
ABSTRACT
Background: p53 is a tumor suppressor protein involved in regulating a wide array of signaling pathways. The role of p53 in the cell is determined by the type of imposed oxidative stress, its intensity and duration. The last decade of research has unravelled a dual nature in the function of p53 in mediating the oxidative stress burden. However, this is dependent on the specific properties of the applied stress and thus requires further analysis.
Methods: A systematic review was performed following an electronic search of Pubmed, Google Scholar, and ScienceDirect databases. Articles published in the English language between January 1, 1990 and March 1, 2017 were identified and isolated based on the analysis of p53 in skeletal muscle in both animal and cell culture models.
Results: Literature was categorized according to the modality of imposed oxidative stress including exercise, diet modification, exogenous oxidizing agents, tissue manipulation, irradiation, and hypoxia. With low to moderate levels of oxidative stress, p53 is involved in activating pathways that increase time for cell repair, such as cell cycle arrest and autophagy, to enhance cell survival. However, with greater levels of stress intensity and duration, such as with irradiation, hypoxia, and oxidizing agents, the role of p53 switches to facilitate increased cellular stress levels by initiating DNA fragmentation to induce apoptosis, thereby preventing aberrant cell proliferation.
Conclusion: Current evidence confirms that p53 acts as a threshold regulator of cellular homeostasis. Therefore, within each modality, the intensity and duration are parameters of the oxidative stressor that must be analyzed to determine the role p53 plays in regulating signaling pathways to maintain cellular health and function in skeletal muscle.
Abbreviations: Acadl: acyl-CoA dehydrogenase, long chain; Acadm: acyl-CoA dehydrogenase, C-4 to C-12 straight chain; AIF: apoptosis-inducing factor; Akt: protein kinase B (PKB); AMPK: AMP-activated protein kinase; ATF-4: activating transcription factor 4; ATM: ATM serine/threonine kinase; Bax: BCL2 associated X, apoptosis regulator; Bcl-2: B cell Leukemia/Lymphoma 2 apoptosis regulator; Bhlhe40: basic helix-loop-helix family member e40; BH3: Borane; Bim: bcl-2 interacting mediator of cell death; Bok: Bcl-2 related ovarian killer; COX-IV: cytochrome c oxidase IV; cGMP: Cyclic guanosine monophosphate; c-myc: proto-oncogene protein; Cpt1b: carnitine palmitoyltransferase 1B; Dr5: death receptor 5; eNOS: endothelial nitric oxide synthase; ERK: extracellular regulated MAP kinase; Fas: Fas Cell surface death receptor; FDXR: Ferredoxin Reductase; FOXO3a: forkhead box O3; Gadd45a: growth arrest and DNA damage-inducible 45 alpha; GLS2: glutaminase 2; GLUT 1 and 4: glucose transporter 1(endothelial) and 4 (skeletal muscle); GSH: Glutathione; Hes1: hes family bHLH transcription factor 1; Hey1: hes related family bHLH transcription factor with YRPW motif 1; HIFI-α: hypoxia-inducible factor 1, α-subunit; HK2: Hexokinase 2; HSP70: Heat Shock Protein 70; H2O2: Hydrogen Peroxide; Id2: inhibitor of DNA-binding 2; IGF-1-BP3: Insulin-like growth factor binding protein 3; IL-1β: Interleukin 1 beta; iNOS: inducible nitric oxide synthase; IRS-1: Insulin receptor substrate 1; JNK: c-Jun N-terminal kinases; LY-83583: 6-anilino-5,8-quinolinedione; inhibitor of soluble guanylate cyclase and of cGMP production; Mdm 2/ 4: Mouse double minute 2 homolog (mouse) Mdm4 (humans); mtDNA: mitochondrial DNA; MURF1: Muscle RING-finger protein-1; MyoD: Myogenic differentiation 1; MyoG: myogenin; Nanog: Nanog homeobox; NF-kB: Nuclear factor-κB; NO: nitric oxide; NoxA: phorbol-12-myristate-13-acetate-induced protein 1 (Pmaip1); NRF-1: nuclear respiratory factor 1; Nrf2: Nuclear factor erythroid 2-related factor 2; P21: Cdkn1a cyclin-dependent kinase inhibitor 1A (P21); P38 MAPK: mitogen-activated protein kinases; p53R2: p53 inducible ribonucleotide reductase gene; P66Shc: src homology 2 domain-containing transforming protein C1; PERP: p53 apoptosis effector related to PMP-22; PGC-1α: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PGM: phosphoglucomutase; PI3K: Phosphatidylinositol-4,5-bisphosphate 3-kinase; PKCβ: protein kinase c beta; PTEN: phosphatase and tensin homolog; PTIO: 2-phenyl-4, 4, 5, 5,-tetramethylimidazoline-1-oxyl 3-oxide (PTIO) has been used as a nitric oxide (NO) scavenger; Puma: The p53 upregulated modulator of apoptosis; PW1: paternally expressed 3 (Peg3); RNS: Reactive nitrogen species; SIRT1: sirtuin 1; SCO2: cytochrome c oxidase assembly protein; SOD2: superoxide dismutase 2; Tfam: transcription factor A mitochondrial; TIGAR: Trp53 induced glycolysis repulatory phosphatase; TNF-a: tumor necrosis factor a; TRAF2: TNF receptor associated factor 2; TRAIL: type II transmembrane protein.
Introduction
Skeletal muscle is a highly malleable tissue, capable of conforming to numerous metabolic and physiological requirements imposed by various stressors. This adaptable property of skeletal muscle is a trait that is essential for regulating cellular homeostasis in response to various stressor intensities and durations. As skeletal muscle comprises approximately 40% of the total body mass, maintaining muscle health and function is imperative to sustaining whole body health and protection against chronic disease [Citation1]. Dysfunctional skeletal muscle, as seen with chronic inactivity, aging, and tissue damage, is a precipitating cause towards conditions such as type II diabetes, cardiovascular disease, and cancer [Citation2–4]. Depending on the imposed stress, various signaling pathways may be activated and/or repressed to ensure that skeletal muscle health is preserved. Therefore, it is essential to understand the molecular mechanisms involved in the maintenance of skeletal muscle health under the demand of various oxidative stressors to ultimately determine future therapeutic modalities.
Oxidative stress is a result of the imbalance between the induction of reactive oxygen species (ROS) and the cells ability to metabolize them [Citation5,Citation6]. Many molecular signaling events characterize oxidative stress, including the production of hydrogen peroxide, superoxide, and peroxynitrite [Citation6–8]. When produced in high quantities, they place a high demand on the detoxification systems, and lead to the disruption of numerous metabolic pathways causing upwards of 200 human diseases [Citation7].
p53 is a well-known tumor suppressor protein and is commonly referred to as the ‘Guardian of the Genome’ for its role as a major determinant of cell fate. p53 regulates the expression of an assortment of genes involved in maintaining homeostasis, including those involved in cell cycle regulation, redox homeostasis (i.e. antioxidant enzyme production), DNA replication and repair, apoptosis, and autophagy [Citation9–13]. Various forms of oxidative stress lead to post-translational modifications of p53, allowing it to regulate genes to cause either beneficial outcomes, such as the upregulation of mitochondrial biogenesis, or more dysfunctional consequences such as cellular senescence and apoptosis [Citation6,Citation13–17]. The divergent effects of p53 likely depend on the degree of oxidative stress imposed. As such, delineating the role of p53 in response to various intensities and durations of oxidative stress and its ensuing regulation of skeletal muscle health is essential to determine the molecular signaling pathways involved in maintaining cellular homeostasis.
With this purpose, we set out to analyze various modalities of imposed oxidative stress and the subsequent signaling pathways activated in skeletal muscle, with a specific focus on the role of p53 in regulating these pathways. This review will describe the signaling mechanisms controlled by various stressors and how, under these influences, p53 ultimately functions to maintain muscle health and function, while additionally focusing on the pathways that lead to pathological consequences.
Methods
A systematic review of the experimental evidence was performed on the role of p53 in regulating oxidative stress in skeletal muscle. Articles published in the English language were evaluated through Pubmed, ScienceDirect, and Google Scholar search engines using the key terms ‘p53’ and ‘Skeletal Muscle’ and ‘Oxidative Stress’, while restricting against ‘Cancer’ and ‘Tumor’. Primary research studies included for comparison involve only animal and cell culture models. Important studies involving human subjects published in this area are discussed where applicable, but not compiled in the data tables for analysis in order to keep the review focused. However, a list of excluded human studies can be found in the appendix. All materials published between January 1, 1990 and March 1, 2017 were included for review.
Studies were initially screened based on title; duplicate articles and articles not publishing original research were excluded. Studies were then screened by a review of abstracts fitting the appropriate inclusion criteria (). If abstracts fit the criteria, a complete full-text analysis was performed using similar inclusion criteria. A total of 578 studies were included for review, and following exclusion, 31 studies remained for further analysis ().
Table 1. Inclusion and exclusion criteria. At the various stages of study analysis for inclusion in review, papers were included and excluded based on the above criteria. At the point of study inclusion, some papers satisfied the requirement of more than one form of oxidative stress.
Figure 1. Layout of systematic analysis for literature inclusion. Various databases were utilized to identify all studies published between 1990 and 2017 in which an animal or cell culture models under imposed oxidative stress were assessed to examine the effects on skeletal muscle and downstream proteomic markers. We screened 578 articles published in the English language; 38 were eligible for critical appraisal resulting in a total of 31 articles to be included in this review.
