
ABSTRACT
Objectives: Inflammation is an important predisposing and progressive factor in chronic kidney disease (CKD). Heterogeneous nuclear ribonucleoprotein K (hnRNPK) is associated with many fundamental cellular processes, but in chronic inflammatory pathologies remains unclear.
Methods: An in vitro peripheral inflammation model was established using lipopolysaccharide (LPS)-stimulated mouse RAW264.7 macrophages, followed by inflammasome activation by ATP treatment. Knockdown of hnRNPK by sihnRNPK and FLICE-like inhibitory protein (FLIP) by siFLIP transfection were achieved in Raw264.7 macrophages. ELISA was used to determine the expression of IL-1β, IL-18 and TNF-α. Real time PCR was applied to detect the mRNA levels of hnRNPK, NOD-like receptors family pyrin domain-containing 3 (NLRP3), FLIP, Caspase-1, IL-1β and IL-18. Western blot and immunofluorescence were performed to detect relevant protein expressions. Co-immunoprecipitation (Co-IP) was used to assess the interaction of hnRNPK with FLIP.
Results: Results showed that LPS plus ATP activated NLRP3 inflammasome, which evidenced by the up-regulation of TNF-α, IL-1β and IL-18. Notably, hnRNPK and FLIP were significantly up-regulated in activated NLRP3 inflammasome of macrophages. HnRNPK or FLIP knockdown significantly suppressed the activation of NLRP3 inflammasome, as reflected by down-regulation of Caspase-1, IL-1β and IL-18. Importantly, hnRNPK could directly bind to FLIP in activated NLRP3 inflammasome.
Discussion: Our findings suggest that hnRNPK could promote the activation of NLRP3 inflammasome by directly binding FLIP, which might provide potential new therapeutic targets for CKD.
GRAPHICAL ABSTRACT
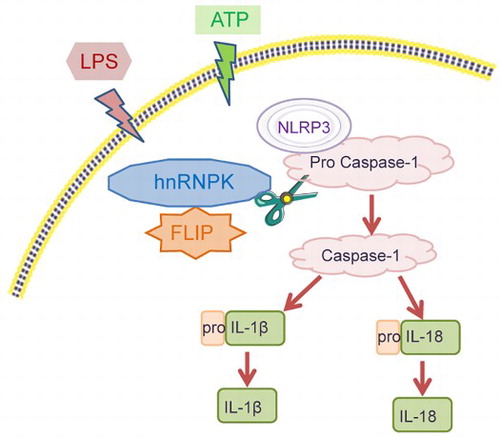
Introduction
Chronic kidney disease (CKD) is a serious health issue affecting kidney structure and function with an increasing prevalence rate worldwide [Citation1]. Most CKD patients share several risk factors, including hypertension, diabetes, obesity, and metabolic alterations [Citation2–4]. Unfortunately, there is no radical cure for CKD, but preserve the residual kidney as far as possible to delay the progression to end-stage renal failure.
Accumulating studies have shown that inflammatory response is an important predisposing and progressive factor in CKD [Citation5–7]. Inflammasomes, multi-protein cytoplasmic complexes, function as pattern recognition receptors to participate in activation of inflammatory caspases, various immune and cellular death pathways [Citation8,Citation9]. By activating caspase-1, inflammasome cleaves pro-inflammatory cytokines IL-1β and IL-18 to their active and secreted forms [Citation10]. NOD-like receptors family pyrin domain-containing 3 (NLRP3) inflammasome is recently considered to be the best characterized inflammasome, which could trigger caspase-1 activation and inflammation factors release in response to diverse stimuli in CKD patients [Citation5,Citation11]. To date, several studies have demonstrated that elevated levels of NLRP3, caspase-8, IL-1β and IL-18 are critical components of inflammatory process in models of kidney disease [Citation12–15].
