed and embedded in paraffin wax. Sections about 5–6 µm thick were made and stained with hematoxylin and eosin for histopathology [Citation20].
Immunohistochemistry of PKB
Immunohistochemistry of paraffin embedded tissues of the heart and kidney was performed after the tissues were obtained from buffered formalin perfused rats. The tissues were processed based on the methods described by Todorich et al. [Citation20] with slight modification. Briefly, paraffin sections were melted at 60◦C in the oven. Dewaxing of the samples in xylene was followed by passage through ethanol at a decreasing concentration (100–80%). Peroxidase quenching in 3% H2O2/methanol was carried out with subsequent antigen retrieval performed by microwave heating in 0.01 M citrate buffer (pH 6.0) to the point of boiling. All the sections were blocked in normal goat serum (10%, HistoMark®, KPL, Gaithersburg, MD, USA) and probed with PKB antibody (Abclonal®, Baltimore, MD, USA), 1:200 overnight at 4°C in a refrigerator. Detection of bound antibody was carried out using biotinylated (goat anti-rabbit, 2.0 µg/mL) secondary antibody and subsequently, streptavidin peroxidase (Horse Radish Peroxidase-streptavidin system) according to manufacturer’s protocol (HistoMark®, KPL, Gaithersburg, MD, USA).
Reaction product was enhanced with diaminobenzidine (DAB, Amresco®, USA) for 6–10 minutes and counterstained with high definition hematoxylin (Enzo®, NY, USA), with subsequent dehydration in ethanol. The slides were covered with coverslips and sealed with DBx mountant. The immuno-positive expressions of intensive regions were viewed starting from low magnification on each slice then with 100 × magnifications using a photo microscope (Olympus) and a digital camera (Toupcam®, Touptek Photonics, Zhejiang, China).
Statistical analysis
All values were expressed as mean ± standard deviation (SD). The test of significance between two groups was estimated by Student’s t-test with P-values < 0.05 considered statistically significant using Graphpad Prism 5.0.
Results
Effect of sodium arsenite on antioxidant status in the heart
There was a significant (P < 0.05) increase in SOD, GST and CAT and GPx in the heart of rats sub-chronically exposed to sodium arsenite compared to the control. On the other hand, following withdrawal of the treatment, significant (P < 0.05) decreases in all aforementioned antioxidant enzymes were observed in all the treatment groups ().
Table 1. Cardiac antioxidant enzyme profile following exposure and withdrawal of sodium arsenite.
Effect of sodium arsenite on antioxidant status in the kidney
After 4 weeks of sodium arsenite exposure, the kidneys of rats showed decreases in GST and GPx relative to the control. However, upon withdrawal of sodium arsenite, a significant increase in the activities of GST and GPx was obtained post withdrawal of the treatment ().
Table 2. Renal antioxidant enzyme profile following exposure and withdrawal of sodium arsenite.
Effect of sodium arsenite on markers of oxidative stress in the heart
Significant decreases in total thiols in all exposed groups compared to control group were observed and this was accompanied by slight decrease in GSH levels and decreased non-protein thiols. A significant decrease in the GSH content was however in all treatment groups relative to the control. Withdrawal of sodium arsenite produced a similar trend of decreased thiols and GSH levels in the heart of rats (). The sub-chronic exposure of rats to sodium arsenite produced significant increases in MDA and H2O2 production in the heart accompanied by significant increases in NO contents in Groups C and D. Sequel to withdrawal of the treatment, a non-significant increase in MDA and H2O2 contents were recorded for all groups, and significant increases in NO contents were recorded for Groups B and C ().
Table 3. Cardiac thiols and markers of oxidative stress following exposure and withdrawal of sodium arsenite.
Effect of sodium arsenite on markers of oxidative stress in the kidney
Increased H2O2 production and significant increases in MDA formation in the kidney of rats sub-chronically exposed to sodium arsenite was observed (). These increases were also recorded post withdrawal. However, NO levels which were slightly increased with treatment and became greatly reduced post withdrawal (). Significant decreases in total thiol and non-protein thiol for all treated rats were observed with slight decreases in GSH levels compared to those of the controls. Thereafter, decreased GSH levels and total thiols were obtained with an increase in non-protein thiol post withdrawal of the sodium arsenite ().