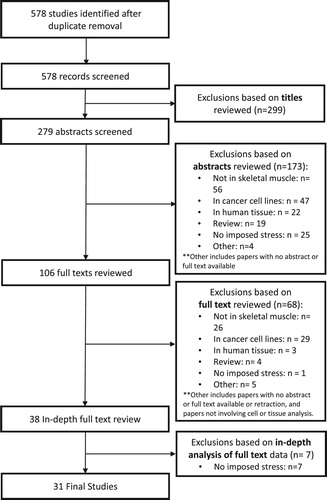
Studies were organized based on the oxidative stress intervention and categorized into one of six groups: exercise, diet modification, tissue manipulation, chemical and medicinal agents, irradiation, and oxygen deprivation. A limited number of studies fulfilled more than one category. Data extraction was performed to assess study methodology, the effect of upstream regulators and downstream targets of p53-regulated signaling pathways affected by the oxidative stress, and the application and relevance of the model. Qualitative analyses were performed to compare within and between interventions on the effectiveness of the model based on the induction of oxidative stress, and the role of p53 and other signaling mechanisms in regulating stress intensity and duration. A meta-analysis was not performed on this data.
Results
The role of p53 in mediating oxidative stress imposed by acute and chronic exercise
Acute and chronic contractile activity triggers a plethora of signals that induce beneficial metabolic and biochemical adaptations to enhance muscle health and performance. For example, the moderate increases in ROS produced by exercise are able to repair and strengthen the oxidative capacity of the cell by increasing mitochondrial content and fuel oxidation, and thus has dissimilar effects compared to the consequences of chronic, high levels of ROS [Citation18–20]. In this review, a total of seven exercise studies (two acute, five chronic) have been analyzed to establish the role of p53 in regulating this physiologically beneficial form of oxidative stress to augment muscle health ().
Table 2. Literature compiled on oxidative stress imposed through various exercise modalities.
One bout of acute exercise is sufficient to initiate transcriptional signaling towards mitochondrial biogenesis, and thus ultimately improves the oxidative capacity of skeletal muscle with the assistance of p53 [Citation21–23]. Increased activity of upstream kinases such as AMPK and p38 MAPK leads to Ser15 phosphorylation of p53, allowing for both nuclear and mitochondrial localization [Citation12,Citation22,Citation24]. In the nucleus, p53 binds to response elements within the promoter of PGC-1α leading to the expression of nuclear genes encoding mitochondrial proteins (NUGEMPs) such as Tfam, COX-IV, SCO2, and AIF [Citation12,Citation17,Citation22–26]. A recovery period of ∼3 h further improves signaling events (i.e. nuclear p53 localization and transcriptional regulation) to enhance mitochondrial biogenesis marker expression in both rodent and human models [Citation24,Citation27,Citation28]. p53 localization to mitochondria enhances mitochondrial biogenesis in both subsarcolemmal and intermyofibrillar populations through its exonuclease activity on mtDNA, and by increasing the expression and binding of Tfam and Polg1 to mtDNA to maintain genomic integrity [Citation12,Citation22,Citation24,Citation29–31].
In contrast to acute exercise, training is classified as repeated bouts of endurance exercise interspersed with recovery sessions over a period of time. The result of chronic exercise is a heightened adaptive state in which the signaling response to each exercise bout is attenuated, including reduced ROS production [Citation3]. This adaptation consists of increased mitochondrial content and improved mtDNA integrity, reduced telomere shortening and consequentially decreased cellular senescent and apoptotic signaling, less emphasis on glycolytic energy utilization and more on oxidative phosphorylation, reduced lactate production, and ultimately improved VO2 max and skeletal muscle performance [Citation11,Citation18,Citation26,Citation32,Citation33]. These beneficial adaptations, observed through the implementation of knockout (KO) models, are dependent on increased p53 nuclear and mitochondrial abundance under variable chronic exercise regimens. Despite the fact that these beneficial alterations in cellular milieu can be optimized to enhance skeletal muscle health based on p53 signaling activation and localization, the absence of p53 does not seem to necessarily hinder the ability to adapt with exercise [Citation11,Citation18,Citation33]. Though there is a reduced exercise capacity in p53 knockout mice, there is a similar increase in mitochondrial content compared to wildtype (WT) mice, indicating that exercise provokes the overlapping of redundant signals to ultimately induce the observed adaptations in mitochondria with training [Citation11].
The subcellular localization of p53 is indispensable for exercise-mediated mtDNA repair and mitochondrial biogenesis, notably through the initiation of the Tfam-Polg1 complex in the mitochondria, independent of the nuclear regulation of TIGAR, SCO2, p53R2, and FDXR expression [Citation26,Citation32]. An interesting controversy arises, however, with human models wherein short interval training (SIE), more than continuous exercise (CE), increases p53 Ser15 phosphorylation and PGC-1α nuclear localization [Citation27]. Indeed, higher training volumes result in increased p53 content, as well as mitochondrial biogenesis and augmented respiration [Citation34]. However, another study reported no differences in signaling or gene expression responses when high and lower intensity exercise regimens were performed once matched for workload [Citation35]. Therefore, further examination into the variable parameters associated with exercise, including sex, age, exercise protocol, and muscle type is required.
The role of p53 in mediating oxidative stress imposed by diet modification
There is ample evidence confirming that the modification of nutritional resources can alter physiological equilibria leading to specific muscle adaptations. As such, diet modification is considered a cellular stressor through its disruption of homeostasis. p53 is an essential upstream regulator of metabolism whereby it specifically transcribes TIGAR, SCO2, HK2, GLS2, and GLUT 1 and 4 genes [Citation36,Citation37]. Upregulation of these genes enhances oxidative phosphorylation for energy production and reduces the emphasis on glycolysis in order to prevent the Warburg effect, a hallmark of cancer metabolism [Citation36,Citation37]. In this review, a total of six studies (two diet modification, two caloric restriction, and two fasting) were analyzed to determine the signaling mechanisms involved with diet modification and how they may be regulated by p53 ().
Table 3. Literature compiled on oxidative stress imposed through various dietary modifications.
Caloric restriction extends longevity by reducing metabolic risk factors including blood pressure, serum fasting glucose, and total cholesterol [Citation38–40]. Furthermore, in humans, reduced carbohydrate/glycogen availability, with the addition of exercise, maximizes beneficial adaptations by inducing a glycogen-mediated effect on p53 signaling leading to increased transcription of mitochondrial biogenesis markers [Citation41–43]. When deprived of dietary nutrients, the body derives glucose from liver glycogen stores which quickly deplete, causing a metabolic switch in peripheral organs such as muscle to utilize fatty acids from adipose tissue as their primary energy source [Citation38,Citation44–46]. This switch in metabolic fuel usage is mediated by numerous upstream regulators, one of which is p53. The upregulation of p53 in response to fasting-induced oxidative stress enhances both antioxidant production and fatty acid oxidation through the specific mechanisms detailed below.
Glucose withdrawal upregulates nitric oxide production and activates ATM kinase through Ser1987 phosphorylation [Citation47–49]. This initiates the cellular stress-response pathway by Ser18 phosphorylation of p53 leading to increased nuclear localization [Citation38,Citation49]. When p53 is bound to PGC-1α in the nucleus, it can control Lipin-1 expression, a regulator of fatty acid metabolism, to increase fatty acid oxidation (FAO) and triglyceride synthesis, as well as upregulate TIGAR to reduce the emphasis on glycolytic energy production [Citation38,Citation49–52]. p53 can also upregulate GLS2 expression to catalyze the conversion of glutamine to glutamate and increase α-ketoglutarate to enhance mitochondrial respiration and ATP production [Citation49,Citation53]. This occurs concomitantly with an enhancement in antioxidant defences through the upregulation of GSH content which directly (reacts with ) and indirectly (revitalizing antioxidants) scavenges ROS [Citation49,Citation53,Citation54]. In addition to the regulation of metabolic fuel pathways, p53 also controls specific cell signaling pathways. When in the nucleus, phosphorylated p53 can interact with PGC-1α to enhance Nrf2 and SOD2 antioxidant expression, concomitant with sestrin activation, to buffer the harmful effects of ROS/RNS to protect the cell during moderate to prolonged fasting states [Citation48,Citation49]. Furthermore, with fasting there is an upregulation of p21 and Gadd45a cellular senescent regulators, and a decrease in apoptotic mRNA such as bok, puma, noxa, and dr5 [Citation44]. Therefore, during fasting or under reduced glucose conditions, p53 leads to an upregulation of antioxidant production, cellular senescence, and FAO, while reducing glycolytic and apoptotic signaling.
On the other hand, the effect of chronic supplementation of metabolic fuels in excess to a normal diet enhances vascular oxidative stress leading to the development of pathological conditions including obesity, diabetes, and coronary artery disease [Citation55,Citation56]. With elevated glucose and fatty acid consumption, there is an increase in endothelial cell p53 expression via reduced Mdm4 negative regulation of p53 [Citation56,Citation57]. p53 accumulation can prevent the phosphorylation of eNOS via Akt downregulation by PTEN, while also reducing the expression of PGC-1α and its downstream targets NRF-1 and Tfam, which are involved in vascular angiogenesis and mitochondrial biogenesis, respectively [Citation56,Citation57]. Reductions in additional PGC-1α-target genes involved in FAO including Acadl, Acadm, and Cpt1b can lead to fat accumulation [Citation58]. p53 upregulation can also reduce insulin sensitivity by downregulating endothelial GLUT1 through direct transcriptional repression, causing impaired glucose uptake which can lead to oxidative stress by preventing substrate availability for energy production [Citation56,Citation59–61]. Interestingly, the deletion of endothelial p53 inhibits the diet-induced downregulation of GLUT1 expression in these cells to improve glucose uptake into skeletal muscle [Citation56]. In addition to reducing GLUT1 expression, p53 has an inhibitory effect on the GLUT4 promoter within skeletal muscle, suggesting that p53 can negatively regulate insulin sensitivity in this tissue and induce insulin resistance [Citation59]. However, with malignancy-induced mutations in the p53 DNA-binding domain, p53 repression on the GLUT1 and 4 promoters is lifted leading to enhanced insulin sensitivity and glucose tolerance to facilitate the Warburg effect for tumorigenesis [Citation37,Citation59].