Heterogeneous nuclear ribonucleoprotein K (hnRNPK) contains three K homology (KH) domains responsible for DNA/RNA binding and take part in the regulation of gene transcription, RNA splicing and mRNA translation [Citation16] and one K protein interactive region (KI) domain responsible for protein-protein interactions [Citation17], which is one of the most extensively studied hnRNP family members [Citation18]. High expression of hnRNPK is associated with tumor formation, development and prognosis, including colorectal adenocarcinoma [Citation19], oral squamous cell carcinoma [Citation20] and pancreatic cancer [Citation21]. Interestingly, a previous study indicated that hnRNPK could mediate insulin inhibition of renal angiotensinogen gene expression and prevention hypertension and kidney injury in diabetic mice [Citation22]. Another research demonstrated that hnRNPK could promote the activation of macrophages by regulating the lipopolysaccharide (LPS)-induced TAK1 mRNA translation, and up-regulate the transcription of TNF-α, IL-1α, IL-1β and IL-10 [Citation23]. FLICE-like inhibitory protein (FLIP) is found to be critical for protecting rheumatoid arthritis synovial macrophages from Fas-mediated apoptosis [Citation24]. A study from Huang et al. [Citation25] showed that reduction of FLIP in macrophages might be an effective therapeutic approach to suppress inflammation depending upon the disease stage. Additionally, it has been reported that hnRNPK could up-regulate the transcriptional level of cellular FLIP to negatively regulate the tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) [Citation26]. Our previous study showed that the protein level of FLIP was positively correlated with hnRNPK. These findings imply that hnRNPK may potentially play a role in chronic inflammatory pathologies by binding FLIP.
Raw264.7 macrophages are commonly used to explore kidney disease related to inflammation in vitro [Citation27,Citation28]. As a main component of the cell wall of gram-negative bacteria, LPS widely used to cause inflammatory response in vitro and in vivo, including kidney disease [Citation29,Citation30]. To validate our hypothesis that hnRNPK binding FLIP in NLRP3 inflammasome activation, we first prepared a peripheral inflammation model with LPS-stimulated Raw264.7 macrophages to investigate whether hnRNPK participates in activation of inflammatory macrophages by regulating cellular inflammation reaction. Moreover, whether FLIP was involved in the regulation of hnRNPK in NLRP3 inflammasome activation was further determined.
Materials and methods
Cell culture and treatment
Raw264.7 macrophages were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured in RPMI-1640 medium (Gibco) with 10% fetal bovine serum (FBS, Gibco) in a humidified incubator containing 5% CO2 at 37°C. For constructing inflammasome, RAW264.7 macrophages were treated with 20 ng/mL LPS (Sigma) for 4 h, followed by 5 nM ATP (Sigma) incubation for 30 min. RAW264.7 macrophages were divided into four groups: Control, LPS, ATP and LPS + ATP.
ELISA
ELISA assay was performed to detect the levels of cytokines released from Raw264.7 macrophages from four groups. In brief, the supernatants were collected and the expression of TNF-α, IL-1β and IL-18 were determined with their respective ELISA kits according to the manufacturer’s instructions (Elabscience). According to the drawn standard curves, the concentration of TNF-α, IL-1β and IL-18 were calculated.
Western blot
Raw264.7 macrophages were washed with phosphate buffer saline (PBS) and lysed with RIPA (Biosharp) for 30 min. A total of 30 μg protein was separated by 10% SDS-PAGE and electro-transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). The membranes were blocked in TBS with 0.2% Tween 20 (TBST) containing 5% (W/V) dried skimmed milk for 1 h at room temperature. Then, the membranes were incubated with primary antibodies against hnRNPK, FLIP, NLRP3, pro Caspase-1, Caspase-1, pro IL-1β, IL-1β, pro IL-18, IL-18 and GAPDH (Abcam) overnight, followed by incubation with corresponding HRP-conjugated secondary antibodies. The membranes were subsequently exposed to enhanced ECL chemiluminescent substrate (Beyotime Biotech). GAPDH was used as an internal control.
Cell immunofluorescence
RAW264.7 macrophages were fixed with 4% paraformaldehyde for 15 min and permeated with 0.3% Triton X-100 for 15 min at room temperature. After washed with PBS for 1 h, the cells were incubated with primary antibodies against hnRNPK, FLIP and NLRP3 (Abcam) at 4 °C overnight. The coverslips were exposed to Alexa Fluor conjugated-secondary antibodies (Cell Signaling Technology) for 1 h in the dark. The nucleus was marked with 4′, 6-Diamidino-2-phenylindole (DAPI). The stained images were observed using an Inverted/Fluorescence Microscope (LIONHEART LX, BioTek).
