
ABSTRACT
Increasing evidence points to the respiratory Complex II (CII) as a source and modulator of reactive oxygen species (ROS). Both functional loss of CII as well as its pharmacological inhibition can lead to ROS generation in cells, with a relevant impact on the development of pathophysiological conditions, i.e. cancer and neurodegenerative diseases. While the basic framework of CII involvement in ROS production has been defined, the fine details still await clarification. It is important to resolve these aspects to fully understand the role of CII in pathology and to explore its therapeutic potential in cancer and other diseases.
Introduction
For decades, reactive oxygen species (ROS) have captivated many researchers because of their intractable nature and both beneficial and detrimental roles in cell physiology and pathology. Mitochondria, the site of cellular respiration, are considered the primary site of endogenous ROS production in most cell types. Depending on their concentration, ROS can initiate diverse cellular actions. At physiological levels, they support signaling pathways involved in cell growth and protection, while their high levels lead to cellular damage followed by cell death. The balance between ROS generation and ROS scavenging needs to be tightly regulated [Citation1–3]. Indeed, mitochondrial ROS production has been connected to numerous pathological conditions including neurodegenerative diseases [Citation4], aging [Citation5], oxidative damage during ischemia/reperfusion injury [Citation6], and cancer [Citation7,Citation8].
Initially, respiratory Complex I (NADH:ubiquinone oxidoreductase, CI) and Complex III (ubiquinol:cytochrome c oxidoreductase, CIII) were considered the main sources of mitochondrial ROS, while the contribution of Complex II (succinate dehydrogenase, SDH, CII) was overlooked [Citation9–11]. Identification of mutations in CII resulting in increased ROS production in cancer and neurodegenerative diseases [Citation12–14] and realization that CII plays a crucial role in ROS production also during the reverse electron transfer (RET) through CI [Citation15–18] changed the traditional view. Presently we know that CII contributes significantly to ROS both directly and indirectly (via RET), with important implications in physiology and disease. Mutations in CII are associated with familiar and sporadic forms of cancer, particularly with pheochromocytoma/paraganglioma (PHEO/PGL), gastrointestinal stromal tumors (GIST), and renal cancer, but also with the Leigh syndrome, a neurodegenerative disease. Moreover, CII inhibitors are cardioprotective in ischemia/reperfusion injury (I/R), and can be also applied to cancer therapy [Citation19].
Complex II structure and function
The research on mitochondrial CII as a source of redox cofactors dates back more than 6 decades [Citation20]. CII is unique in linking the tricarboxylic acid (TCA) cycle and the respiratory chain. CII catalyzes the oxidation of succinate to fumarate, which does not directly contribute to the generation of the proton motive force, but concurrently transfers two electrons derived from this reaction to membrane-bound ubuiquinone. Ubiquinone is thereby reduced to ubiquinol, which fuels CIII and CIV. Interestingly, unlike the other members of the respiratory chain, none of CII subunit is encoded by mitochondrial DNA [Citation21].
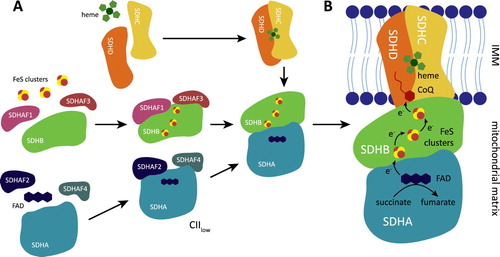
Human CII consists of four subunits, SDHA-D (). SDHA is the largest subunit and contains an active site with covalently bound flavine adenine dinucleotide (FAD), which removes electrons from succinate. SDHB carries three linearly aligned iron–sulfur clusters that mediate electron transfer to the ubiquinone molecule located in the ubiquinone-binding (Q) site, jointly formed by SDHB, SDHC, and SDHD. Subunits SDHC and SDHD anchor CII to the mitochondrial inner membrane [Citation22,Citation23]. Four assembly factors participate in CII biogenesis [Citation24–27]. SDHAF2 and SDHAF4 are involved in the maturation of SDHA, and may facilitate covalent flavinylation [Citation24,Citation27,Citation28]. SDHAF1, assisted by SDHAF3, promotes insertion of Fe-S clusters into SDHB [Citation25,Citation26]. Mature SDHA is attached to mature SDHB to be linked to membrane-bound SDHC/SDHD. While the crystal structure of CII was described more than 10 years ago [Citation21,Citation22], additional alternative assembly forms were discovered recently in bacteria [Citation29–31]. One such alternative CII, designated CIIlow and containing SDHA, SDHAF2, and SDHAF4, was identified also in mammalian cells. CIIlow is induced in the absence of SDHB or during respiratory dysfunction and may orchestrate cellular adaptations to energy stress [Citation32].
Figure 1. Complex II assembly and its metabolic activity. (A) CII is assembled from four structurally different subunits SDHA-D. SDHA, with covalently bound FAD, requires two assembly factors SDHAF2 and SDHAF4 that assist with SDHA maturation and flavinylation. SDHB contains three iron-sulphur (FeS) clusters, and two additional assembly factors SDHAF1 and SDHAF3 are needed for their insertion. Dimer of SDHA/B is then linked to transmembrane subunits SDHC/D. Under some conditions, an alternative assembly species of CII consisting of SDHA, SDHAF2, and SDHAF4 (described as CIIlow) is stabilized and has an independent biological function. (B) Metabolic activity of the mature CII. CII promotes oxidation of succinate to fumarate. Electrons from succinate are removed by FAD in SDHA and then passed to FeS clusters of SDHB subunit. FeS cofactors transfer the electrons to ubiquinone (CoQ) within the ubiquinone binding (Q) site formed by SDHB, SDHC, and SDHD.