Another dietary modification includes the addition of toxic substances into the diet, such as alcohol in excessive quantities, which can lead to reduced contractile proteins (myosin, desmin, actin, troponin) in the muscle itself causing alcoholic myopathy, defined by cramps, impaired muscle strength, difficulties in gait, and reduced lean tissue mass [Citation62–64]. Interestingly, the expected increase in the expression of p53 and Bcl-2-family proteins with increasing ethanol dosage does not occur. Instead, there is an activation of the proto-oncogene c-myc by acetaldehyde, specifically leading to HSP70 binding and initiation of the unfolded protein response and apoptotic cellular stress pathways, either dependent or independent of p53 [Citation62,Citation65,Citation66]. Excess consumption of almost any macronutrient can cause debilitating consequences, and therefore illuminating the toxic pathways activated in mediating this oxidative stress, as well as those that may combat it, are essential to developing therapeutic modalities for these diet-induced pathologies.
The role of p53 in mediating oxidative stress imposed by pathological tissue manipulation
In contrast to the effect of healthy physical activity on skeletal muscle, this tissue is also subject to reduced activity levels brought about by trauma, denervation, or immobilization. With the knowledge that p53 plays a role in regulating overall cellular homeostasis, the intensity of the various physical manipulations of this kind may lead to alternate functions required of p53 to mediate or attenuate this stress. A total of five studies have been selected to assess the potential variations in p53-mediated regulation of muscle adaptations under these conditions ().
Table 4. Literature compiled on oxidative stress imposed through various modalities of tissue manipulation.
The most common manipulations of muscle include crush injury, tissue autografting, disease, and burn injury which can lead to direct skeletal muscle atrophy, or indirect atrophy through immobilization [Citation67–72]. Tissue crush injury, involving a significant disruption to muscle structure but preserving blood and nerve supply, leads to impaired myotube formation, a greater percentage of necrotic tissue, and increased desmin protein which is exaggerated in the absence of p53 [Citation68,Citation70]. However, p53 does not seem to be required for muscle regeneration following injury since its other family members, p63 and p73, compensate for its loss [Citation70]. Major trauma or full-thickness burn injury can induce significant metabolic dysfunction (hyperglycemia, insulin resistance, lactate production) which contributes to inflammation, increased catabolism, and ultimately muscle wasting [Citation71,Citation73]. Though p53 is known to regulate the inflammatory response and apoptosis, its role in mediating the cellular responses to major trauma has not yet been examined. One study revealed that three days-post burn injury increased the cytokine-mediated activation of iNOS to produce NO as a defence mechanism, leading to the inactivation of SIRT1 and the subsequent activation of p65 NF-kB and p53 [Citation71]. This activation initiates the inflammatory response by increasing TNF-α and IL-1β expression, while concomitantly increasing insulin resistance and apoptosis respectively, to induce muscle wasting [Citation71,Citation74]. Another important and more common cause of muscle atrophy is immobilization. The immobilization-induced increase in p53 allows it to function as a key ATF-4-independent mediator of muscle atrophy, leading to direct p21 activation and subsequent tissue atrophy of all fiber types through cell cycle-dependent mechanisms [Citation68]. Additionally, results from hindlimb suspension models of inactivity have shown increased cytosolic Id2 protein and p53 levels, which play a role in apoptosis-related atrophy [Citation74,Citation75]. Interestingly, atrophic stimuli affect skeletal muscle fiber types differently, with cachexia-associated atrophy leading to a greater reduction in slow fiber type size, while fast fiber types are more susceptible to atrophy when p53 is removed, possibly through the upregulation of the atrogin-1 ubiquitin ligase-mediated pathway [Citation68,Citation74].
Additional roles for p53 can be observed in disease settings that induce pathological muscle degeneration, such as seen with diabetes and cancer cachexia. A precursor to many diseases, diabetes leads to cellular stress by reducing proteasomal degradation of p53 through the inhibition of the negative Mdm2/Akt upstream regulatory pathway, leading to an abnormal accumulation of p53 and the induction of apoptosis [Citation67]. Interestingly, increased p53 expression in adipose tissue appears to assist in reducing adiponectin levels, causing the development of insulin resistance and type II diabetes [Citation67,Citation76]. On the other hand, with cancer cachexia, TNF-α-mediated inhibition of myogenin and MyoD, and activation of p53-dependent cell death proteins, leads to the inhibition of myogenic differentiation via upregulation of atrogin-1 to enhance apoptosis and atrophy [Citation68,Citation77,Citation78]. Therefore, understanding the global role of p53 within the organism and how it contributes to muscle wasting with pathological tissue manipulations such as these may lead to the identification of more effective therapeutics. Treatment modalities such as statins (atorvastatin) for cardiovascular disease and diabetes, or progestagens for cancer cachexia, in addition to physical rehabilitation during hospitalization, can ultimately function to reduce muscle wasting symptomatology and associated comorbidities [Citation67,Citation79].
The role of p53 in mediating oxidative stress imposed by oxygen deprivation
Cellular respiration, an essential metabolic process for energy generation, requires oxygen as the terminal electron acceptor in oxidative phosphorylation to produce ATP energy. Low oxygen tension denotes a hypoxic microenvironment which may be induced by mechanisms such as ischemia or high-altitude exposure, causing detrimental physiological consequences [Citation80–82]. p53, a regulator of mitochondrial metabolism, is suspected to play a major role in compensating for this insult and may be able to provide protective adjustments against cellular damage. We examined two studies that were designed to elucidate the role of p53 in regulating the cellular responses to hypoxic stress ().
Table 5. Literature compiled on oxidative stress imposed through oxygen deprivation.
Hypoxia, defined as ∼1% of physiological oxygen volume in skeletal muscle following an ischemic insult, represses myogenesis through numerous transcriptome changes [Citation80,Citation83]. One study found that hypoxia upregulated 641 genes involved in the cell cycle and in metabolism (HIF1-α and glycolysis), and downregulated 224 genes involved in protein catabolism and muscle organ development [Citation80]. Of the specific genes upregulated, p53 expression was enhanced, with further augmentation of its downstream cell cycle, metabolic, and antioxidant signaling pathways to protect against oxidative-induced cell death [Citation80,Citation84].
It is well known that hypoxia inhibits myogenin and myogenic differentiation [Citation81,Citation85]. Recently, the mechanisms regulating this pathway have been elucidated and are dependent on p53 activation in a time-sensitive manner. Increased Ser37, Ser46, and Ser392 phosphorylation and decreased Lys373 acetylation occur at 5-h of anoxic exposure (complete oxygen deprivation), with additional phosphorylation at Ser6, Ser9, Ser15, and Ser20 at 20 h [Citation84]. These post-translational modifications enhance p53 nuclear localization where it functions as a transcription factor of numerous negative regulators of myogenic differentiation, including those involved in cell cycle arrest, DNA repair, and apoptosis, which were observed to increase following anoxia [Citation84]. Furthermore, p53 is necessary for the induction of the helix–loop–helix transcription factor Bhlhe40 which inhibits myogenin-driven terminal differentiation by reducing the binding of MyoD to the MyoG promoter [Citation80]. Under hypoxic conditions specifically, p53 can upregulate Bhlhe40 expression to reduce the differentiation of myoblasts [Citation80]. This repression of differentiation is additionally impacted by Notch activation, which translocates to the nucleus to reduce the activation of MyoD by creating interfering Hes1 and Hey1 heterodimers [Citation85–87]. Repression is further accelerated by MyoD degradation through Bhlhe40 disruption of its binding to the myogenin promoter and through inhibition of the PI3K cell cycle progression and survival pathway [Citation85,Citation86,Citation88,Citation89]. Therefore, p53 plays a role in regulating the repression of myogenesis under hypoxic exposure. This may serve to allocate time towards upregulating stress–response genes involved in cell cycle repair and maintenance to enhance the prospect for cell survival and to transition the organism into hypometabolism with chronic oxygen deprivation.
The role of p53 in mediating oxidative stress imposed by radiation
Ionizing radiation energy is an extreme stress signal which, depending on the length of exposure, can lead to cell death via two mechanisms. The first is through p53-dependent and -independent initiation of apoptosis which serves as a protective mechanism to eliminate heavily damaged or mutated cells [Citation90]. The second mechanism is through the induction of irreversible G1/S cell cycle arrest which is dependent on ATM and p53, as well as its downstream target p21 [Citation90,Citation91]. Though the effect of radiation-induced death has been studied in many tissues, whether similar mechanisms are activated in skeletal muscle remains unknown. Further, the radiation dose and exposure time required to activate these signaling pathways in muscle tissue are under-discussed. Therefore two studies, measuring the effects of irradiation (5 Gy) exposure and its activation of the p53 signaling pathway were examined in this review ().
Table 6. Literature compiled on oxidative stress imposed through irradiation.
The role of p53 on skeletal muscle differentiation following DNA damage is a field of interest because of its implications for the regenerative capacity of muscle. Previous work has revealed that p53 maintains the genetic stability of mouse embryonic stem (MES) cells following DNA damage by repressing Nanog, a cancer-stem cell marker, to then regulate and promote differentiation [Citation92,Citation93]. Regulation of MES cells is of key importance since skeletal muscle originates through myogenesis from the mesoderm, one of the three primary germ layers. Depending on the exposure time and intensity, p53 activation is required for cytokine-mediated inhibition of myogenic differentiation via TNF-α-driven apoptotic augmentation, leading to muscle atrophy [Citation77,Citation92,Citation94]. Therefore, analysis of the regulation of key myogenic regulatory factors by p53 was examined under irradiation stress. The results indicate a direct role for p53 transcriptional repression of myogenin, with the likely purpose of ensuring adequate time for DNA damage repair and chromosomal segregation [Citation92]. Furthermore, a time course was evaluated leading to the conclusion that a two-phase conditional regulation by p53 exists. Under non-ionizing radiation, p53 exerts low to minimal repression of the myogenin promoter, whereas under greater stress duration, p53 switches to become a negative regulator of myogenin to prevent aberrant cell proliferation [Citation92].