Table 4. Renal thiols and markers of oxidative stress following exposure and withdrawal of sodium arsenite.
Effect of sodium arsenite on plasma creatinine and urea levels
Slight changes in plasma creatinine levels were observed with a significantly increased level recorded for Group B compared with the control. These values were however significantly reduced after withdrawal for 4 weeks. Decreases in plasma urea levels were obtained with sub-chronic exposure to sodium arsenite which changed to increased levels after a switch to tap water was effected ().
Table 5. Plasma creatinine and urea profile following exposure and withdrawal of sodium arsenite.
Immunohistochemistry of PKB
A lower expression of protein kinase B (PKB) in a dose dependent fashion was observed in the heart and kidneys of all exposed groups to sodium arsenite compared with the control ( and ). Following withdrawal of arsenic for 4 weeks, lower expressions of PKB were observed compared with the control in the heart of experimental rats. On the other hand, no visible immuno-positive reaction of PKB was observed in the kidneys of rats in groups C and D following withdrawal ( and ).
Figure 1. Immunohistochemistry of protein kinase B (PKB) in the heart of rats exposed to sodium arsenite for 4 weeks. (A) Control: shows positive and high expression of PKB; (B) 10 mg/kg NaAsO2: shows lower expression of PKB than control: (C) 20 mg/kg NaAsO2 shows lower expression of PKB than control; (D) 40 mg/kg NaAsO2: shows no expression of PKB. The result is indicative that exposure of rats to NaAsO2 reduced the expressions of survival protein (Akt/PKB). Scale bar (for A, B, C and D) = 5.04 × 3.87 mm. The slides were counterstained with high definition hematoxylin and viewed ×400 objectives.
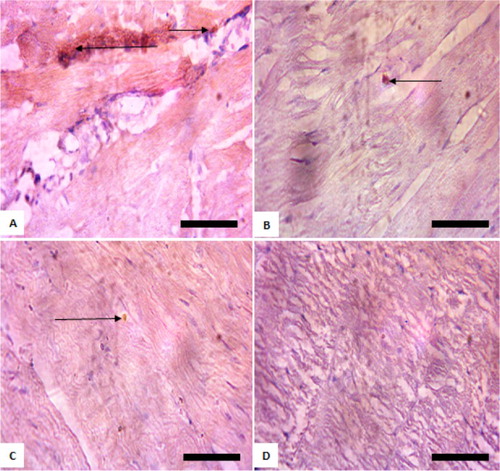
Figure 2. Immunohistochemistry of protein kinase B (PKB) in the kidneys of rats exposed to sodium arsenite for 4 weeks. (A) Control: shows positive expression of PKB; (B) 10 mg/kg NaAsO2 shows lower expression of PKB than control; (C) 20 mg/kg NaAsO2 shows no expression PKB; (D) 40 mg/kg NaAsO2: shows no expression PKB. The result is indicative that exposure of rats to NaAsO2 reduced the expressions of survival protein (Akt/PKB). Scale bar (for A, B, C, and D) = 5.04 × 3.87 mm. The slides were counterstained with high definition hematoxylin and viewed ×400 objectives.
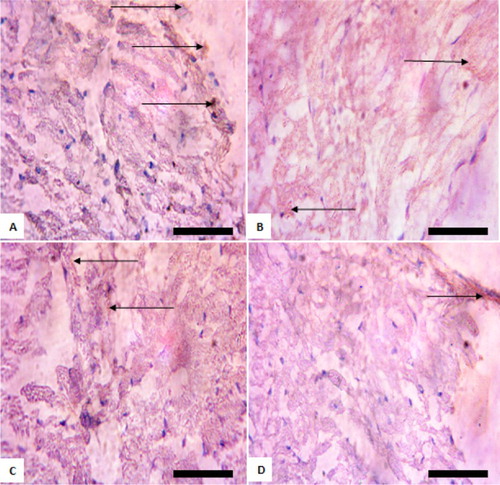
Figure 3. Immunohistochemistry of protein kinase B (PKB) in the heart of rats after withdrawal of sodium arsenite exposure for 4 weeks. (A) Control: shows positive and high expression of PKB; (B) 10 mg/kg NaAsO2: shows lower expression of PKB than control; (C) 20 mg/kg NaAsO2 shows lower expression of PKB than control; (D) 40 mg/kg NaAsO2: shows lower expression of PKB than control. The result shows that following withdrawal of NaAsO2, the expressions of the survival protein (Akt/PKB) are still lower than the control. Scale bar (for A, B, C, and D) = 5.04 × 3.87 mm. The slides were counterstained with high definition hematoxylin and viewed ×400 objectives.