Complex II and generation of ROS
In recent years, it has become apparent that CII has an important role in ROS production. This role is either direct, when ROS are generated at CII, or indirect, when ROS are produced at other sites from electrons supplied by CII (). The indirect role was described first, because of the old observation that isolated mitochondria produce large amounts of ROS in the presence of high concentrations (≥5 mM) of succinate, the substrate of CII [Citation15–17,Citation33,Citation34], implicating CII in the process. This is due to RET, when succinate-derived electrons from CII reduce the ubiquinone pool, and electrons are forced backwards from ubiquinone towards CI, where vast quantities of ROS are formed. It was suggested that also CII can produce ROS under these conditions, but this is somewhat controversial and may be tissue-specific [Citation35].
Figure 2. Complex II contributes to ROS production in both physiological and pathophysiological conditions. (A) In the presence of high concentrations of succinate, CII does not produce ROS directly but can contribute to indirect ROS generation via reverse electron transfer (RET) by forcing the electrons onto CI. (B) At lower, physiological succinate concentration, succinate molecule passes the electrons to FAD forming FADH2 which is then able to react with oxygen within the unoccupied succinate binding site, therefore directly forming ROS. (C) The ROS generating ability of reduced FAD is significantly increased when the Q site is blocked by an inhibitor. In contrast, succinate binding site inhibitors block ROS production. (D) Incorrect assembly or damage to CII subunits can induce ROS formation either via reduced FAD or possibly via exposed FeS clusters.
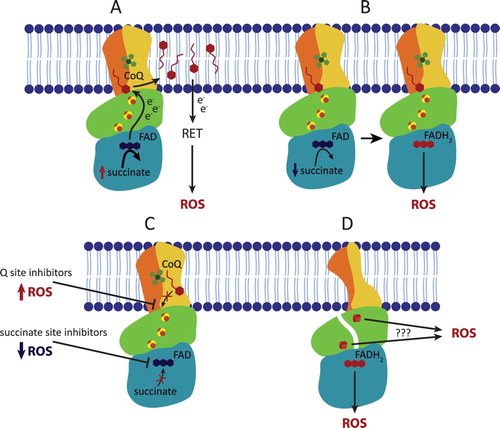
The direct role of CII in ROS production went long unrecognized. This is because the primary ROS producing site in CII, FAD in SDHA, cannot generate ROS when succinate concentration is high (≥5 mM). The mechanism of succinate-mediated inhibition of ROS production at FAD is not entirely clear, but succinate may block access of oxygen to FAD [Citation36]. Respiratory measurements are traditionally performed at 5-10 mM succinate, which masks FAD contribution to ROS production. At 0.5 mM succinate, a concentration similar to normal intracellular succinate levels, the contribution of CII’s FAD to ROS generation can be substantial when electron transport through CII is blocked at the Q site or further downstream (at CIII, for example), suggesting that ROS is produced when FAD is reduced, but the active site is not occupied [Citation36,Citation37]. Under specific conditions, ROS generation was also observed at the Q site [Citation38], however, this is likely infrequent in mammalian CII.
Inhibitors of CII show ambivalent effect on mitochondrial ROS production depending on substrate supply, membrane potential and overall metabolic activity of the cell as well as intracellular succinate concentration [Citation39,Citation40]. Specific inhibitors of CII bind either to the succinate-binding site, i.e. oxaloacetate and malonate (reviewed in [Citation14]), or to the Q site, i.e. thenoyltrifluoroacetone (TTFA) [Citation22], atpenin [Citation41], α−tocopheryl succinate [Citation42], or mitochondrially targeted vitamin E succinate (MitoVES) [Citation43–45]. Generally, succinate-binding site inhibitors suppress ROS production from CII as they block FAD, while Q site inhibitors stimulated ROS generation as they reduce FAD by blocking electron transfer to ubiquinone. However, in intact cells only intermediate affinity (TTFA, MitoVES) Q site inhibitors induce ROS (and cell death), while high-affinity Q site inhibitors such as atpenin do not induce ROS [Citation45]. The explanation is that the plasma membrane is impermeable for succinate, and succinate rapidly accumulates when high-affinity Q site inhibitors are employed, canceling ROS production from FAD. This is perhaps the case for atpenin A5, which immediately blocks all CII molecules in a cell, so that succinate cannot be consumed. Lower affinity Q site inhibitors do not occupy all CII molecules at the same time, which keeps succinate down, allowing ROS production at FAD of those CII molecules that have the Q site blocked. Indeed, atpenin treatment [Citation46], unlike TTFA and MitoVES, do not induce ROS-mediated cell death [Citation45], and atpenin is quite well tolerated by cultured cells. Finally, both the succinate-binding site and the Q site inhibitors suppress ROS production under high succinate concentrations during RET [Citation47] as they all prevent the transfer of electrons from succinate via CII to the ubiquinone pool.