Cell transfection
Three different small interfering RNA sequences targeting hnRNPK (sihnRNPK-1: 5′-GAGGAAUAAUUGGUGUUAAUU-3′, sihnRNPK-2: 5′-GGGAGAUCUAAUGGCUUAUUU-3′ and sihnRNPK-3: 5′-GCCCAUCAGAAUGGCAAAUUU-3′), three different siRNA targeting FLIP (siFLIP-1: 5′-CCUCCUGGAUAGCUUAAGUUU-3′, siFLIP-2: 5′-CACCUGGUUUCUGAUUAUAUU-3′ and siFLIP-3: 5′-GGCCCAACAUCAAGACUAUUU-3′) and negative control siRNA (si-NC: 5′-UUCUCCGAACGUGUCACGUTT-3′) were synthesized by RiboBio (Guangzhou, China). Knockdown of hnRNPK or FLIP was achieved using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Briefly, RAW264.7 macrophages were cultured in 6-well plates at 37 °C overnight and transfected with different sihnRNPK or siFLIP at a final concentration of 100 nM for 48 h, followed by LPS and ATP treatment. Subsequently, the macrophages were collected for the in vitro experiments.
Real time PCR
Total RNA was extracted from macrophages using Trizol reagent (TAKARA). After RNA quantification using spectrophotometry, cDNA was synthesized using a Reverse Aid first strand cDNA synthesis kit (DBI Bioscience). Quantitative real time PCR analysis was performed on Stratagene Real-Time System (AgilentA) using SYBR Green PCR reagent kit (DBI Bioscience). The specific primer sequences were as follows: hnRNPK: forward 5′-CAGCTCCCGCTCGAATCTG-3′ and reverse 5′-ACCCTATCAGGTTTTCCTCCAA-3′; FLIP: forward 5′-TTACACAGGCAGAGGCAAGA-3′ and reverse 5′-GCTGGACTGGGTGTACTTCT-3′; NLRP3: forward 5′-GTGTTGTCAGGATCTCGCAT-3′ and reverse 5′-CTGCAAGTTACACTGTGGGT-3′; Caspase-1: forward 5′-CATCTTTCTCCGAGGGTTGG-3′ reverse 5′-TGTGGTCCCACATATTCCCT-3′; IL-1β: forward 5′-AATGCCACCTTTTGACAGTGATG-3′ and reverse 5′-TGTGCTGCTGCGAGATTTG-3′; IL-18: forward 5′-GACTCTTGCGTCAACTTCAAGG-3′ and reverse 5′-CAGGCTGTCTTTTGTCAACGA-3′; β-actin: forward 5′-CATTGCTGACAGGATGCAGA-3′ and reverse 5′-CTGCTGGAAGGTGGACAGTGA-3′. The gene expression was quantified using 2−ΔΔCT method with β-actin as internal control.
Co-immunoprecipitation (Co-IP)
Endogenous Co-IP was performed to validate the interaction between hnRNPK and FLIP. Briefly, harvested macrophages from control, LPS + ATP + si-NC or LPS + ATP + si-hnRNPK groups were dealt with lysis buffer. Total protein was incubated with specific antibodies against FLIP on shaking tables, followed by incubation with protein G-sepharose beads (Thermo Fisher Scientific, Waltham, MA, USA). Then, the immunoprecipitated samples were washed with pre-cooling PBS for three times (5 min each time), followed by western blotting as described previously [Citation31].
Statistical analysis
All experiments were repeated three times. Statistical analysis was performed using SPSS version 20.0 (SPSS Inc., Chicago, IL, USA). Quantitative data were presented as the means ± standard deviation (SD). Differences between two groups were analyzed using an unpaired t test and among groups were assessed by one-way analysis of variance (ANOVA) with Shapiro-Wilk test. The results were considered to be statistically significant when p values less than 0.05.