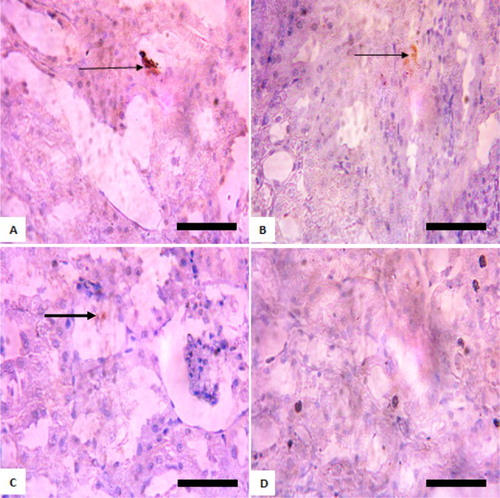
Figure 4. Immunohistochemistry of protein kinase B (PKB) in the kidneys of rats after withdrawal of sodium arsenite exposure for 4 weeks. (A) Control: shows positive and high expression of PKB; (B) 10 mg/kg NaAsO2 shows lower expression of PKB than control; (C) 20 mg/kg NaAsO2: shows no expression of PKB; (D) 40 mg/kg NaAsO2: shows no expression of PKB. The result shows that following withdrawal of NaAsO2, the expressions of the survival protein (Akt/PKB) are still lower than the control. Scale bar (for A, B, C, and D) = 5.04 × 3.87 mm. The slides were counterstained with high definition hematoxylin and viewed ×400 objectives.
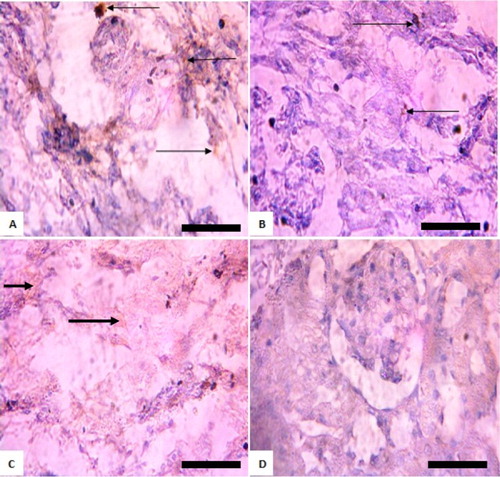
Histopathology
Histological examination of the heart of rats revealed varying pathology after exposure to sodium arsenite at various doses ranging from mild to severe pyknosis and necrosis of myocardial cells as well as loss of cardiac architecture (). At post withdrawal, these anomalies were still evident (). Also, in the kidneys, pathologies observed included peritubular infiltration by inflammatory cells, mild to disseminated congestion of vessels and mild tubular necrosis ( and ).
Figure 5. Photomicrograph showing heart of rats exposed to sodium arsenite orally for 4 weeks. (A) Control shows no significant lesion; (B) 10 mg/kg NaAsO3 mild pyknosis and necrotic cells (black arrows); (C) 20 mg/kg NaAsO3 reveals loss of striations and pyknotic cells (black arrows) and (D) 40 mg/kg NaAsO3 with loss of structural architecture, severe pyknosis and necrosis of cells (black arrows). Scale bar (for A, B, C and D) = 2.15 × 2.79 mm Plates are stained with H and E stains and viewed with ×100 objectives.