The paradox of ROS production from CII
It has been shown that functional loss of CII can lead to succinate accumulation and ROS generation in cells [Citation19]. Guzy et al. found that pharmacological inhibition of CII or silencing of SDHB can lead to ROS production and ROS-dependent stabilization of hypoxia-inducible factor-α [Citation48], while others ascribed this effect to the accumulation of succinate [Citation49]. Similarly, CII dysfunction, increased ROS formation, and mtDNA mutability were observed in a yeast model with mutated SDHB [Citation50]. Mutations in the SDHC subunit of CII in fibroblasts from a transgenic mouse enhance ROS generation due to dysfunction of mitochondrial respiration [Citation51]. Similarly, downregulation of the expression of the SDHC subunit in hepatocellular carcinoma was linked to increased cancer cell growth and metastasis due to elevated ROS production with subsequent nuclear factor-κB signaling [Citation52]. A study using hamster fibroblasts revealed that mutation in SDHD resulted in elevated ROS production [Citation53]. A similar effect on the production of ROS and instability of DNA was observed in yeast mutant of SDH [Citation54].
These observations are puzzling given recent strong evidence for FAD in SDHA being the principal site of ROS production in the mature mammalian CII, coming both from isolated mitochondria and from intact cells [Citation36,Citation37,Citation45]. We face the following paradox. Mutations and/or manipulations that interfere with CII and therefore favor reduced FAD will also increase intracellular succinate to concentration over 5 mM which is incompatible with ROS production from FAD in mammalian CII. Indeed, PHEO/PGL-associated mutations in the SDHC subunit that stimulate ROS at low (0.5 mM) succinate levels in isolated mitochondria often do not stimulate ROS in intact cells [Citation45]. There are several relevant aspects that should be considered when thinking about CII-derived ROS in pathology. When wild-type CII alleles are present (heterozygous mutations, incomplete knockdown), these will control succinate levels to some degree to allow ROS production at FAD by mutated CII. Indeed, inherited PHEO/PGL-associated germline mutations are heterozygous during tumor development. Yeast results could perhaps be explained by a different behavior of mammalian/Escherichia coli CII compared to Saccharomyces cerevisiae CII with respect to ROS production. While the amount of ROS produced at different succinate concentrations follows the typical bell-shaped curve for human and E. coli CII (with a maximum at about 0.5 mM succinate, corresponding to a typical concentration in normal cells) [Citation36,Citation47,Citation55], this is not the case for S. cerevisiae CII. In yeast, ROS production at CII is succinate-insensitive and the likely source is the Q site [Citation56,Citation57]. For this reason, yeast CII may not be the optimal model to study ROS-related aspects of CII-dependent tumorigenesis.
Improperly assembled CII, for example incorrect insertion of FeS clusters into SDHB, can result in increased ROS [Citation26]. Yet, Maklashina et al. showed that free E. coli SDHA flavoproteins have only minor catalytic activity and generate little or no ROS. Their results suggest that the iron–sulfur protein SDHB in CII is necessary for robust catalytic activity and ROS generation by incomplete CII [Citation58]. This could explain how CII could produce ROS to amplify the apoptotic response. In this scenario, SDHA/SDHB subcomplex disengages from the membrane-bound SDHC/SDHD, and superoxide is formed [Citation59]. The precise site of superoxide generation was not identified, but it could possibly originate from the exposed FeS clusters of SDHB that would be insensitive to succinate inhibition. This raises the possibility that CII mutations, which can alter CII conformation (particularly in SDHB), could allow ROS production even in the presence of accumulated intracellular succinate, circumventing the FAD paradox.
CII in disease
Isolated defects of CII are relatively rare, accounting for approximately 2% of all respiratory chain deficiency diagnoses [Citation60]. Still, accumulating evidence links SDHx mutations to pathology of the nervous system and the brain. Deficiency of CII is recognized to cause encephalomyopathy in childhood and optic atrophy in adulthood [Citation61]. Jain-Ghai et al. reviewed 37 clinical cases of CII deficiency, concluding that neurological findings, abnormal brain imaging, and developmental delay were the most common manifestation of CII defects, regardless of the large variation in the phenotype [Citation62]. Chronic administration of 3-nitropropionic acid (3-NPA), an irreversible inhibitor of succinate dehydrogenase, replicates the neuropathologic and clinical features of Huntington disease (HD) in nonhuman primates [Citation63]. Later it was shown that patients with HD have severe defects of CII in caudate nucleus [Citation64], which can mediate striatal cell death and neurodegeneration mimicking the development of HD [Citation65]. On the other hand, Naseri et al. recently measured an elevated SDH activity in HD patient lymphoblasts [Citation66], pointing to a possible compartment-specific CII regulation.
One of the rare cases of documented autosomal inheritance of SDHA subunit defect was linked to bilateral optic atrophy, ocular movement disorder, progressive polyneuropathy, psychiatric involvement, and cardiomyopathy [Citation60]. Mutations in SDHA, SDHB and SDHAF1 were reported in leukodystrophy [Citation67], Leigh syndrome and cardiomyopathy [Citation23,Citation68–70], and infantile leukoencephalopathy [Citation25]. Recently, a case of encephalomyopathy has been connected to a recessive germline mutation in SDHD subunit [Citation71]. Moreover, assembly factor SDHAF4 was implicated in neuroprotection, possibly decreasing ROS generated by the free SDHA subunit [Citation27]. Hence, ROS production via CII may play a role in neurodegenerative processes.