Coordinated commitment to cell division is clearly monitored by p53-mediated intrinsic and extrinsic stress signals, such as DNA damage, hypoxia, low levels of ribosomal biogenesis, or oncogene activation, which introduces infidelity to cellular division through the inhibition of specific signals. Repression of such signals including the IGF-1-Akt and mTOR pathways allows for the coordinated activation of autophagy and apoptosis in a tissue-specific manner [Citation95–98]. Ultraviolet radiation can slow ribosomal biogenesis and DNA damage, the effects of which lead to p53 activation and subsequent upregulation of downstream targets such as PTEN and IGF-BP3 [Citation95,Citation96]. These proteins function to shut down the IGF-1–AKT–mTOR signaling mechanisms in skeletal muscle to coordinate autophagy and apoptosis [Citation95,Citation96]. These results have implications for chronic physiological stress accumulating throughout a lifetime and indicate a role for persistent p53 repression of myogenin and cellular energy regulating pathways (IGF-1–Akt–mTOR) that may cumulatively lead to the development of myopathic diseases and aging overtime, and consequentially inducing muscle atrophy [Citation92,Citation95].
The role of p53 in mediating oxidative stress imposed by chemical or medicinal agents
To further understand the activation of p53 and its downstream signaling pathways, various naturally occurring and synthetically manufactured drugs have been examined. These chemical agents, depending on whether they are direct or indirect oxidizing agents, trigger specific mechanisms to combat oxidative stress. Therefore, to determine how these oxidizing agents lead to variable signaling mechanisms, particularly stress signals activated by p53, a total of 11 studies (5 direct oxidizing agents and 6 indirect oxidizing agents) were analyzed ().
Table 7. Literature compiled on oxidative stress imposed through various types of chemical or medicinal oxidizing agents.
Direct oxidizing agents crucially impact metabolism and cellular homeostasis by acting as the final electron acceptor for many reactions. Numerous pharmacologic agents have been naturally discovered or synthetically constructed that fit the description of a direct oxidizing agent and consequentially play an important role in inducing or mediating oxidative stress through the activation of p53. Harmful oxidizing agents, such as H2O2, increase p53 expression which can activate kinases PKCβ and JNK to phosphorylate p66Shc on its Ser36 residue, allowing for its translocation to the mitochondria [Citation99–101]. Once in the mitochondria, p66Shc functions as a redox enzyme to amplify oxidative stress by provoking structural changes, such as mitochondrial outer membrane permeabilization, to further increase H2O2 generation and the release of cytochrome c[Citation99,Citation100]. p66Shc, when activated by H2O2, also inhibits the ERK signaling pathway, leading to inappropriate actin cytoskeleton polymerization and irregular glucose transport into skeletal muscle [Citation99,Citation102,Citation103]. Under this form of oxidative stress, ERK is also known for abrogating the access of FOXO3a to DNA-binding sites by phosphorylating its threonine and serine residues [Citation92,Citation104]. Specific FOXOs that are activated with H2O2 include the JNK-mediated Thr447 and Thr452 phosphorylation of FOXO3a and FOXO4, respectively, which can then upregulate pro-apoptotic proteins Fas, TRAIL, and Bim [Citation104–106]. Phosphorylation of p53 under H2O2 oxidative stress additionally leads to the transcription of pro-apoptotic proteins, including Bax, BH3-only family members, and PERP [Citation99,Citation104,Citation107,Citation108]. Antimycin A, another inducer of oxidative stress, functions by inhibiting the mitochondrial respiratory chain at complex III [Citation105]. This activates alternative stress-resistant pathways through the SIRT1-mediated de-acetylation of p53 and concomitant FOXO activation [Citation105,Citation109]. Furthermore, this enhances the expression of ROS-detoxifying enzymes such as SOD2 and catalase [Citation105,Citation109]. Snake venom neotoxin/cardiotoxin, another direct oxidative stress inducer, elevates ROS to induce p38 MAPK-mediated Ser33 phosphorylation of p53 [Citation110,Citation111]. This leads to persistent senescence through Notch and Wnt signaling [Citation88,Citation110,Citation111]. Interestingly, certain agents such as resveratrol, α-B-crystallin, E2 (17β-estradiol), and testosterone can protect against the H2O2-mediated p53 induction of apoptosis by enhancing the de-acetylation of p53 by SIRT1 or by binding certain pro-apoptotic family members [Citation99,Citation104,Citation105,Citation112]. p53 de-acetylation inhibits the transcription and subsequent synthesis of caspase 3 and Bax, and downregulates Puma and PERP which are upstream activators of Bax-dependent apoptosis [Citation99,Citation104,Citation105,Citation112]. This ultimately reduces ROS generation and p53-induced apoptosis.
Indirect oxidizing agents function through additional and alternate pathways to ultimately lead to similar results as direct oxidizing agents. Synthetic agents, in particular, have been created to mediate ROS production and regulate p53 signaling pathways depending on the pathology. For example, in diabetic hyperglycemia, p53 accumulates and activates downstream genes involved in myocyte death [Citation113,Citation114]. Desferrioxiamine, a hypoxia mimetic, leads to a similar accumulation of p53 as seen with diabetes, ultimately to activate apoptosis [Citation67]. Atorvastatin, on the other hand, is a medicinal statin used to treat diabetes and high cholesterol, and serves to reduce p53 accumulation by increasing IRS-1/Akt phosphorylation of Mdm2 to upregulate p53 ubiquitination and degradation [Citation67,Citation115].
Specific indirect oxidizing agents, such as PTIO, LY-83583, and buthionine sulfoximine, induce oxidative stress through the upregulation of NO/iNOS/nNOS and/or GSH depletion pathways [Citation48,Citation73,Citation116]. As a first response compensatory mechanism, the antioxidant pathway is upregulated to control this redox imbalance before stress can consume the cell and initiate apoptosis. The antioxidant defence system is activated by the inflammatory cascade initiated by GSH decrements and increased NO/cGMP which leads to the activation of p53 [Citation48,Citation73,Citation116]. Activated p53 is a result of either increased inhibition of its negative regulators Mdm2 and SIRT1, or through Cys124 S-nitrosylation of p53 due to NO flux, allowing it to localize to the nucleus [Citation48,Citation73,Citation116]. Once in the nucleus, p53 binds to the PGC-1α promoter to enhance its transcription [Citation48,Citation116,Citation117]. Both p53 and PGC-1α then act synergistically to co-activate the Nrf2-mediated antioxidant response to buffer harmful ROS/RNS [Citation48,Citation117]. However, as stress progresses and cannot be alleviated by increased antioxidant production, pre-apoptotic pathways such as the inflammation pathway, are activated. Specifically, the p53 gene target PW1 physically interacts with TRAF2 to activate the cytokine TNF-α–NF-kB pathway under chemically induced oxidative stress [Citation68,Citation118]. This provokes protein degradation and prevents muscle differentiation by upregulating the ubiquitin-proteasome pathway via atrogin-1, and additionally initiating apoptosis by enhancing bax mitochondrial localization [Citation68,Citation78,Citation119]. Depending on the length of chemical activation, chronic exposure can lead to SIRT1-mediated de-acetylation of PGC-1α, p53-mediated regulation of damage associated molecular patterns (DAMPs), or increased binding between p53 and MURF1 to trigger ubiquitination [Citation48,Citation73]. These changes ultimately lead to progressive inflammation, premature atrophy, and cell death [Citation48,Citation73,Citation116,Citation120]. For example, chronic exposure to environmental contaminants such as polycyclic aromatic hydrocarbon benzo(α)pyrene and C60 fullerene pollutants act to initially increase antioxidant defences by increasing glutathione levels [Citation121,Citation122]. However, as the oxidative stress consumes these defences, these pollutants switch to act as genotoxins by producing hydroxyl and superoxide anion radicals that react with DNA, triggering DNA strand breaks which then activate p53-mediated induction of apoptosis [Citation121,Citation122]. Therefore, both direct and indirect oxidizing chemicals provide insight on the p53-mediated regulation of low to high levels of oxidative stress, and elucidate some of the mechanisms activated with various drugs.
Summary and conclusion of cited studies
Oxidative stress in skeletal muscle is a result of impaired redox homeostasis due to either the overproduction of ROS, or a compromised antioxidant defence system. ROS overproduction, caused by either endogenous or exogenous sources, can lead to damaged proteins, lipids, and DNA, inevitably causing pathologies such as metabolic (type II diabetes and cancer), neuromuscular (dystrophies), neural (Alzheimer’s disease) or cardiovascular diseases, as well as promoting muscle aging [Citation123,Citation124]. Endogenous sources of ROS, such as those produced by exercise, as well as exogenous sources, such as excess nutrient availability, radiation exposure, hypoxia, and tissue manipulations, can lead to p53 subcellular localization for the regulation of specific signaling pathways to alleviate the imposed oxidative stress and restore cellular homeostasis ().