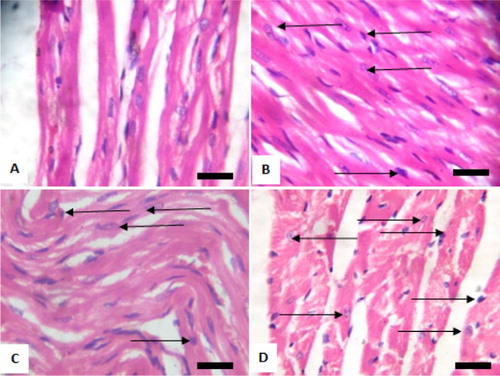
Figure 6. Photomicrograph showing heart of rats post withdrawal of sodium arsenite for 4 weeks. (A) Control; (B) 10 mg/kg NaAsO3 mild myocardial damage with pyknotic cells (black arrows); (C) 20 mg/kg NaAsO3 moderate myocardial damage with pyknotic cells (black arrows) and (D) 40 mg/kg NaAsO3 severe myocardial damage with pyknotic cells (black arrows). Scale bar (for A, B, C and D) = 2.15 × 2.79 mm. Plates are stained with H and E stained and viewed with ×100 objectives.
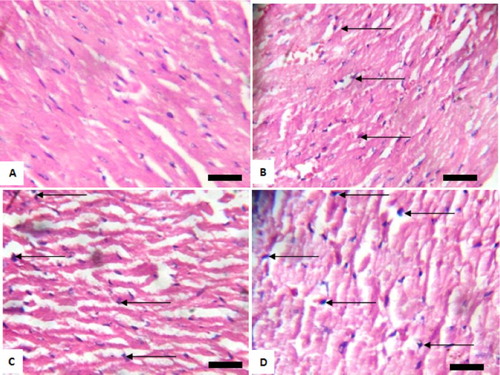
Figure 7. Photomicrograph showing kidney of rats exposed to sodium arsenite orally for 4 weeks. (A) Control shows focal hemorrhagic lesion (blue arrow); (B) 10 mg/kg NaAsO2 with peritubular infiltration by inflammatory cells (black arrow) and mild tubular necrosis (green arrow); (C) 20 mg/kg NaAsO2 showing congestion of vessel (blue arrow) and (D) 40 mg/kg NaAsO2 showing congestion of vessels (blue arrow) and peritubular infiltration by inflammatory cells (black arrow). Scale bar (for A, B, C and D) = 2.15 × 2.79 mm. Plates are stained with H and E stains and viewed with 100 objectives.
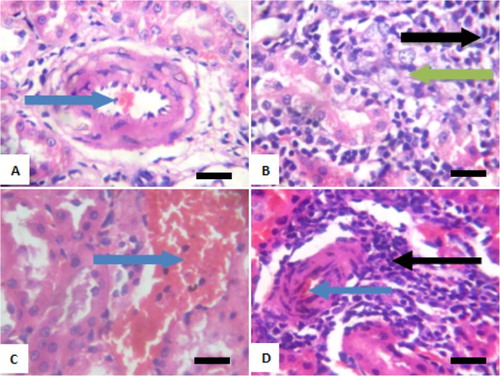
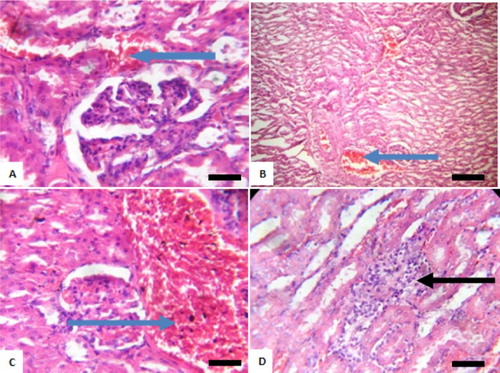
Figure 8. Photomicrograph showing kidneys of rats post withdrawal of sodium arsenite for 4 weeks. (A) Control with mild congestion of blood vessel (blue arrow); (B) 10 mg/kg NaAsO2 shows congestion of vessel (blue arrow); (C) 20 mg/kg NaAsO2 with marked disseminated congestion of blood vessel (blue arrow) and (D) 40 mg/kg NaAsO2 shows focal area of peritubular inflammation (black arrow). Scale bar (A, B, C and D) = 2.15 × 2.79 mm. Plates are stained with H and E stains and viewed with ×100 objectives.
Discussion
Arsenic intoxication over a 4-week period caused oxidative stress in both cardiac and renal tissues of rats as revealed by significant increase in MDA, H2O2 generation and thiol levels. Arsenic is reported to cause oxidative stress by increasing ROS and reactive nitrogen species (RNS) production which interact with membrane lipids to generate lipid peroxides leading to cellular damage [Citation21,Citation22]. Although, it is still unclear how these reactive species are produced [Citation23], however, Yamanaka et al. [Citation24] theorized that the metabolism of arsenic by the body involves the formation of intermediary arsine species which may react with oxygen to generate ROS and initiate the widely accepted free radical chain reaction mechanism of lipid peroxidation. MDA is a product of lipid peroxidation whose levels indicate damage to membrane lipids of arsenic exposed rats [Citation25].