SDHx defects show a strong association with tumorigenesis, and SDHx genes are considered tumor suppressors. Germline mutations in subunits SDHA-D, as well as assembly factor SDHAF2, were recognized to cause familial PHEO/PGL [Citation13,Citation23,Citation72]. Further, SDH dysregulation is linked to GIST oncogenesis [Citation23,Citation73] and renal carcinoma [Citation74,Citation75], but less frequently. In addition, the familiar SDHx defects are connected to PTEN mutation-negative Cowden syndrome, associated with breast, thyroid, and endometrial neoplasias [Citation76].
Unlike in neurological disorders and cancer, in other pathologies, the direct genetic link to CII has not been established. However, evidence is emerging for the role of mitochondrial ROS in obesity [Citation77–79], insulin resistance/diabetes [Citation79,Citation80], cardiovascular diseases [Citation81], and non-alcoholic fatty liver disease [Citation79,Citation82,Citation83]. With regard to CII/ROS, skeletal muscle biopsies from patients with obesity and diabetes showed changes in CII activity [Citation78,Citation84,Citation85]. Also visceral adipose tissue in obese patients exhibits decreased CII activity compared to subcutaneous adipose tissue which can be restored in vitro by addition of the mitochondria-specific oxidant scavenger mito-TEMPO [Citation77]. Chemical inhibition of CI and CII by amiodarone followed by increased ROS production may result in steatohepatitis [Citation86]. Moreover, Fazakerley et al. have suggested that loss of mitochondrial CoQ can drive adipocyte insulin resistance most likely via CII-dependent mitochondrial ROS production [Citation87]. Altogether, CII should be considered when searching for novel therapeutic approaches in metabolic disorders.
Targeting CII/ROS as a therapeutic approach
Mitochondrial malfunction and increased ROS production are relevant in aging, neurodegenerative diseases, obesity, diabetes, and cancer [Citation88,Citation89]. ROS can be countered by antioxidants, but the therapeutic application of antioxidants has yielded disappointing results, possibly because only a small fraction of these compounds are taken up by mitochondria [Citation88]. Hence, mitochondrial targeting was employed to accumulate antioxidants within mitochondria [Citation89]. One of the best characterized mitochondria-targeted antioxidants is mitochondrially targeted coenzyme Q (MitoQ) containing the triphenylphosphonium (TPP+) moiety (reviewed in [Citation89,Citation90]). In mitochondria, the reduced form of MitoQ is oxidized, followed by its rapid re-reduction at CII, which was documented to act as a protective mechanism in different cell models of mitochondrial oxidative stress [Citation91] and neuroprotection [Citation92]. Furthermore, MitoQ was studied in metabolic syndrome and proved to be effective against hypercholesterolemia, hypertriglyceridemia, mtDNA oxidative damage, hyperglycemia, and hepatic steatosis (reviewed in [Citation83]).
Mitochondrial ROS production is involved in I/R injury, and CII inhibitors exert protective effects in different I/R models by suppressing RET [Citation93–96]. Mitochondria-targeted tanshinone IIA, a new CII inhibitor, was developed and showed to be protective in I/R oxidative injury [Citation97]. A similar effect was shown for the ferulic acid derivative hmy-paa (3-(4-hydroxy-3-methoxyphenyl)-N-(1H-pyrazol-3-yl)acrylamide) [Citation98]. This is because during the ischemic phase of I/R accumulated succinate is quickly oxidized upon oxygen availability, resulting in massive RET and ROS generation at CI. CII inhibitors, such as malonate, that prevent electron transfer through CII to the ubiquonine pool, therefore, prevent RET and ROS production, are protective during I/R or cold ischemia [Citation99,Citation100]. However, it has also been proposed that CII-dependent reserved respiratory capacity affords cardioprotection during cardiomyocyte recovery from hypoxia [Citation101].
Mills et al. showed that CII-induced ROS production by RET is involved in LPS-stimulated macrophage activity. Succinate-dependent ROS generation was observed, resulting in pro-inflammatory responses, while inhibition of CII by malonate promoted an anti-inflammatory outcome [Citation102]. Interventions at CII can thus regulate inflammation which is associated with numerous metabolic and cardiovascular disorders [Citation103]. Cardioprotective effects of diazoxide was linked directly to inhibition of SDH [Citation104]. In addition, inhibition of CII with 3-NPA reduced glucose-stimulated insulin secretion and ROS production, thereby offering new directions in treatment of cell damage in diabetes [Citation105]. Conversely, while most therapies are focused on inhibiting CII and ROS production, stimulation of SDH activity by succinate administration drives production of ROS and thermogenic respiration in brown adipose tissue, which may stimulate protection against diet-induced obesity and improve glucose tolerance [Citation106]. These findings suggest that targeting CII and CII-driven ROS production may broaden the potential treatment of metabolic disorders.