Results
hnRNPK and FLIP were significantly up-regulated in LPS + ATP-activated NLRP3 inflammasome of macrophages
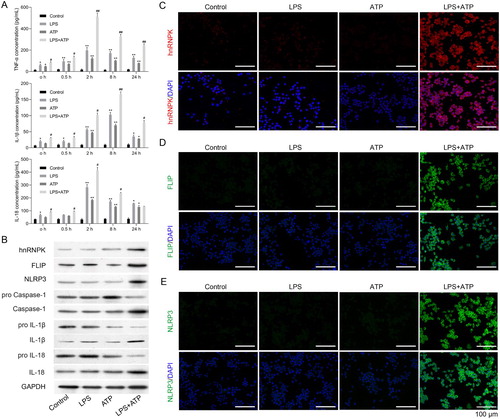
To investigate the role of hnRNPK in NLRP3 inflammasome activation associated with CDK progression, macrophage inflammasome was constructed in RAW264.7 macrophages by LPS with ATP treatment. ELISA assay indicated that LPS single stimulation raised the TNF-α, IL-1β and IL-18 expressions in supernatant of RAW264.7 macrophages (p < 0.05, p < 0.01). ATP single treatment also enhanced the TNF-α, IL-1β and IL-18 expressions in supernatant of RAW264.7 macrophages (p < 0.05, p < 0.01). But the effects of ATP treatment on TNF-α, IL-1β and IL-18 in RAW264.7 macrophages were weaker than LPS stimulation. ATP + LPS treatment induced a rapid release of TNF-α, IL-1β and IL-18 at different time points (A, p < 0.05, p < 0.01). Moreover, the results of western blot displayed that the protein levels of hnRNPK, FLIP and NLRP3 in ATP treatment of LPS-primed macrophages was obviously up-regulated compared with LPS or ATP single group (B). The protein expressions of Caspase-1, IL-1β and IL-18 were also remarkably raised in ATP treatment of LPS-primed macrophages, which were accompanied with the decreased expressions of pro-Caspase-1, pro-IL-1β and pro-IL-18. Furthermore, similar results were found in immunofluorescence, which showed that the expression levels of hnRNPK, NLRP3 and FLIP were all elevated after stimulated by LPS+ ATP treatment (C–E).
Figure 1. hnRNPK and FLIP were significantly up-regulated in LPS + ATP-activated NLRP3 inflammasome of macrophages. RAW264.7 macrophages were incubated with LPS, followed by ATP incubation (A) Inflammatory factors TNF-α, IL-1β and IL-18 expressions were measured at different time points via ELISA. *p < 0.05, **p < 0.01 vs. Control; #p < 0.05, ##p < 0.01 vs. LPS; (B) The protein expressions of hnRNPK, FLIP, NLRP3, pro Caspase-1, Caspase-1, pro IL-1β, IL-1β, pro IL-18 and IL-18 in different groups were evaluated by western blot. The expression and distribution of (C) hnRNPK, (D) FLIP and (E) NLRP3 were determined in four groups were detected via immunofluorescence.
hnRNPK knockdown significantly suppressed NLRP3 inflammasome activation
Since hnRNPK was up-regulated in LPS + ATP-activated NLRP3 inflammasome, we thus performed loss-of-function assay in RAW264.7 macrophages. As shown in A, three different siRNA sequences were designed to silence hnRNPK. The sihnRNPK-1, sihnRNPK-2 and sihnRNPK-3 significantly suppressed the expression of hnRNPK in LPS+ ATP induced macrophages. Notably, sihnRNPK-2 transfection produced the most obvious inhibitory effects, thus sihnRNPK-2 was selected for the subsequent experiments. Next, we further detected the effects of hnRNPK on activation of NLRP3 inflammasome. Real time PCR demonstrated that the mRNA levels of hnRNPK, FLIP, NLRP3, Caspase-1, IL-1β and IL-18 were significantly increased in LPS + ATP-induced macrophages compared with control group (p < 0.01), but remarkably decreased after hnRNPK knockdown (p < 0.01) (B–C). The western blot (A) and immunofluorescence analysis of hnRNPK (B), FLIP (C) and NLRP3 (D) also confirmed a significant decrease of hnRNPK, FLIP and NLRP3 expressions in the hnRNPK silencing NLRP3 inflammasome. Besides, after hnRNPK knockdown, the protein levels of Caspase-1, IL-1β and IL-18 were also reduced in LPS + ATP-induced macrophages (A).