The concomitant decrease in renal antioxidant enzymes is indicative of the body’s inability to cope with the excessive ROS production over its inherent antioxidant capability viz; oxidative stress. Antioxidant enzymes and thiols form the antioxidant defence system of the body. The sub-chronic exposure to sodium arsenite gave rise to a decrease in renal SOD, GST, GPx and CAT of rats. This was accompanied by a significant decrease in thiol levels, although GSH levels were slightly reduced. These lowered levels revealed the precipitation of oxidative stress due to sub-chronic sodium arsenite intoxication in rats. The GSH is an important intracellular antioxidant defence molecule in the body; protecting against cytotoxicity due to xenobiotics by conjugation and hydro-reactive intermediates [Citation26]. The decrease in renal GSH levels can be attributed to its exhaustion by the kidneys which participate in elimination of arsenic. GSH is also utilized as a substrate by GPx and GST in xenobiotic conjugation and elimination of toxic electrophiles.
Cardiac antioxidant enzymes (SOD, GPx, GST and CAT) levels however were elevated in this study. These findings are linked to the interference of arsenic in cell signaling pathway of NRF2/KEAP1 (nuclear factor erythroid 2-related factor 2/Kelch-like ECH-associated protein 1). This, it does by regulating NRF2/KEAP1 via increasing ROS mediated activation of PI3K (phosphatidylinositol-4, 5-biphosphate 3-kinase) and reducing the formation of NRF2/KEAP1/CUL3 (Cullin 3) degradation complex. This leads to an increase in the activity of NRF2. NRF2 activation induces cytoprotective gene expression and high activity is known to evoke subsequent expression of detoxifying enzymes, mostly mediated by glutathione conjugation such as GST and GPx [Citation27].
On withdrawal of sodium arsenite to allow for recovery in rats, there were significant decreases in cardiac antioxidant enzymes; SOD, CAT, GPx and GST which is in line with the absence of arsenic which upregulated NRF2 activity and masked oxidative stress. This was accompanied by slight differences in GSH and NPSH although, total thiols were significantly reduced. Recovery was evident in the slight decrease in MDA and H2O2 levels as opposed to the significantly increased levels observed during the period of exposure. After 4 weeks of withdrawal, renal SOD, GST, GPx and CAT indicated recovery in the rats as revealed by slightly increased levels and with slight differences in these antioxidant enzymes. This shows the body’s capability to ward off oxidative stress and restore balance in the system is slowly being regained. This recovery was also observed in the close levels of GSH and thiols of recovering rats and control group.
Due to the severity of insult on the kidneys; particularly due to its participation in arsenic elimination from the body, the significant decrease in renal NO levels indicative of hypertension is not strange. The decrease in NO bioavailability has been associated with hypertension and derangement in vascular endothelium [Citation28,Citation29]. From the present study, the rats did not recover from the reduced tissue NO bioavailability. This is however suggestive of persistent hypertension even after withdrawal from arsenic intoxication. The struggle for recovery was also shown by increased MDA and slight differences in H2O2 levels in the kidneys. Decreased plasma creatinine levels with slight increases in urea can be attributed to previous injury during exposure to sodium arsenite from which complete recovery was yet to occur. It can however be inferred that recovery from renal damage due to arsenic exposure might not be possible perhaps if exposure to human subject occurred.
Immunohistochemistry of PKB in cardiac and renal tissues of rats exposed to sodium arsenite for 4 weeks revealed a decreasing expression of PKB compared to control with increasing dose of sodium arsenite. PKB regulates cell survival and metabolism in the body. It ensures survival of cells by transcription of pro-survival genes [Citation30]. The decrease in PKB expression was attributable to oxidative stress and damage to the cell with indications towards apoptosis. PKB phosphorylates BAD a pro-apoptotic member of the Bcl2 family series causing it to dissociate and lose its pro-apoptotic function [Citation31,Citation32]. Therefore, decreased expression of PKB in this study correlates with the oxidative stress revealed via biochemical assays. Recovery was noticed in the cardiac tissues as revealed by increased expressions of PKB. However, in renal tissues recovery was observed only in rats that were exposed to 10 mg/kg of sodium arsenite. It is safe to speculate that given more time post withdrawal, the tissues may recover in all groups.