It has been proposed that CII may function as a general sensor for apoptosis [Citation59,Citation107], making CII a regulator of cell death [Citation108]. Indeed, blockade of the Q site of CII can induce apoptosis by stimulating ROS production from FAD. The amplitude of cell death is directly proportional to the amount of CII-produced ROS [Citation45]. Thus, CII can be targeted for cancer therapy, and efficient experimental anti-cancer agents directed at the Q site have been developed [Citation42–44,Citation109]. The list of potential chemicals to manipulate CII and CII-dependent ROS has been recently updated, including α-TOS, mitoVES, 3-bromopyruvate, malonate, 3-NPA, TTFA, atpenins, lonidamine, and DT-010 as possible candidates for cancer therapy [Citation110]. In addition to the direct effect on cancer cells, some agents also reduce tumor angiogenesis [Citation111,Citation112]. Furthermore, non-toxic doses of the Q site inhibitor TTFA sensitize cancer cells to cell death regulated by other drugs [Citation113], suggesting that CII has potential in combinational cancer therapy. This is in line with CII being an important player in cell death induction. Additionally, it has recently been shown that tumors carrying SDHB mutations produce more ROS and accumulate iron, and disruption of redox hemostasis by ascorbic acid to induce cell death seems to be a promising tool for the treatment of SDHB-mutated PGL/PHEO [Citation114].
Conclusions
Accumulating evidence suggests that CII is an important and underestimated source and modulator of ROS in physiological and pathophysiological conditions that can be manipulated to both induce and suppress cell death, depending on the scenario. Since literature on the therapeutic application of CII modulation in cancer, neurodegeneration, and other pathologies is still fractional, a better understanding of the basic mechanisms of ROS regulation by CII in disease may lead to new therapeutic approaches.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002 Jan;82(1):47–95.
- Zorov DB, Bannikova SY, Belousov VV, et al. Reactive oxygen and nitrogen species: friends or foes? Biochem Biokhimiia. 2005 Feb;70(2):215–221.
- Andreyev AY, Kushnareva YE, Murphy AN, et al. Mitochondrial ROS metabolism: 10 years later. Biochem Biokhimiia. 2015 May;80(5):517–531.
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006 Oct 19;443(7113):787–795.
- Boveris A, Navarro A. Brain mitochondrial dysfunction in aging. IUBMB Life. 2008 May;60(5):308–314.
- Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. 2007 Aug;1767(8):1007–1031.
- Costa A, Scholer-Dahirel A, Mechta-Grigoriou F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin Cancer Biol. 2014 Apr;25:23–32.
- Assi M. The differential role of reactive oxygen species in early and late stages of cancer. Am J Physiol Regul Integr Comp Physiol. 2017 Dec 1;313(6):R646–r653.
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009 Jan 1;417(1):1–13.
- Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, et al. Mitochondria and reactive oxygen species. Free Radic Biol Med. 2009 Aug 15;47(4):333–343.
- Drose S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol. 2012;748:145–169.
- Yankovskaya V, Horsefield R, Tornroth S, et al. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003 Jan 31;299(5607):700–704.
- Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta. 2011 Nov;1807(11):1432–1443.
- Iverson TM, Maklashina E, Cecchini G. Structural basis for malfunction in complex II. J Biol Chem. 2012 Oct 12;287(42):35430–35438.
- Votyakova TV, Reynolds IJ. Deltapsi(m)-dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001 Oct;79(2):266–277.
- Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002 Mar;80(5):780–787.
- Muller FL, Liu Y, Abdul-Ghani MA, et al. High rates of superoxide production in skeletal-muscle mitochondria respiring on both complex I- and complex II-linked substrates. Biochem J. 2008 Jan 15;409(2):491–499.
- Treberg JR, Quinlan CL, Brand MD. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J Biol Chem. 2011 Aug 5;286(31):27103–27110.
- Ralph SJ, Moreno-Sanchez R, Neuzil J, et al. Inhibitors of succinate: quinone reductase/complex II regulate production of mitochondrial reactive oxygen species and protect normal cells from ischemic damage but induce specific cancer cell death. Pharm Res. 2011 Nov;28(11):2695–2730.
- Beinert H. Spectroscopy of succinate dehydrogenases, a historical perspective. Biochim Biophys Acta. 2002 Jan 17;1553(1-2):7–22.
- Cecchini G. Function and structure of complex II of the respiratory chain. Annu Rev Biochem. 2003;72:77–109.
- Sun F, Huo X, Zhai Y, et al. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005 Jul 1;121(7):1043–1057.
- Bezawork-Geleta A, Rohlena J, Dong L, et al. Mitochondrial complex II: At the crossroads. Trends Biochem Sci. 2017 Apr;42(4):312–325.
- Hao HX, Khalimonchuk O, Schraders M, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009 Aug 28;325(5944):1139–1142.
- Ghezzi D, Goffrini P, Uziel G, et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet. 2009 Jun;41(6):654–656.
- Na U, Yu W, Cox J, et al. The LYR factors SDHAF1 and SDHAF3 mediate maturation of the iron-sulfur subunit of succinate dehydrogenase. Cell Metab. 2014 Aug 5;20(2):253–266.
- Van Vranken JG, Bricker DK, Dephoure N, et al. SDHAF4 promotes mitochondrial succinate dehydrogenase activity and prevents neurodegeneration. Cell Metab. 2014 Aug 5;20(2):241–252.
- Belt K, Van Aken O, Murcha M, et al. An assembly factor promotes assembly of flavinated SDH1 into the succinate dehydrogenase complex. Plant Physiol. 2018 Aug;177(4):1439–1452.
- Sharma P, Maklashina E, Cecchini G, et al. Crystal structure of an assembly intermediate of respiratory complex II. Nat Commun. 2018;9(1):274.