Figure 2. p53 subcellular localization induced by oxidative stressors. p53 is normally maintained at low levels in the cytosol by its negative regulator Mdm2. When Mdm2 dimerizes, p53 is poly-ubiquitinated and targeted for degradation by the 26S proteasome [Citation67,Citation115]. With the induction of stress, p53 post-translational modifications allow for its subcellular redistribution. Exercise. Muscular contractions induce the activation of numerous signals, such as kinase phosphorylation, increased ROS production, enhanced cytosolic calcium concentration, and an increased AMP: ATP ratio, ultimately leading to p53 Ser15 phosphorylation [Citation12,Citation17,Citation22]. Once activated, p53 localizes to the nucleus to increase NUGEMP transcription, and to the mitochondria to regulate mtDNA transcription. p53 additionally regulates energy metabolism and DNA repair pathways [Citation26,Citation32,Citation36,Citation37,Citation42,Citation52]. Diet modification. With caloric restriction/glucose withdrawal there is an upregulation of NO which activates ATM kinase to phosphorylate p53 (Ser18) leading to its nuclear localization [Citation38,Citation49]. In the nucleus, p53 regulates energy metabolism, antioxidant, cellular senescent, and apoptotic pathways [Citation38,Citation44,Citation47–50,Citation53,Citation54]. Tissue manipulation. With immobilization and cachexia-mediated atrophy, there is p53-mediated transcriptional activation and repression of cell cycle arrest and myogenic genes, respectively [Citation68,Citation75]. With burn injuries, there is an increase in cytokine-mediated activation of iNOS to produce NO which inhibits SIRT1 leading to the activation of p53 as well as increased TNF-α and atrogin 1 expression to induce muscle wasting [Citation68,Citation71,Citation77,Citation78]. Oxygen deprivation. Acute and prolonged hypoxia induce p53 phosphorylation at numerous sites to enhance the repression of myogenic differentiation and upregulate cell cycle arrest and apoptotic pathways [Citation80,Citation84,Citation86–88]. Irradiation. Under IR stress, TNF-α driven apoptosis is induced through p53 activation, to enhance nuclear apoptotic signaling pathways in addition to increasing p66Shc phosphorylation to initiate MOMP [Citation92,Citation101]. Direct/indirect oxidizing agents. Oxidizing agents such as H2O2 lead to the activation of cell death pathways mediated by phosphorylated p53 to initiate persistent senescence and apoptosis [Citation107,Citation108,Citation111]. Furthermore, p53 activates kinases to phosphorylate p66Shc, inhibit ERK signaling, prevent cytoskeleton polymerization, and to induce MOMP [Citation99–101,Citation103]. With indirect oxidizing agents, NO/iNOS/nNOS pathways are upregulated to activate p53 by reducing the inhibition by its negative regulator SIRT1 and by S-nitrosylation of p53. This enhances nuclear localization for antioxidant and apoptotic transcription with increasing stress, and increases atrogin-1 mediated ubiquitin-proteasome activation [Citation48,Citation73,Citation78,Citation116,Citation117].
![Figure 2. p53 subcellular localization induced by oxidative stressors. p53 is normally maintained at low levels in the cytosol by its negative regulator Mdm2. When Mdm2 dimerizes, p53 is poly-ubiquitinated and targeted for degradation by the 26S proteasome [Citation67,Citation115]. With the induction of stress, p53 post-translational modifications allow for its subcellular redistribution. Exercise. Muscular contractions induce the activation of numerous signals, such as kinase phosphorylation, increased ROS production, enhanced cytosolic calcium concentration, and an increased AMP: ATP ratio, ultimately leading to p53 Ser15 phosphorylation [Citation12,Citation17,Citation22]. Once activated, p53 localizes to the nucleus to increase NUGEMP transcription, and to the mitochondria to regulate mtDNA transcription. p53 additionally regulates energy metabolism and DNA repair pathways [Citation26,Citation32,Citation36,Citation37,Citation42,Citation52]. Diet modification. With caloric restriction/glucose withdrawal there is an upregulation of NO which activates ATM kinase to phosphorylate p53 (Ser18) leading to its nuclear localization [Citation38,Citation49]. In the nucleus, p53 regulates energy metabolism, antioxidant, cellular senescent, and apoptotic pathways [Citation38,Citation44,Citation47–50,Citation53,Citation54]. Tissue manipulation. With immobilization and cachexia-mediated atrophy, there is p53-mediated transcriptional activation and repression of cell cycle arrest and myogenic genes, respectively [Citation68,Citation75]. With burn injuries, there is an increase in cytokine-mediated activation of iNOS to produce NO which inhibits SIRT1 leading to the activation of p53 as well as increased TNF-α and atrogin 1 expression to induce muscle wasting [Citation68,Citation71,Citation77,Citation78]. Oxygen deprivation. Acute and prolonged hypoxia induce p53 phosphorylation at numerous sites to enhance the repression of myogenic differentiation and upregulate cell cycle arrest and apoptotic pathways [Citation80,Citation84,Citation86–88]. Irradiation. Under IR stress, TNF-α driven apoptosis is induced through p53 activation, to enhance nuclear apoptotic signaling pathways in addition to increasing p66Shc phosphorylation to initiate MOMP [Citation92,Citation101]. Direct/indirect oxidizing agents. Oxidizing agents such as H2O2 lead to the activation of cell death pathways mediated by phosphorylated p53 to initiate persistent senescence and apoptosis [Citation107,Citation108,Citation111]. Furthermore, p53 activates kinases to phosphorylate p66Shc, inhibit ERK signaling, prevent cytoskeleton polymerization, and to induce MOMP [Citation99–101,Citation103]. With indirect oxidizing agents, NO/iNOS/nNOS pathways are upregulated to activate p53 by reducing the inhibition by its negative regulator SIRT1 and by S-nitrosylation of p53. This enhances nuclear localization for antioxidant and apoptotic transcription with increasing stress, and increases atrogin-1 mediated ubiquitin-proteasome activation [Citation48,Citation73,Citation78,Citation116,Citation117].](/cms/asset/72db3d19-7387-41d4-9595-776aac41a823/yrer_a_1416773_f0002_oc.jpg)
Depending on the type, duration, and intensity of the imposed stress, various signaling pathways are activated within specific organelles, such as the mitochondria or nucleus, to mitigate the cellular damage caused by these non-physiological levels of ROS. Specifically, p53 is deemed to be of key importance in regulating these cellular stress responses. The studies outlined in this review confirm a dual ability for p53 activation of specific signaling mechanisms, dependent on the intensity and length of the oxidative stress. With low levels of oxidative stress induced by dietary restriction, non-ionizing radiation, hypoxia, and oxidizing agents, there is an upregulation of p53-mediated activation of the antioxidant enzymes Nrf2, SOD2, sestrins, and catalase. Furthermore, cell cycle arrest and cellular senescent proteins p21 and Gadd45a are upregulated, concomitant with increased autophagy to maintain genomic integrity as well as a healthy organelle pool, respectively. Interestingly, metabolic modifications additionally occur exemplified by increased TIGAR, PGM, and GLS2 expression which reduce glycolysis and pyruvate levels, while increasing Lipin-1 to upregulate FAO. Surprisingly, with exercise, p53 enhances cellular function by increasing mitochondrial biogenesis and maintaining mtDNA utility.
Yet with greater and prolonged levels of stress, signaling pathways that exacerbate oxidative dysfunction, such as those involved in inflammatory cascades and cell death, are intensified. As observed with chronic nutrient abundance, anoxia/hypoxia, and burn injuries, there is increased expression of inflammatory proteins and cytokines including c-myc, TNF-α, IL-1β, Bhlhe4, and Notch. Immobilization of muscle on the other hand, a mechanism solely dependent on duration, is characterized by increased ATF4, p21, and atrogin-1 expression to cause tissue atrophy. With even greater levels of stress induced by radiation and chronic exposure to oxidizing agents, there is an increase in numerous signaling pathways including TNF-α-PWI-caspase-Bax, PTEN, FOXOs, p66Shc, MURF1, and IGF-BP3, which initiate the accumulation of harmful free radical species leading to cell death. In these cases, p53 acts as a pro-oxidant to further exacerbate the stress load. This culminates in the inhibition of preventative oxidative signaling pathways and enhances the caspase cascade to initiate apoptosis. Interestingly, this duality function of p53 is observed with specific oxidative stress sources such as radiation, nutrient regulation, hypoxia, and oxidizing/non-oxidizing agents, but not with exercise and tissue manipulation.
This review outlines for the first time the various modalities of imposed oxidative stress in skeletal muscle that analyze the regulation of p53 and its numerous downstream signaling pathways involved in maintaining cellular homeostasis (). Clearly the integration of p53 into regulatory networks that combat oxidative stress has allowed this protein to earn its title as the ‘Guardian of the Genome’. This exciting avenue of research concerning the regulation of oxidative stress by varying mechanisms activated by p53 will significantly contribute to existing knowledge on how p53 can be therapeutically targeted to treat numerous disorders and diseases for which oxidative stress is a major contributory factor.
Figure 3. p53 nuclear and mitochondrial localization and regulation of signaling mechanisms with oxidative stress. With the induction of stress, p53 post-translational modifications allow it to localize to various cellular compartments to regulate cellular homeostasis. Exercise. Once phosphorylated, p53 localizes to the mitochondria to assist Tfam and Polγ binding to mtDNA, and to increase 16S rRNA production to enhance mitochondrial biogenesis [Citation12,Citation17,Citation24,Citation26,Citation29,Citation30]. p53 also translocates to the nucleus to increase PGC-1α expression and activate NUGEMPs transcription [Citation12,Citation17,Citation22–26,Citation48]. Furthermore, p53 can regulate glycolysis by reducing glycolytic energy production and enhancing the pentose phosphate, fatty acid oxidation, and oxidative phosphorylation energy pathways [Citation26,Citation36,Citation37,Citation42,Citation52]. Diet Modification. With diet modifications (DM), there is a p53-mediated upregulation of energy metabolism genes to reduce glycolysis and upregulate oxidative phosphorylation [Citation36,Citation37]. With caloric restriction (CR) or glucose withdrawal, p53 co-localizes in the nucleus with PGC-1α to enhance Lipin-1 expression for increased FAO. p53 upregulates enzymes for mitochondrial substrate provision, increases antioxidant defenses, increases cellular senescent and cell cycle arrest genes, and decreases apoptotic gene transcription [Citation38,Citation44,Citation47–50,Citation53,Citation54]. On the other hand, excess caloric intake leads to p53 transcriptional repression (exemplified by red X) of GLUT 1 and GLUT 4 genes, reduces PGC-1α and NUGEMP activation, and impairs FAO gene transcription, leading to impaired energy utilization and fat accumulation [Citation56–59,Citation61]. Tissue Manipulation. With immobilization there is an p53-mediated transcription of cell cycle arrest genes as well as an increase in Id2 to decrease DNA-binding and transcriptional activity [Citation68,Citation75]. Oxygen Deprivation. Hypoxia induces p53 phosphorylation at numerous sites leading to cell cycle arrest and apoptotic transcriptional activation. Irradiation. Under IR stress, p53 increases p66Shc phosphorylation, to coordinate autophagy and apoptosis, in order to regulate cellular integrity [Citation101,Citation103]. Direct/Indirect Oxidizing Agents. Oxidizing agents, such as H2O2, lead to the activation of cell death pathways mediated by phosphorylated p53 to initiate persistent senescence and to enhance transcription of pro-apoptotic proteins such as Bax and PERP [Citation107,Citation108,Citation110,Citation111]. With indirect oxidizing agents, p53 nuclear localization increases antioxidant transcription. With prolonged exposure apoptosis can be induced [Citation48].