Histological examination revealed pathology of various severities in the heart after exposure to sodium arsenite. Arsenic toxicity in this study caused pyknosis and necrosis of myocardial cells. Severity of pathology was dose dependent with loss of structural architecture after exposure to 40 mg/kg sodium arsenite. With withdrawal of arsenic, pathology remained as heart was yet to fully recover or compensate for the arsenic-induced damage to heart. Chen and colleagues [Citation33] reported abnormal ultra-structural changes in cardiac tissues exposed to arsenic. Next to the risk of cancer with arsenic exposure, a dose dependent relationship has been established between arsenic exposure, mortality and cardiovascular diseases especially heart diseases [Citation33]. Exposure to arsenic caused pathological changes in the kidneys of rats ranging from peritubular infiltration by inflammatory cells, congestion of vessels to tubular necrosis in the present study. Inflammatory cell infiltration can be attributed to the sub-chronic arsenic exposure which increases platelet aggregation [Citation34]. This can also be correlated with the resultant decreased NO levels in the kidneys owing to the reaction between NO and ROS generated by arsenic toxicity leading to the formation of peroxynitrite which has been implicated in the upregulation of inflammatory mediators [Citation5]. The kidney participates in the elimination of arsenic causing detection of high levels of arsenic due to accumulation in the kidneys thus making it a primary target for arsenic-induced toxicity. From this study, we report for the first time the involvement of protein kinase B (Akt/PKB) signaling and oxidative stress in sodium arsenite-induced cardiovascular and renal dysfunction in experimental rat model. However, the limitations of our study is how to extrapolate the data obtained in this study to human pathology at the risk of exposure and the lack of data presenting molecular changes that might have been induced by the changes in oxidative status/biochemical changes reported in rats following arsenic exposure. Also, determination of message proteins levels of the respective enzymes mediating oxidative stress would have given good correlative changes in the activities reported. Altogether, this aspect of the study will be of a future planned study to follow-up on the observed trend of withdrawal-induced reversal of the changes in the parameters reported as well as how the signaling molecules – protein, enzymes and genes recover following withdrawal and that the extrapolation of results from such a study can be related to human at the risk of exposure.
In conclusion, arsenic-induced cardio-renal toxicity results from sub-chronic exposure to sodium arsenite in rats by oxidative stress in a dose dependent manner. However, withdrawal from arsenic exposure shows promise of a possible reversal over time although the question of the extent of recovery or full recovery will be the subject of another study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Dr. Ademola Adetokunbo Oyagbemi (DVM, MSc., PhD, FCVSN) is a biochemist and toxicologist at the Department of Veterinary Physiology, Biochemistry and Pharmacology, Faculty of Veterinary Medicine, University of Ibadan, Nigeria.
Dr. Temidayo Olutayo Omobowale (DVM, PhD, FCVSN) is a senior lecturer and consultant at Veterinary Internal Medicine Unit at the Department of Veterinary Medicine, Faculty of Veterinary Medicine, University of Ibadan, Nigeria.
Dr. Ebunoluwa Racheal Asenuga (DVM, MSc.) is a lecturer and a biochemist at the Department of Veterinary Medicine, Faculty of Veterinary Medicine, University of Benin, Benin City, Nigeria.
Dr. Grace Onyeche Ochigbo (DVM, MSc.) is a toxicologist and postgraduate student at the Department of Veterinary Physiology, Biochemistry and Pharmacology, Faculty of Veterinary Medicine, University of Ibadan, Nigeria.
Dr. Abiola Olumuyiwa Adejumobi (DVM, MSc.) is a lecturer at Veterinary Internal Medicine Unit at the Department of Veterinary Medicine, Faculty of Veterinary Medicine, University of Ibadan, Nigeria.
Adeolu Alex Adedapo (DVM, PhD FIIA) is a professor of Pharmacology at the Department of Veterinary Physiology, Biochemistry and Pharmacology, Faculty of Veterinary Medicine, University of Ibadan, Nigeria.