- Sharma P, Maklashina E, Cecchini G, et al. Maturation of the respiratory complex II flavoprotein. Curr Opin Struct Biol. 2019 Mar 6;59:38–46.
- Maher MJ, Herath AS, Udagedara SR, et al. Crystal structure of bacterial succinate:quinone oxidoreductase flavoprotein SdhA in complex with its assembly factor SdhE. Proc Natl Acad Sci USA. 2018 Mar 20;115(12):2982–2987.
- Bezawork-Geleta A, Wen H, Dong L, et al. Alternative assembly of respiratory complex II connects energy stress to metabolic checkpoints. Nat Commun. 2018 Jun 7;9(1):2221.
- Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. 2008 Dec;1147:37–52.
- Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). J Biol Chem. 2004 Sep 17;279(38):39414–39420.
- Moreno-Sanchez R, Hernandez-Esquivel L, Rivero-Segura NA, et al. Reactive oxygen species are generated by the respiratory complex II–evidence for lack of contribution of the reverse electron flow in complex I. FEBS J. 2013 Feb;280(3):927–938.
- Quinlan CL, Orr AL, Perevoshchikova IV, et al. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012 Aug 3;287(32):27255–27264.
- Siebels I, Drose S. Q-site inhibitor induced ROS production of mitochondrial complex II is attenuated by TCA cycle dicarboxylates. Biochim Biophys Acta. 2013 Oct;1827(10):1156–1164.
- Bonke E, Zwicker K, Drose S. Manganese ions induce H2O2 generation at the ubiquinone binding site of mitochondrial complex II. Arch Biochem Biophys. 2015 Aug 15;580:75–83.
- Drose S, Hanley PJ, Brandt U. Ambivalent effects of diazoxide on mitochondrial ROS production at respiratory chain complexes I and III. Biochim Biophys Acta. 2009 Jun;1790(6):558–565.
- Drose S, Bleier L, Brandt U. A common mechanism links differently acting complex II inhibitors to cardioprotection: modulation of mitochondrial reactive oxygen species production. Mol Pharmacol. 2011 May;79(5):814–822.
- Miyadera H, Shiomi K, Ui H, et al. Atpenins, potent and specific inhibitors of mitochondrial complex II (succinate-ubiquinone oxidoreductase). Proc Natl Acad Sci USA. 2003 Jan 21;100(2):473–477.
- Dong LF, Low P, Dyason JC, et al. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene. 2008 Jul 17;27(31):4324–4335.
- Dong LF, Jameson VJ, Tilly D, et al. Mitochondrial targeting of vitamin E succinate enhances its pro-apoptotic and anti-cancer activity via mitochondrial complex II. J Biol Chem. 2011 Feb 4;286(5):3717–3728.
- Dong LF, Jameson VJ, Tilly D, et al. Mitochondrial targeting of alpha-tocopheryl succinate enhances its pro-apoptotic efficacy: a new paradigm for effective cancer therapy. Free Radic Biol Med. 2011 Jun 1;50(11):1546–1555.
- Kluckova K, Sticha M, Cerny J, et al. Ubiquinone-binding site mutagenesis reveals the role of mitochondrial complex II in cell death initiation. Cell Death Dis. 2015 May 7;6:e1749.
- Mbaya E, Oules B, Caspersen C, et al. Calcium signalling-dependent mitochondrial dysfunction and bioenergetics regulation in respiratory chain complex II deficiency. Cell Death Differ. 2010 Dec;17(12):1855–1866.
- Drose S. Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre- and postconditioning. Biochim Biophys Acta. 2013 May;1827(5):578–587.
- Guzy RD, Sharma B, Bell E, et al. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008 Jan;28(2):718–731.
- Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005 Jan;7(1):77–85.
- Goffrini P, Ercolino T, Panizza E, et al. Functional study in a yeast model of a novel succinate dehydrogenase subunit B gene germline missense mutation (C191Y) diagnosed in a patient affected by a glomus tumor. Hum Mol Genet. 2009 May 15;18(10):1860–1868.
- Ishii T, Miyazawa M, Onodera A, et al. Mitochondrial reactive oxygen species generation by the SDHC V69E mutation causes low birth weight and neonatal growth retardation. Mitochondrion. 2011 Jan;11(1):155–165.
- Li J, Liang N, Long X, et al. SDHC-related deficiency of SDH complex activity promotes growth and metastasis of hepatocellular carcinoma via ROS/NFkappaB signaling. Cancer Lett. 2019 Oct 1;461:44–55.
- Owens KM, Aykin-Burns N, Dayal D, et al. Genomic instability induced by mutant succinate dehydrogenase subunit D (SDHD) is mediated by O2(-*) and H2O2. Free Radic Biol Med. 2012 Jan 1;52(1):160–166.
- Chang YL, Hsieh MH, Chang WW, et al. Instability of succinate dehydrogenase in SDHD polymorphism connects reactive oxygen species production to nuclear and mitochondrial genomic mutations in yeast. Antioxid Redox Signal. 2015 Mar 1;22(7):587–602.
- Messner KR, Imlay JA. Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J Biol Chem. 2002 Nov 8;277(45):42563–42571.
- Guo J, Lemire BD. The ubiquinone-binding site of the Saccharomyces cerevisiae succinate-ubiquinone oxidoreductase is a source of superoxide. J Biol Chem. 2003 Nov 28;278(48):47629–47635.