Figure 2. hnRNPK knockdown decreased the expressions of FLIP, NLRP3, Caspase-1, IL-1β and IL-18 in LPS + ATP-activated NLRP3 inflammasome. (A) RAW264.7 macrophages were transfected with three different siRNAs targeting hnRNPK, followed by incubation with LPS and ATP, the mRNA and protein expressions of hnRNPK were detected by real-time PCR and western blot. **p < 0.01 vs. si-NC. RAW264.7 macrophages were transfected with sihnRNPK-2, followed by incubation with LPS and ATP, the mRNA expressions of hnRNPK, FLIP, NLRP3 (B), as well as Caspase-1, IL-1β, IL-18 (C) were detected by real-time PCR. ##p < 0.01 vs. Control, **p < 0.01 vs. LPS + ATP + si-NC.
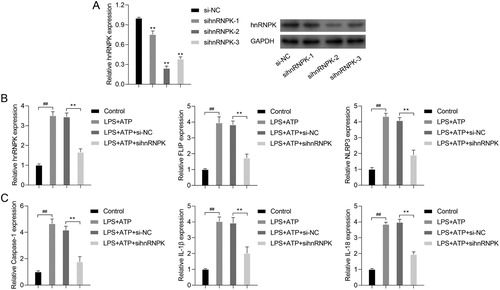
Figure 3. hnRNPK knockdown decreased activated NLRP3 inflammasome related protein levels and bound to FLIP. RAW264.7 macrophages were transfected with si-hnRNPK, followed by incubation with LPS and ATP. (A) The protein expressions of hnRNPK, FLIP, NLRP3, pro Caspase-1, Caspase-1, pro IL-1β, IL-1β, pro IL-18 and IL-18 were evaluated by western blot. The expression and distribution of (B) hnRNPK, (C) FLIP and (D) NLRP3 were determined via immunofluorescence. (E) The binding relationship between hnRNPK and FLIP were detected by co-IP.
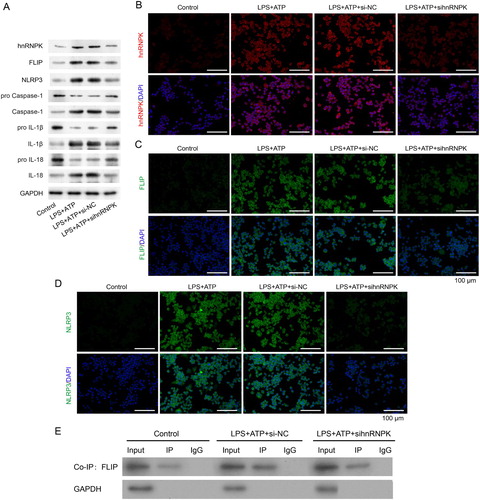
hnRNPK binds to FLIP in LPS + ATP-activated NLRP3 inflammasome
The above results showed that FLIP was down-regulated after hnRNPK knockdown, we thus speculated that FLIP might interact with hnRNPK. To confirm our hypothesis, hnRNPK-FLIP binding was detected by co-IP in LPS + ATP-activated NLRP3 inflammasome. As depicted in E, the interaction of FLIP and hnRNPK was weakened after hnRNPK knockdown in LPS + ATP-activated NLRP3 inflammasome. These results suggest that hnRNPK might affect the activation of NLRP3 inflammasome by directly interacting with FLIP.