Momoh Audu Yakubu (PhD) is a professor at the Department of Environmental and Interdisciplinary Sciences, COSET, Texas Southern University, 3100 Cleburne Avenue, Houston, 77004, Texas, USA.
ORCID
Momoh Audu Yakubu http://orcid.org/0000-0002-6072-2473
References
- Lu TH, Tseng TJ, Su CC, et al. Arsenic induces reactive oxygen species-caused neuronal cell apoptosis through JNK/ERK-mediated mitochondria-dependent and GRP 78/CHOP-regulated pathways. Toxicol Lett. 2014;224:130–140. doi: 10.1016/j.toxlet.2013.10.013
- Adil M, Kandhare AD, Ghosh P, et al. Sodium arsenite-induced myocardial bruise in rats: ameliorative effect of naringin via TGF-β/Smad and Nrf/HO pathways. Chem Biol Interact. 2016;253:66–77. doi: 10.1016/j.cbi.2016.05.015
- Smedley PL, Kinniburgh DG, Macdonald DMJ, et al. Arsenic associations in sediments from the loess aquifer of La Pampa, Argentina. Appl Geochem. 2005;20:989–1016. doi: 10.1016/j.apgeochem.2004.10.005
- Roy D, Gaur P, Verma N, et al. Bioremediation of arsenic (III) from water using Baker yeast Saccharomyces cerevisiae. Int J Environ Bioremed Biodegrad. 2013;1:14–19.
- Singh AP, Goel RK, Kaur T. Mechanisms pertaining to arsenic toxicity. Toxicol Int. 2011;18(2):87–93. doi: 10.4103/0971-6580.84267
- Yang P, He XQ, Peng L, et al. The role of oxidative stress in hormesis induced by sodium arsenite in human embryo lung fibroblast (HELF) cellular proliferation model. J Toxicol Environ Health A. 2007;70:976–983. doi: 10.1080/15287390701290832
- Zhang Z, Wang X, Cheng S, et al. Reactive oxygen species mediate arsenic induced cell transformation and tumorigenesis through Wnt/β-catenin pathway in human colorectal adenocarcinoma DLD1 cells. Toxicol Appl Pharmacol. 2011;256:114–121. doi: 10.1016/j.taap.2011.07.016
- Jiang X, Chen C, Zhao W, et al. Sodium arsenite and arsenic trioxide differently affect the oxidative stress, genotoxicity and apoptosis in A549 cells: an implication for the paradoxical mechanism. Environ Toxicol Pharmacol. 2013;36:891–902. doi: 10.1016/j.etap.2013.08.002
- PHS (Public Health Service). Public health service policy on humane care and the use of laboratory animals. Washington (DC): US Department of Health and Humane Services; 2015; p. 99–158.
- Gornal AG, Bardawill JC, David MM. Determination of serum proteins by means of Biuret reaction. J Biol Chem. 1949;177:751–766.
- Beutler EO, Duron B, Kelly M. Improved method for the determination of blood glutathione. J Lab Clin Med. 1963;61:882–888.
- Rotruck JT, Pope AL, Ganther HE, et al. Selenium: biochemical role as a component of glutathione peroxidase. Sci. 1973;179:588–590. doi: 10.1126/science.179.4073.588
- Habig WH, Pabst MJ, Jacoby WB. Glutathione-S-transferase activity: the enzymic step in mercapturic acid formation. J Biol Chem. 1974;249:130–139.
- Sinha KA. Colorimetric assay of catalase. Anal Biochem. 1972;47:389–394. doi: 10.1016/0003-2697(72)90132-7
- Misra HP, Fridovich I. The role of superoxide anion in the autooxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem. 1972;247:3170–3175.
- Oyagbemi AA, Omobowale TO, Akinrinde AS, et al. Lack of reversal of oxidative damage in renal tissues of lead acetate-treated rats. Environ Toxicol. 2015;30:1235–1243. doi: 10.1002/tox.21994
- Varshney R, Kale RK. Effect of calmodulin antagonists on radiation induced lipid peroxidation in microsomes. Int J Biol. 1990;158:733–741.