- Szeto SS, Reinke SN, Sykes BD, et al. Ubiquinone-binding site mutations in the Saccharomyces cerevisiae succinate dehydrogenase generate superoxide and lead to the accumulation of succinate. J Biol Chem. 2007 Sep 14;282(37):27518–27526.
- Maklashina E, Rajagukguk S, Iverson TM, et al. The unassembled flavoprotein subunits of human and bacterial complex II have impaired catalytic activity and generate only minor amounts of ROS. J Biol Chem. 2018 May 18;293(20):7754–7765.
- Lemarie A, Huc L, Pazarentzos E, et al. Specific disintegration of complex II succinate:ubiquinone oxidoreductase links pH changes to oxidative stress for apoptosis induction. Cell Death Differ. 2011 Feb;18(2):338–349.
- Courage C, Jackson CB, Hahn D, et al. SDHA mutation with dominant transmission results in complex II deficiency with ocular, cardiac, and neurologic involvement. Am J Med Genet A. 2017 Jan;173(1):225–230.
- Rustin P, Rotig A. Inborn errors of complex II–unusual human mitochondrial diseases. Biochim Biophys Acta. 2002 Jan 17;1553(1-2):117–122.
- Jain-Ghai S, Cameron JM, Al Maawali A, et al. Complex II deficiency–a case report and review of the literature. Am J Med Genet A. 2013 Feb;161a(2):285–294.
- Brouillet E, Hantraye P, Ferrante RJ, et al. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA. 1995 Jul 18;92(15):7105–7109.
- Gu M, Gash MT, Mann VM, et al. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol. 1996 Mar;39(3):385–389.
- Benchoua A, Trioulier Y, Zala D, et al. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated Huntingtin. Mol Biol Cell. 2006 Apr;17(4):1652–1663.
- Naseri NN, Bonica J, Xu H, et al. Novel metabolic Abnormalities in the tricarboxylic acid cycle in Peripheral cells from Huntington's disease patients. PLoS One. 2016;11(9):e0160384–e0160384.
- Alston CL, Davison JE, Meloni F, et al. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J Med Genet. 2012 Sep;49(9):569–577.
- Bourgeron T, Rustin P, Chretien D, et al. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 1995 Oct;11(2):144–149.
- Parfait B, Chretien D, Rotig A, et al. Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum Genet. 2000 Feb;106(2):236–243.
- Pagnamenta AT, Hargreaves IP, Duncan AJ, et al. Phenotypic variability of mitochondrial disease caused by a nuclear mutation in complex II. Mol Genet Metab. 2006 Nov;89(3):214–221.
- Jackson CB, Nuoffer JM, Hahn D, et al. Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. J Med Genet. 2014 Mar;51(3):170–175.
- Pang Y, Liu Y, Pacak K, et al. Pheochromocytomas and paragangliomas: from genetic diversity to targeted therapies. Cancers. 2019;11(4):436.
- Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA. 2011 Jan 4;108(1):314–318.
- Neumann HPH, Pawlu C, Pęczkowska M, et al. Distinct clinical features of paraganglioma Syndromes associated With SDHB and SDHD Gene mutations. JAMA. 2004;292(8):943–951.
- Ricketts C, Woodward ER, Killick P, et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100(17):1260–1262.
- Ni Y, Zbuk KM, Sadler T, et al. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet. 2008 Aug;83(2):261–268.
- Ngo DTM, Sverdlov AL, Karki S, et al. Oxidative modifications of mitochondrial complex II are associated with insulin resistance of visceral fat in obesity. Am J Physiol Endocrinol Metab. 2019 Feb 1;316(2):E168–e177.
- He J, Watkins S, Kelley DE. Skeletal muscle lipid content and oxidative enzyme activity in relation to muscle fiber type in type 2 diabetes and obesity. Diabetes. 2001 Apr;50(4):817–823.
- Bonomini F, Rodella LF, Rezzani R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015;6(2):109–120.
- Maslov LN, Naryzhnaya NV, Boshchenko AA, et al. Is oxidative stress of adipocytes a cause or a consequence of the metabolic syndrome? J Clin Transl Endocrinol. 2019 Mar;15:1–5.
- Sverdlov AL, Elezaby A, Qin F, et al. Mitochondrial reactive oxygen species mediate Cardiac Structural, functional, and mitochondrial Consequences of diet-induced metabolic Heart disease. J Am Heart Assoc. 2016 Jan;5(1):e002555. DOI:10.1161/JAHA.115.002555.
- Pessayre D, Berson A, Fromenty B, et al. Mitochondria in steatohepatitis. Semin Liver Dis. 2001;21(1):57–69.
- Ajith TA. Role of mitochondria and mitochondria-targeted agents in non-alcoholic fatty liver disease. Clin Exp Pharmacol Physiol. 2018;45(5):413–421.
- Oberbach A, Bossenz Y, Lehmann S, et al. Altered fiber distribution and fiber-specific glycolytic and oxidative enzyme activity in skeletal muscle of patients with type 2 diabetes. Diabetes Care. 2006 Apr;29(4):895–900.
- Monaco CMF, Hughes MC, Ramos SV, et al. Altered mitochondrial bioenergetics and ultrastructure in the skeletal muscle of young adults with type 1 diabetes. Diabetologia. 2018;61(6):1411–1423.