FLIP knockdown imitated the effects of hnRNPK silencing on the activation of NLRP3 inflammasome
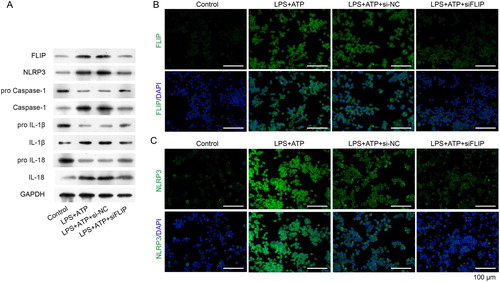
Our previous study showed that FLIP was positively correlated with hnRNPK (data not shown). Based on the physical interaction between FLIP and hnRNPK, we further investigated the biological role of FILP in LPS + ATP-activated NLRP3 inflammasome. First, we designed three siRNA sequences to specific silence FLIP in RAW264.7 macrophages, as confirmed by real time PCR and western blot analysis (A, p < 0.01). It was worthy noted that siFLIP-3 generated the strongest silencing effects on FLIP expression in all the three siRNA sequences (p < 0.01), which was utilized for the subsequent analysis. Consistently with hnRNPK knockdown, silencing FLIP significantly down-regulated the expression of FLIP, NLRP3 and Caspase-1, as well as the inflammatory factors (IL-1β and IL-18), as determined by, real time PCR (B–C, p < 0.01), western blot (A) and immunofluorescence (B and C), respectively. These results demonstrated that FLIP might be involved in the regulatory effect of hnRNPK on NLRP3 inflammasome activation.
Figure 4. FLIP knockdown decreased the expressions of FLIP, NLRP3, Caspase-1, IL-1β and IL-18 in LPS + ATP-activated NLRP3 inflammasome. (A) RAW264.7 macrophages were transfected with three different siRNA targeting FLIP, followed by incubation with LPS and ATP. the mRNA and protein expressions of FLIP were detected by real-time PCR and western blot. **p < 0.01vs. si-NC. RAW264.7 macrophages were transfected with si-FLIP-3, followed by incubation with LPS and ATP, the mRNA expressions of FLIP, NLRP3 (B), as well as Caspase-1, IL-1β, IL-18 (C) were detected by real-time PCR. ##p < 0.01 vs. Control, **p < 0.01 vs. LPS + ATP + si-NC.
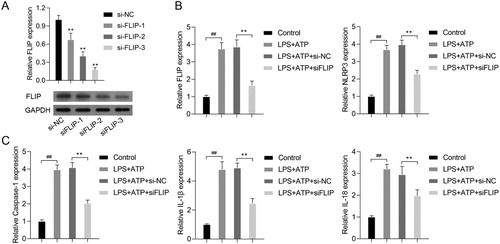
Figure 5. FLIP knockdown decreased activated NLRP3 inflammasome related protein levels. RAW264.7 macrophages were transfected with si-FLIP, followed by incubation with LPS and ATP. (A) The protein expressions of FLIP, NLRP3, pro Caspase-1, Caspase-1, pro IL-1β, IL-1β, pro IL-18 and IL-18 were measured by western blot. (B–C) The expression and distribution of FLIP and NLRP3 were detected by immunofluorescence staining assay.
Discussion
Increasing evidences show that the endotoxin model using LPS has been widely applied to explore the regulatory mechanism of the inflammatory response in several diseases, including acute lung injury [Citation32] and neurodegenerative diseases [Citation33], as well as CKD [Citation34]. Hwang et al. [Citation35] reported that LPS-stimulated Raw264.7 macrophages accelerated pro-inflammatory and pro-fibrotic cytokines, which contributed to accelerate chronic kidney diseases. Nagase et al. [Citation36] indicated that LPS-stimulated Raw264.7 could be serving as an in vitro cell model to simulate renal inflammation and injury. In the present study, in vitro LPS-stimulated mouse RAW264.7 macrophages was used to investigate the regulatory mechanism of hnRNPK and FLIP in NLRP3 inflammasome activation. We found that LPS induced the production of TNF-α, IL-1β and IL-18 in the culture medium of RAW264.7 macrophages. Moreover, LPS plus ATP activated the NLRP3 inflammasome, reflected by up-regulation of TNF-α, IL-1β and IL-18. Consistently, NLRP3 inflammasome consists of NRLP3 and caspase-1 which is implicated in the pathogenesis of inflammatory diseases by elevating the secretion of pro-inflammatory cytokines [Citation37–39]. In addition, loss of NLRP3 significantly reduced inflammation and tubulointerstitial fibrosis in mice after unilateral ureteral obstruction [Citation40]. Herein, we found that both the NLRP3 and Caspase-1 expressions were increased after LPS plus ATP treatment, which further confirmed the activation of NLRP3 inflammasome.