- Woff SF. Ferrous ion oxidation in the presence of ferric ion indicator xylenol Orange for measurement of hydrogen peroxides. Methods Enzymol. 1994;233:182–189. doi: 10.1016/S0076-6879(94)33021-2
- Olaleye SB, Adaramoye OA, Erigbali PP. Lead exposure increases oxidative stress in the gastric mucosa of HCl/ethanol-exposed rats. World J Gastroent. 2007;13:5121–5126. doi: 10.3748/wjg.v13.i38.5121
- Drury RA, Wallington EA, Cancerson R. Carlton’s histopathological techniques. 4th ed. Oxford/London/New York: Oxford University Press; 1976.
- Valko M, Jomova K, Rhodes CJ, et al. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch Toxicol. 2016;90:1–37. doi: 10.1007/s00204-015-1579-5
- Wang F, Zhou X, Liu W, et al. Arsenite-induced ROS/RNS generation causes zinc loss and inhibits the activity of poly(ADP-ribose) polymerase-1. Free Radic Biol Med. 2013;61:249–256. doi: 10.1016/j.freeradbiomed.2013.04.019
- Manna P, Sinha M, Sil PC. Arsenic-induced oxidative myocardial injury: protective role of arjunolic acid. Arch Toxicol. 2008;82:137–149. doi: 10.1007/s00204-007-0272-8
- Yamanaka K, Mizol M, Kato K, et al. Oral administration of dimethylarsinic acid, a main metabolite of inorganic arsenic, in mice promotes skin tumorigenesis initiated by dimethylbenz(a)anthracene with or without ultraviolet B as a promoter. Biol Pharm Bull. 2001;24:510–514. doi: 10.1248/bpb.24.510
- Spickett CM. The lipid peroxidation product 4-hydroxy-2-nonenal: advances in chemistry and analysis. Redox Biol. 2013;1:145–152. doi: 10.1016/j.redox.2013.01.007
- Most P, Papenbrock J. Possible roles of plant sulfurtransferases in detoxification of cyanide, reactive oxygen species, selected heavy metals and arsenate. Molecules. 2015;20:1410–1423. doi: 10.3390/molecules20011410
- Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Rad Biol Med. 2015;88:93–100. doi: 10.1016/j.freeradbiomed.2015.06.006
- Todorich B, Olopade JO, Surguladze N, et al. The mechanism of vanadium-mediated developmental hypomyelination is related to destruction of oligodendrocyte progenitors through a relationship with ferritin and iron. Neurotox Res. 2011;19:361–373. doi: 10.1007/s12640-010-9167-1
- Gonçalves-Rizzi VH, Possomato-Vieira JS, Sales Graça TU, et al. Sodium nitrite attenuates hypertension-in-pregnancy and blunts increases in soluble FMS-like tyrosine kinase-1 and in vascular endothelial growth factor. Nitric Oxide. 2016;57:71–78. doi: 10.1016/j.niox.2016.05.004
- Faissner A, Heck N, Dobbertin A, et al. DSD-1-proteoglycan/phosphacan and receptor protein tyrosine phosphatase-beta isoforms during development and regeneration of neural tissues. Adv Exp Med Biol. 2006;557:25–53. doi: 10.1007/0-387-30128-3_3
- Zhao EY, Efendizade A, Cai L, et al. The role of Akt (protein kinase B) and protein kinase C in ischemia-reperfusion injury. Neurol Res. 2016;38:301–308. doi: 10.1080/01616412.2015.1133024
- Li Y, Chen YM, Sun MM, et al. Inhibition on apoptosis induced by elevated hydrostatic pressure in retinal ganglion cell-5 via laminin upregulating β1-integrin/focal adhesion kinase/protein kinase B signaling pathway. Chin Med J (Engl). 2016;129:976–983. doi: 10.4103/0366-6999.179785
- Chen Y, Factor-Litvak P, Howe GR, et al. Arsenic exposure from drinking water and mortality from cardiovascular disease in Bangladesh: prospective cohort study. Br Med J. 2011;342:d2431. doi: 10.1136/bmj.d2431
- Li SW, Sun X, He Y, et al. Assessment of arsenic trioxide in the heart of Gallus gallus: alterations of oxidative damage parameters, inflammatory cytokines, and cardiac enzymes. Environ Sci Pollut Res Int. 2017;24(6):5781–5790. doi: 10.1007/s11356-016-8223-7