- Fromenty B, Fisch C, Berson A, et al. Dual effect of amiodarone on mitochondrial respiration. Initial protonophoric uncoupling effect followed by inhibition of the respiratory chain at the levels of complex I and complex II. J Pharmacol Exp Ther. 1990 Dec;255(3):1377–1384.
- Fazakerley DJ, Chaudhuri R, Yang P, et al. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife. 2018;7:e32111.
- Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656.
- Smith RA, Murphy MP. Mitochondria-targeted antioxidants as therapies. Discov Med. 2011 Feb;11(57):106–114.
- Cocheme HM, Kelso GF, James AM, et al. Mitochondrial targeting of quinones: therapeutic implications. Mitochondrion. 2007 Jun;7(Suppl):S94–S102.
- James AM, Sharpley MS, Manas AR, et al. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem. 2007 May 18;282(20):14708–14718.
- Zhou J, Wang H, Shen R, et al. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am J Transl Res. 2018;10(6):1887–1899.
- Ozcan C, Bienengraeber M, Dzeja PP, et al. Potassium channel openers protect cardiac mitochondria by attenuating oxidant stress at reoxygenation. Am J Physiol Heart Circ Physiol. 2002 Feb;282(2):H531–H539.
- Ozcan C, Terzic A, Bienengraeber M. Effective pharmacotherapy against oxidative injury: alternative utility of an ATP-sensitive potassium channel opener. J Cardiovasc Pharmacol. 2007 Oct;50(4):411–418.
- Wojtovich AP, Brookes PS. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol. 2009 Mar;104(2):121–129.
- Valls-Lacalle L, Barba I, Miro-Casas E, et al. Selective inhibition of succinate dehydrogenase in reperfused myocardium with intracoronary malonate reduces infarct size. Sci Rep. 2018 Feb 5;8(1):2442.
- Zhao YP, Wang F, Jiang W, et al. A mitochondrion-targeting tanshinone IIA derivative attenuates myocardial hypoxia reoxygenation injury through a SDH-dependent antioxidant mechanism. J Drug Target. 2019 Sep;27(8):896–902.
- Wang F, Peng Q, Liu J, et al. A novel ferulic acid derivative attenuates myocardial cell hypoxia reoxygenation injury through a succinate dehydrogenase dependent antioxidant mechanism. Eur J Pharmacol. 2019 Aug 5;856:172417.
- Chouchani ET, Pell VR, Gaude E, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014 Nov 20;515(7527):431–435.
- Martin JL, Costa ASH, Gruszczyk AV, et al. Succinate accumulation drives ischaemia-reperfusion injury during organ transplantation. Nat Metab. 2019;1:966–974.
- Pfleger J, He M, Abdellatif M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015 Jul 30;6:e1835.
- Mills EL, Kelly B, Logan A, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016 Oct 6;167(2):457–470. e13.
- Forrester SJ, Kikuchi DS, Hernandes MS, et al. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122(6):877–902.
- Anastacio MM, Kanter EM, Makepeace C, et al. Cardioprotective mechanism of diazoxide involves the inhibition of succinate dehydrogenase. Ann Thorac Surg. 2013 Jun;95(6):2042–2050.
- Edalat A, Schulte-Mecklenbeck P, Bauer C, et al. Mitochondrial succinate dehydrogenase is involved in stimulus-secretion coupling and endogenous ROS formation in murine beta cells. Diabetologia. 2015;58(7):1532–1541.
- Mills EL, Pierce KA, Jedrychowski MP, et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature. 2018 Aug;560(7716):102–106.
- Grimm S. Respiratory chain complex II as general sensor for apoptosis. Biochim Biophys Acta. 2013 May;1827(5):565–572.
- Hwang MS, Rohlena J, Dong LF, et al. Powerhouse down: complex II dissociation in the respiratory chain. Mitochondrion. 2014 Nov;19(Pt A):20–28.
- Guo L, Shestov AA, Worth AJ, et al. Inhibition of mitochondrial complex II by the Anticancer Agent lonidamine. J Biol Chem. 2016 Jan 1;291(1):42–57.
- Pozza E D, Dando I, Pacchiana R, et al. Regulation of succinate dehydrogenase and role of succinate in cancer. Semin Cell Dev Biol. 2020 Feb;98:4–14.
- Rohlena J, Dong LF, Kluckova K, et al. Mitochondrially targeted alpha-tocopheryl succinate is antiangiogenic: potential benefit against tumor angiogenesis but caution against wound healing. Antioxid Redox Signal. 2011 Dec 15;15(12):2923–2935.
- Dong LF, Swettenham E, Eliasson J, et al. Vitamin E analogues inhibit angiogenesis by selective induction of apoptosis in proliferating endothelial cells: the role of oxidative stress. Cancer Res. 2007 Dec 15;67(24):11906–11913.
- Kruspig B, Valter K, Skender B, et al. Targeting succinate:ubiquinone reductase potentiates the efficacy of anticancer therapy. Biochim Biophys Acta. 2016 Aug;1863(8):2065–2071.
- Liu Y, Pang Y, Zhu B, et al. Therapeutic targeting of SDHB-mutated pheochromocytoma/paraganglioma with pharmacologic ascorbic acid. Clin Cancer Res. 2020 Mar. DOI:10.1158/1078-0432.CCR-19-2335.