![Figure 3. p53 nuclear and mitochondrial localization and regulation of signaling mechanisms with oxidative stress. With the induction of stress, p53 post-translational modifications allow it to localize to various cellular compartments to regulate cellular homeostasis. Exercise. Once phosphorylated, p53 localizes to the mitochondria to assist Tfam and Polγ binding to mtDNA, and to increase 16S rRNA production to enhance mitochondrial biogenesis [Citation12,Citation17,Citation24,Citation26,Citation29,Citation30]. p53 also translocates to the nucleus to increase PGC-1α expression and activate NUGEMPs transcription [Citation12,Citation17,Citation22–26,Citation48]. Furthermore, p53 can regulate glycolysis by reducing glycolytic energy production and enhancing the pentose phosphate, fatty acid oxidation, and oxidative phosphorylation energy pathways [Citation26,Citation36,Citation37,Citation42,Citation52]. Diet Modification. With diet modifications (DM), there is a p53-mediated upregulation of energy metabolism genes to reduce glycolysis and upregulate oxidative phosphorylation [Citation36,Citation37]. With caloric restriction (CR) or glucose withdrawal, p53 co-localizes in the nucleus with PGC-1α to enhance Lipin-1 expression for increased FAO. p53 upregulates enzymes for mitochondrial substrate provision, increases antioxidant defenses, increases cellular senescent and cell cycle arrest genes, and decreases apoptotic gene transcription [Citation38,Citation44,Citation47–50,Citation53,Citation54]. On the other hand, excess caloric intake leads to p53 transcriptional repression (exemplified by red X) of GLUT 1 and GLUT 4 genes, reduces PGC-1α and NUGEMP activation, and impairs FAO gene transcription, leading to impaired energy utilization and fat accumulation [Citation56–59,Citation61]. Tissue Manipulation. With immobilization there is an p53-mediated transcription of cell cycle arrest genes as well as an increase in Id2 to decrease DNA-binding and transcriptional activity [Citation68,Citation75]. Oxygen Deprivation. Hypoxia induces p53 phosphorylation at numerous sites leading to cell cycle arrest and apoptotic transcriptional activation. Irradiation. Under IR stress, p53 increases p66Shc phosphorylation, to coordinate autophagy and apoptosis, in order to regulate cellular integrity [Citation101,Citation103]. Direct/Indirect Oxidizing Agents. Oxidizing agents, such as H2O2, lead to the activation of cell death pathways mediated by phosphorylated p53 to initiate persistent senescence and to enhance transcription of pro-apoptotic proteins such as Bax and PERP [Citation107,Citation108,Citation110,Citation111]. With indirect oxidizing agents, p53 nuclear localization increases antioxidant transcription. With prolonged exposure apoptosis can be induced [Citation48].](/cms/asset/3dd6e755-6350-44f2-b234-dbcc03aa6c66/yrer_a_1416773_f0003_oc.jpg)
Acknowledgements
We thank our colleagues around the world who have dedicated their expertise towards pursuing an understanding of p53 in skeletal muscle.
Disclosure statement
The authors of this paper have no conflicts of interest to disclose.
Notes on contributors
Kaitlyn Beyfuss attended McMaster University and graduated in 2014 with an Honours Bachelor degree in Life Sciences. She then successfully obtained a clinical research assistant position at Sunnybrook Health Sciences Centre specializing in surgical oncology. Her interest in pursuing wet lab research led her to obtain a Master’s of Science position in Dr. David Hood’s lab focusing on mitochondrial biogenesis and exercise, with further specialization in p53 signaling. She is currently completing the second and final year of her Master’s degree.
Dr. David Hood attended Queen’s University and graduated with a degree in Physical and Health Education. His interest in pursuing an academic career led him to complete his M.Sc. in Exercise Physiology at Dalhousie University. Following this, he completed his Ph.D. at the State University of New York with Dr. Ronald Terjung on muscle amino acid metabolism with contractile activity. He then pursued a post-doctoral fellowship with Dr. Dirk Pette investigating mitochondrial gene expression in muscle. He was successful in obtaining a faculty position at the Department of Physical Education at York University, and initiated the Muscle Health Research Centre.
ORCID
Kaitlyn Beyfuss http://orcid.org/0000-0003-0147-6767
Additional information
Funding
References
- Drake JC, Wilson RJ, Yan Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J 2016;30(1):13–22. doi: 10.1096/fj.15-276337
- Carter HN, Chen CWC, Hood DA. Mitochondria, muscle health, and exercise with advancing age. Physiology. 2015;30(3):208–223. doi: 10.1152/physiol.00039.2014
- Ljubicic V, Joseph AM, Adhihetty PJet al. Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging. 2009;1(9): 818–830. doi: 10.18632/aging.100083
- Warburton DE, Charlesworth S, Ivey A, et al. A systematic review of the evidence for Canada's physical activity guidelines for adults. Int J Behav Nutr Phys Act. 2010;7: 39. doi: 10.1186/1479-5868-7-39
- Valko M, Leibfritz D, Moncol J, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1): 44–84. doi: 10.1016/j.biocel.2006.07.001
- Liu B, Chen Y, St. Clair DK. Ros and p53: a versatile partnership. Free Radical Bio Med. 2008;44(8):1529–1535. doi: 10.1016/j.freeradbiomed.2008.01.011
- Hybertson BM, Gao B, Bose SK, et al. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med. 2011;32(4–6):234–246. doi: 10.1016/j.mam.2011.10.006
- Schieber M, Chandel NS. Ros functions in redox signaling and oxidative stress. Curr Biol. 2014;24(10):R453–R462. doi: 10.1016/j.cub.2014.03.034
- Holley AK, Clair DK S. Watching the watcher: regulation of p53 by mitochondria. Future Oncol. 2009;5(1):117–130. doi: 10.2217/14796694.5.1.117
- Levine AJ, Feng Z, Mak TW, et al. Coordination and communication between the p53 and IGF-1-AKT-mTOR signal transduction pathways. Genes Dev. 2006;20:267–75. doi: 10.1101/gad.1363206
- Saleem A, Adhihetty PJ, Hood DA. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol Genomics. 2009;37(1):58–66. doi: 10.1152/physiolgenomics.90346.2008
- Saleem A, Carter HN, Hood DA. P53 is necessary for the adaptive changes in cellular milieu subsequent to an acute bout of endurance exercise. Am J Physiol Cell Physiol. 2014;306(3):C241–C249. doi: 10.1152/ajpcell.00270.2013
- Smeenk L, van Heeringen SJ, Koeppel M, et al. Characterization of genome-wide p53 binding sites upon stress response. Nucleic Acids Res. 2008;36(11):3639–3654. doi: 10.1093/nar/gkn232
- Rotblat B, Melino G, Knight RA. Nrf2 and p53: januses in cancer? Oncotarget. 2012;3(11):1272–1283. doi: 10.18632/oncotarget.754
- Liu D, Xu Y. P53, oxidative stress, and aging. Antioxid Redox Signal 2011;15(6):1669–1678. doi: 10.1089/ars.2010.3644
- Marino G, Niso-Santano M, Baehrecke EH, et al. Self-consumption: the interplay of autophagy and apoptosis. Nature Rev. 2014;15:81–94. doi: 10.1038/nrm3735
- Ljubicic V, Joseph AM, Saleem A, et al. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: effects of exercise and aging. Biochim Biophys Acta. 2010;1800(3):223–234. doi: 10.1016/j.bbagen.2009.07.031
- Qi Z, He J, Zhang Y, et al. Exercise training attenuates oxidative stress and decreases p53 protein content in skeletal muscle of type 2 diabetic goto-kakizaki rats. Free Radic Biol Med. 2011;50(7):794–800. doi: 10.1016/j.freeradbiomed.2010.12.022
- Steinbacker P, Eckl P. Impact of oxidative stress on exercising skeletal muscle. Biomolecules. 2015;5(2):356–377. doi: 10.3390/biom5020356
- Li X, Fang P, Mai J, et al. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol. 2013;6:19. doi: 10.1186/1756-8722-6-19
- Chen YW, Nader GA, Baar KR, et al. Response of rat muscle to acute resistance exercise defined by transcriptional and translational profiling. J Physiol. 2002;545:27–41. doi: 10.1113/jphysiol.2002.021220
- Saleem A, Carter HN, Iqbal S, et al. Role of p53 within the regulatory network controlling muscle mitochondrial biogenesis. Exerc Sport Sci Rev. 2011;39(4):199–205.