Previous studies have demonstrated that hnRNPK directly related to proliferation, migration and autophagy in several diseases [Citation41–43]. However, it remains unclear whether hnRNPK plays an important role in NLRP3 inflammasome in LPS induced RAW264.7 macrophages. In our study, we found that hnRNPK was significantly up-regulated in activated NLRP3 inflammasome. Knockdown of hnRNPK remarkably alleviated LPS + ATP-induced increased expressions of NLRP3, Caspase-1, IL-1β and IL-18. Bomsztyk et al. [Citation18] has demonstrated that hnRNPK localizes in both the nucleus and cytoplasm and preferentially recognizes poly-C sequences of target RNAs through its three repeats of K homology domains (KH1-3), which play a central role in many fundamental cellular processes, such as cell proliferation, apoptosis and differentiation [Citation44,Citation45]. Similar to our results, Lichtnekert et al. [Citation23] found that hnRNPK contributed to the activation of macrophages by up-regulating the transcription of TNF-α, IL-1A, IL-1B and IL-10. These findings suggested that hnRNPK was closely related to the activation of NLRP3 inflammasome in LPS + ATP-stimulated mouse RAW264.7 macrophages and might play regulatory role in the progression of CKD.
Notably, co-IP assay further showed that hnRNPK directly interacts with FLIP in LPS + ATP-stimulated mouse RAW264.7 macrophages. Moreover, FLIP knockdown imitated the effects of hnRNPK knockdown on the expression of NLRP3, Caspase-1, IL-1β and IL-18 in LPS + ATP-stimulated mouse RAW264.7 macrophages. These was consistent with the previous study, which showed that reduction of FLIP in macrophages could selectively suppress inflammation in arthritis [Citation25]. Taken together, these findings suggested that the connection of hnRNPK-FLIP induced the activation of NLRP3 inflammasome in LPS + ATP-stimulated mouse RAW264.7 macrophages via prompting Caspase-1-medicated IL-1β and IL-18 activation (Graphical abstract).
In conclusion, our study shows that hnRNPK directly binds FLIP to regulate the NLRP3 inflammasome activation in response to LPS + ATP stimulation, which might be a potential regulatory mechanism underlying the progression to CKD.
Author’s contribution statement
Junxia Feng, Hongyan Li, Wei Sun and Yunfang Zhang: Conception and design, financial support, manuscript writing. Jingchun Li and Ping Meng: Administrative support, Collection and assembly of data. Lina Wang, Chunli Liu and Shili Zhao: Data analysis and data interpretation. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article.
Additional information
Funding
References
- Burckhardt P, Nagin D, Vijayasarathy VPR, et al. Multi-trajectory modeling to predict acute kidney injury in chronic kidney disease patients. AMIA Annu Symp Proc AMIA Symp. 2018;2018:1196–1205. PubMed PMID: 30815162; PubMed Central PMCID: PMC6371306.
- Kjeldsen SE. Hypertension and cardiovascular risk: general aspects. Pharmacol Res. 2018 Mar;129:95–99. doi:10.1016/j.phrs.2017.11.003. PubMed PMID: 29127059.
- Maaten JMT, Damman K, Verhaar MC, et al. Connecting heart failure with preserved ejection fraction and renal dysfunction: the role of endothelial dysfunction and inflammation. Eur J Heart Fail. 2016 Jun;18(6):588–598. doi:10.1002/ejhf.497. PubMed PMID: 26861140.
- Boudoulas KD, Triposkiadis F, Parissis J, et al. The cardio-renal interrelationship. Prog Cardiovasc Dis. 2017 May–Jun;59(6):636–648. doi:10.1016/j.pcad.2016.12.003. PubMed PMID: 27993608.
- Granata S, Masola V, Zoratti E, et al. NLRP3 inflammasome activation in dialyzed chronic kidney disease patients. PloS one. 2015;10(3):e0122272. doi:10.1371/journal.pone.0122272. PubMed PMID: 25798846; PubMed Central PMCID: PMC4370586.