- Matoba S, Kang JG, Patino WD, et al. P53 regulates mitochondrial respiration. Science. 2006;312(5780):1650–1653. doi: 10.1126/science.1126863
- Saleem A, Hood DA. Acute exercise induces tumour suppressor protein p53 translocation to the mitochondria and promotes a p53-tfam-mitochondrial DNA complex in skeletal muscle. J Physiol. 2013;591(14):3625–3636. doi: 10.1113/jphysiol.2013.252791
- Donahue RJ, Razmara M, Hoek JB, et al. Direct influence of the p53 tumor suppressor on mitochondrial biogenesis and function. FASEB J 2001;15:635–644. doi: 10.1096/fj.00-0262com
- Park JY, Wang PY, Matsumoto T, et al. P53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ Res. 2009;105(7):705–712. doi: 10.1161/CIRCRESAHA.109.205310
- Granata C, Oliveira RS, Little JP, et al. Sprint-interval but not continuous exercise increases PGC-1α protein content and p53 phosphorylation in nuclear fractions of human skeletal muscle. Sci Rep. 2017;7:44227. doi: 10.1038/srep44227
- Tachtsis B, Smiles WJ, Lane SC, et al. Acute endurance exercise induced nuclear p53 abundance in human skeletal muscle. Front Physiol. 2016;7:144. doi: 10.3389/fphys.2016.00144
- Achanta G, Sasaki R, Feng L, et al. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. EMBO J 2005;24:3482–3492. doi: 10.1038/sj.emboj.7600819
- Bakhanashvili M, Grinberg S, Bonda E, et al. Excision of nucleoside analogs in mitochondria by p53 protein. AIDS. 2009;23:779–788. doi: 10.1097/QAD.0b013e328329c74e
- Yoshida Y, Izumi H, Torigoe T, et al. P53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA. Cancer Res 2003;63:3729–3734.
- Safdar A, Khrapko K, Flynn JM, et al. Exercise-induced mitochondrialp53 repairs mtDNA mutations in mutator mice. Skelet Muscle. 2016;6:7. doi: 10.1186/s13395-016-0075-9
- Siu PM, Always SE. Subcellular responses of p53 and Id2 in fast and slow skeletal muscle in response to stretch-induced overload. J Appl Physiol (1985). 2005;99(5):1897–1904. doi: 10.1152/japplphysiol.00374.2005
- Granata C, Oliveira RS, Little JP, et al. Mitochondria adaptations to high-volume exercise training are rapidly reversed after a reduction in training volume in human skeletal muscle. FASEB J 2016;30(10):3413–3423. doi: 10.1096/fj.201500100R
- Bartlett JD, Hwa Joo C, Jeong TS, et al. Matched work high-intensity interval and continuous running induce similar increases in PGC-1α mRNA, AMPK, p38, and p53 phosphorylation in human skeletal muscle. J Appl Physiol (1985). 2012;112(7):1135–43. doi: 10.1152/japplphysiol.01040.2011
- Puzio-Kuter AM. The role of p53 in metabolic regulation. Genes Cancer. 2011;2(4):385–391. doi: 10.1177/1947601911409738
- Heiden MG V, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809
- Schupp M, Chen F, Briggs ER, et al. Metabolite and transcriptome analysis during fasting suggest a role for the p53-Ddit4 axis in major metabolic tissues. BMC Genomics. 2013;14:758. doi: 10.1186/1471-2164-14-758
- Canto C, Auwerx J. Calorie restriction: is AMPK a key sensor and effector? Physiology. 2011;14:214–224. doi: 10.1152/physiol.00010.2011
- Varady KA, Hellerstein MK. Alternate-day fasting and chronic disease prevention: a review of human and animal trials. Am J Clin Nutr. 2007;14:7–13. doi: 10.1093/ajcn/86.1.7
- Bartlett JD, Louhelainen J, Iqbal Z, et al. Reduced carbohydrate availability enhances exercise-induced p53 signaling in human skeletal muscle: implications for mitochondrial biogenesis. Am J Physiol Regul Integr Comp Physiol. 2013;30(46):R450–R458. doi: 10.1152/ajpregu.00498.2012
- Bartlett JD, Close GL, Drust B, et al. The emerging role of p53 in exercise metabolism. Sports Med. 2014;44(3):303–9. doi: 10.1007/s40279-013-0127-9
- Camera DM, Hawley JA, Coffey VG. Resistance exercise with low glycogen increases p53 phosphorylation and PGC-1α mRNA in skeletal muscle. Eur J Appl Physiol. 2015;115(6):1185–94. doi: 10.1007/s00421-015-3116-x
- Edwards MG, Anderson RM, Yuan M, et al. Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC Genomics. 2007;8:80. doi: 10.1186/1471-2164-8-80
- Cahill GF, Jr. Fuel metabolism in starvation. Annu Rev Nutr. 2006;14: 1–22. doi: 10.1146/annurev.nutr.26.061505.111258
- Zechner R, Zimmermann R, Eichmann TO, et al. Fat signals–lipases and lipolysis in lipid metabolism and signaling. Cell Metab 2012;14:279–291. doi: 10.1016/j.cmet.2011.12.018
- Aquilano K, Baldelli S, Cardaci S, et al. Nitric oxide is the primary mediator of cytotoxicity induced by GSH depletion in neuronal cells. J Cell Sci. 2011;124:1043–1054. doi: 10.1242/jcs.077149
- Aquilano K, Baldelli S, Pagliei B, et al. P53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid Redox Signal. 2013;18(4):386–399. doi: 10.1089/ars.2012.4615
- Assaily W, Rubinger DA, Wheaton K, et al. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol Cell. 2011;44(3):491–501. doi: 10.1016/j.molcel.2011.08.038
- Finck BN, Gropler MC, Chen Z, et al. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005
- Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009;28:3015–3026. doi: 10.1038/emboj.2009.242
- Bensaad K, Tsuruta A, Selak MA, et al. Tigar, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036
- Hu W, Zhang C, Wu R, et al. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. 2010;106(16):7455–7460. doi: 10.1073/pnas.1001006107
- Espinosa-Diez C, Miguel V, Mennerich D, et al. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015;6:183–197. doi: 10.1016/j.redox.2015.07.008
- Holloszy JO, Booth FW. Biochemical adaptations to endurance exercise in muscle. . Annu Rev Physiol. 1976;38:273–291. doi: 10.1146/annurev.ph.38.030176.001421
- Yokoyama M, Okada S, Nakagomi A, et al. Inhibition of endothelial p53 improves metabolic abnormalities related to dietary obesity. Cell Rep. 2014;7(5):1691–1703. doi: 10.1016/j.celrep.2014.04.046
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 2009;96:4240–4245. doi: 10.1073/pnas.96.8.4240
- Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/MCB.20.5.1868-1876.2000
- Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64:2627–2633. doi: 10.1158/0008-5472.CAN-03-0846
- Russell SJ, Kahn CR. Endocrine regulation of ageing. Nat Rev Mol Cell Biol. 2007;8:681–691. doi: 10.1038/nrm2234
- Luo X, Wu J, Jing S, et al. Hyperglycemic stress and carbon stress in diabetic glucotoxicity. Aging Dis. 2016;7(1):90–110. doi: 10.14336/AD.2015.0702
- Nakahara T, Hashimoto K, Hirano M, et al. Acute and chronic effects of alcohol exposure on skeletal muscle c-myc, p53, and Bcl-2 mRNA expression. Am J Physiol Endocrinol Metab. 2003;285(6):E1273–E1281. doi: 10.1152/ajpendo.00019.2003
- Preedy VR, Adachi J, Ueno Y, et al. Alcoholic skeletal muscle myopathy: definitions, features, contribution of neuropathy, impact and diagnosis. Eur J Neurol. 2001;8:677–687. doi: 10.1046/j.1468-1331.2001.00303.x
- Reilly ME, McKoy G, Mantle D, et al. Protein and mRNA levels of the myosin heavy chain isoforms I, IIa, IIx and IIb in type I and type II fibre predominant rat skeletal muscles in response to chronic alcohol feeding. J Muscle Res Cell Motil. 2000;21:763–773. doi: 10.1023/A:1010336624154
- Taira T, Negishi Y, Kihara F, et al. c-myc protein complex binds to two sites in human hsp70 promoter region. Biochim Biophys Acta. 1992;1130:166–174. doi: 10.1016/0167-4781(92)90524-4
- Vafa O, Wade M, Kern S, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–1044. doi: 10.1016/S1097-2765(02)00520-8
- Morimoto Y, Bando YK, Shigeta T, et al. Atorvastatin prevents ischemic limb loss in type 2 diabetes: role of p53. J Atherscler Thromb. 2011;18(3):200–208. doi: 10.5551/jat.6437
- Schwarzkopf M, Coletti D, Sassoon D, et al. Muscle cachexia is regulated by a p53-PW1/Peg3-dependent pathway. Genes Dev 2006;20:3440–3452. doi: 10.1101/gad.412606
- Fox DK, Ebert SM, Bongers KS, et al. Atf4 mediate distinct and additive pathways to skeletal muscle atrophy during limb immobilization. Am J Physiol Endocrinol Metab. 2014;307:E245–E261. doi: 10.1152/ajpendo.00010.2014
- White JD, Collins R, Vermuelen R, et al. The role of p53 in vivo during skeletal muscle post-natal development and regeneration: studies in p53 knockout mice. Int J Dev Biol. 2002;46:577–582.
- Nakazawa H, Chang K, Shinozaki S, et al. Inos as a driver of inflammation and apoptosis in mouse skeletal muscle after burn injury: possible involvement of Sirt1 s-nitrosylation mediated acetylation of p65 NF-kB and p53. PLOS ONE. 2017;12(1):1–18. doi: 10.1371/journal.pone.0170391
- Wang X, Hu Z, Hu J, et al. Insulin resistance accelerates muscle protein degradation: activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. J Endocrin. 2006;147(9):4160–4168. doi: 10.1210/en.2006-0251
- Shinozaki S, Chang K, Sakai M, et al. Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase SIRT1 to increase acetylation and activation of p53 and p65. Sci Signal. 2015;7(351):ra106. doi: 10.1126/scisignal.2005375
- Desplanches D, Mayet MH, Sempore B, et al. Structural and functional responses to prolonged hindlimb suspension in rat muscle. J Appl Physiol. 1987;63:558–563. doi: 10.1152/jappl.1987.63.2.558
- Siu PM, Pistilli EE, Murlasits Z, et al. Hindlimb unloading increases muscle content of cytosolic but not nuclear Id2 and p53 protein in young adult and aged rats. J Appl Physiol (1985). 2006;100(3):907–916. doi: 10.1152/japplphysiol.01012.2005
- Stefan N, Vozarova B, Funahashi T, et al. Plasma adiponectin concentration is associated with skeletal muscle insulin receptor tyrosine phosphorylation, and low plasma concentration precedes a decrease in whole-body insulin sensitivity in humans. Diabetes. 2002;51:1884–1888. doi: 10.2337/diabetes.51.6.1884
- Szalay K, Razga Z, Duda E. Tnf inhibits myogenesis and downregulates the expression of myogenic regulatory factors myoD and myogenin. Eur J Cell Biol. 1997;74:391–398.
- Gomes MD, Lecker SH, Jagoe RT, et al. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci. 2001;98:14440–14445. doi: 10.1073/pnas.251541198
- Tazi EM, Errihani H. Treatment of cachexia in oncology. Indian J Palliat Care. 2010;16(3):129–137. doi: 10.4103/0973-1075.73644
- Wang C, Liu W, Liu Z, et al. Hypoxia inhibits myogenic differentiation through p53 protein-dependent induction of Bhlhe40 protein. J Biol Chem. 2015;290(50):29707–29716. doi: 10.1074/jbc.M115.688671
- Yun Z, Lin Q, Giaccia AJ. Adaptive myogenesis under hypoxia. Mol Cell Biol. 2005;25:3040–3055. doi: 10.1128/MCB.25.8.3040-3055.2005
- Hoppeler H, Vogt M. Muscle tissue adaptations to hypoxia. J Exp Biol. 2001;204:3133–3139.
- Greenbaum AR, Etherington PJ, Manek S, et al. Measurements of oxygenation and perfusion in skeletal muscle using multiple microelectrodes. J Muscle Res Cell Motil. 1997;18:149–159. doi: 10.1023/A:1018653521686
- Zhang J, Biggar KK, Storey KB. Regulation of p53 by reversible post-transcriptional and post-transcriptional mechanisms in liver and skeletal muscle of an anorexia tolerant turtle, trachemys scripta elegans. Gene. 2013;513:147–155. doi: 10.1016/j.gene.2012.10.049
- Carlo A D, Mori R D, Martelli F, et al. Hypoxia inhibits myogenic differentiation through accelerated myod degradation. J Biol Chem. 2004;279:16332–16338. doi: 10.1074/jbc.M313931200
- Gustafsson MV, Zheng X, Pereira T, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010
- Buas MF, Kabak S, Kadesch T. Inhibition of myogenesis by notch: evidence for multiple pathways. J Cell Physiol. 2009;218(1):84–93. doi: 10.1002/jcp.21571
- Liu W, Wen Y, Bi P, et al. Hypoxia promotes satellite cell self-renewal and enhances the efficiency of myoblast transplantation. Development. 2012;139:2857–2865. doi: 10.1242/dev.079665
- Majmundar AJ, Skuli N, Mesquita RC, et al. O-2 regulates skeletal muscle progenitor differentiation through phosphatidylinositol 3-kinase/AKT signaling. Mol Cell Biol. 2012;32:36–49. doi: 10.1128/MCB.05857-11
- Little JB. Principal cellular and tissue effects of radiation. In: Kufe DW, Pollock RE, Weichselbaum RR, et al., editors. Holland-Frei cancer medicine. 6th edition. Hamilton (ON): BC Decker; 2003.
- Little JB, Nagasawa H. Effect of confluent holding on potentially lethal damage repair, cell cycle progression, and chromosomal aberrations in human normal and ataxia-telangiectasia fibroblasts. Radiat Res. 1985;101:81–93. doi: 10.2307/3576305
- Yang ZJP, Kenzelmann Broz D, Noderer WL, et al. P53 suppresses muscle differentiation at the myogenin step in response to genotoxic stress. Cell Death Differ.. 2015;22:560–573. doi: 10.1038/cdd.2014.189
- Lin T, Chao C, Saito S, et al. P53 induces differentiation of mouse embryonic stem cells by suppressing nanog expression. Nat Cell Biol. 2015;7:165–171. doi: 10.1038/ncb1211
- Schwarzkopf M, Coletti D, Marazzi G, et al. Chronic p53 activity leads to skeletal muscle atrophy and muscle stem cell perturbation. Basic Appl Myol. 2009;18:131–138.
- Feng Z, Hu W, de Stanchina E, et al. The regulation of AMPK β1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67(7): 3043–3053. doi: 10.1158/0008-5472.CAN-06-4149
- Feng Z, Zhang H, Levine AJ, et al. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102
- Levine AJ, Hu W, Feng Z. The p53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13:1027–1036. doi: 10.1038/sj.cdd.4401910
- Jones RG, Plas DR, Kubek S, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027
- Pronsato L, Milanesi L. Effect of testosterone on the regulation of p53 and p66Shc during oxidative stress damage in C2C12 cells. Steroids. 2016;106:41–54. doi: 10.1016/j.steroids.2015.12.007
- Giorgio M, Migliaccio E, Orsini F, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;22:221–233. doi: 10.1016/j.cell.2005.05.011
- Le S, Connors TJ, Maroney AC. C-Jun N-terminal kinase specifically phosphorylates p66ShcA at serine 36 in response to ultraviolet irradiation. J Biol Chem. 2001;276:48332–48336. doi: 10.1074/jbc.M106612200
- Natalicchio A, De Stefano F, Perrini S, et al. Involvement of the p66Shc protein in glucose transport regulation in skeletal muscle myoblasts. Am J Physiol Endocrinol Metab. 2009;296:E228–E237. doi: 10.1152/ajpendo.90347.2008
- Natalicchio A, Tortosa F, Perrini S, et al. p66Shc, a multifaceted protein linking Erk signalling, glucose metabolism, and oxidative stress. Arch Physiol Biochem. 2011;117:116–124. doi: 10.3109/13813455.2011.562513
- Colla A L, Vasconsuelo A, Milanesi L, et al. 17β-Estradiol protects skeletal myoblasts from apoptosis through p53, Bcl-2, and FoxO families. J Cell Biochem. 2017;118:104–115. doi: 10.1002/jcb.25616
- Hori YS, Kuno A, Hosoda R, et al. Regulation of FOXOs and p53 by SIRT1 modulators under oxidative stress. PLOS ONE. 2013;8(9):e73875. doi: 10.1371/journal.pone.0073875
- Essers MA, Weijzen S, de Vries-Smits AM, et al. Foxo transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476
- Attardi LD, Reczek EE, Cosmas C, et al. Perp, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev. 2000;14(6):704–18.
- Oda E, Ohki R, Murasawa H, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2011;288:1053–1058. doi: 10.1126/science.288.5468.1053
- Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–2487. doi: 10.1242/jcs.001222
- Roux I L, Konge J, Le Cam L, et al. Numb is required to prevent p53-dependent senescence following skeletal muscle injury. Nat Commun. 2015;6:8528. doi: 10.1038/ncomms9528
- Xu Y, Li N, Xiang R, et al. Emerging roles of the p38 MAPK and PI3 K /AKT /mTOR pathways in oncogene-induced stress. Trends Biochem Sci. 2014;39(6):268–276. doi: 10.1016/j.tibs.2014.04.004
- Liu S, Li J, Tao Y, et al. Small heat shock protein αB-crystallin binds to p53 to sequester its translocation to mitochondria during hydrogen peroxide-induced apoptosis. Biochem Bioph Res Co. 2007;354:109–114. doi: 10.1016/j.bbrc.2006.12.152
- Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455
- Fiordaliso F, Leri A, Cesselli D, et al. Hyperglycemia activates p53 and p53-regulated genes leading to myocyte cell death. Diabetes. 2001;50(10):2363–75. doi: 10.2337/diabetes.50.10.2363
- Ogawara Y, Kishishita S, Obata T, et al. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277:21843–21850. doi: 10.1074/jbc.M109745200
- Baldelli S, Ciriolo MR. Altered S-nitrosylation of p53 is responsible for impaired antioxidant response in skeletal muscle during aging. Aging. 2016;8(12): 3450–3462. doi: 10.18632/aging.101139
- Irrcher I, Ljubicic V, Kirwan AF, et al. Amp activated protein kinase-regulated activation of the PGC-1alpha promoter in skeletal muscle cells. PLOS One. 2008;3:e3614. doi: 10.1371/journal.pone.0003614
- Relaix F, Wei XJ, Wu X. Peg3/Pw1is an imprinted gene involved in the TNF–NFKB signal transduction pathway. Nat Genet. 1998;18:287–291. doi: 10.1038/ng0398-287
- Tisdale MJ. Loss of skeletal muscle in cancer: biochemical mechanisms. Front Biosci. 2001;6:D164–D174.
- Zheng Y, Gardner SE, Clarke MC. Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler Thromb Vasc Biol. 2011;31:2781–2786. doi: 10.1161/ATVBAHA.111.224907
- Di Y, Aminot Y, Schroeder DC, et al. Integrated biological responses and tissue-specific expression of p53 and ras genes in marine mussels following exposure to benzo(a)pyrene and C60 fullerenes, either alone or in combination. Mutagenesis. 2017;32:77–90. doi: 10.1093/mutage/gew049
- Kamat JP, Devasagayam TP, Priyadarsini KI, et al. Oxidative damage induced by the fullerene C60 on photosensitization in rat liver microsomes. Chem Biol Interact. 1998;114: 145–159. doi: 10.1016/S0009-2797(98)00047-7
- Saada YB, Zakharova V, Chernyak B, et al. Control of DNA integrity in skeletal muscle under physiological and pathological conditions. Cell Mol Life Sci. 2017;4(19):3439–3449. doi: 10.1007/s00018-017-2530-0
- Alfadda AA, Sallam RM. Reactive oxygen species in health and disease. J Biomed Biotech. 2012;2012:936486. doi: 10.1155/2012/936486