- Mihai S, Codrici E, Popescu ID, et al. Inflammation-related mechanisms in chronic kidney disease prediction, progression, and outcome. J Immunol Res. 2018;2018:2180373. doi:10.1155/2018/2180373. PubMed PMID: 30271792; PubMed Central PMCID: PMC6146775.
- Liu C, Li H. Correlation of the severity of chronic kidney disease with serum inflammation, osteoporosis and vitamin D deficiency. Exp Ther Med. 2019 Jan;17(1):368–372. doi:10.3892/etm.2018.6916. PubMed PMID: 30651805; PubMed Central PMCID: PMC6307476.
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002 Aug;10(2):417–426. PubMed PMID: 12191486.
- Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13(4):325–332.
- Bugyei-Twum A, Abadeh A, Thai K, et al. Suppression of NLRP3 inflammasome activation ameliorates chronic kidney disease-induced cardiac fibrosis and diastolic dysfunction. Sci Rep. 2016 Dec 21;6:39551. doi:10.1038/srep39551. PubMed PMID: 28000751; PubMed Central PMCID: PMC5175152.
- Wang L, Ma J, Guo C, et al. Danggui buxue tang attenuates tubulointerstitial fibrosis via suppressing NLRP3 inflammasome in a rat model of unilateral ureteral obstruction. Biomed Res Int. 2016;2016:9368483. doi:10.1155/2016/9368483. PubMed PMID: 27872860; PubMed Central PMCID: PMC5107862.
- Melnikov VY, Ecder T, Fantuzzi G, et al. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J Clin Invest. 2001 May;107(9):1145–1152. doi:10.1172/JCI12089. PubMed PMID: 11342578; PubMed Central PMCID: PMC209282.
- Gurung P, Anand PK, Malireddi RK, et al. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol. 2014 Feb 15;192(4):1835–1846. doi:10.4049/jimmunol.1302839. PubMed PMID: 24453255; PubMed Central PMCID: PMC3933570.
- Melnikov VY, Faubel S, Siegmund B, et al. Neutrophil-independent mechanisms of caspase-1- and IL-18-mediated ischemic acute tubular necrosis in mice. J Clin Invest. 2002 Oct;110(8):1083–1091. doi:10.1172/JCI15623. PubMed PMID: 12393844; PubMed Central PMCID: PMC150794.
- Parikh CR, Edward A, Marek A, et al. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J Am Soc Nephrol – JASN. 2005;16(10):3046.
- Siomi H, Matunis MJ, Michael WM, et al. The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucleic Acids Res. 1993 Mar 11;21(5):1193–1198. PubMed PMID: 8464704; PubMed Central PMCID: PMC309281.
- Ostareck-Lederer A, Ostareck DH, Rucknagel KP, et al. Asymmetric arginine dimethylation of heterogeneous nuclear ribonucleoprotein K by protein-arginine methyltransferase 1 inhibits its interaction with c-Src. J Biol Chem. 2006 Apr 21;281(16):11115–11125. doi:10.1074/jbc.M513053200. PubMed PMID: 16492668.
- Bomsztyk K, Denisenko O, Ostrowski J. hnRNP K: one protein multiple processes. BioEssays: News Rev Mol Cell Develop Biol. 2004 Jun;26(6):629–638. doi:10.1002/bies.20048. PubMed PMID: 15170860.
- Klimek-Tomczak K, Mikula M, Dzwonek A, et al. Editing of hnRNP K protein mRNA in colorectal adenocarcinoma and surrounding mucosa. Br J Cancer. 2006 Feb 27;94(4):586–592. doi:10.1038/sj.bjc.6602938. PubMed PMID: 16404425; PubMed Central PMCID: PMC2361188.
- Roychoudhury P, Chaudhuri K. Evidence for heterogeneous nuclear ribonucleoprotein K overexpression in oral squamous cell carcinoma. Br J Cancer. 2007 Aug 20;97(4):574–575. author reply 576. doi:10.1038/sj.bjc.6603911. PubMed PMID: 17667925; PubMed Central PMCID: PMC2360341.
- He D, Huang C, Zhou Q, et al. hnRNPK/miR-223/FBXW7 feedback cascade promotes pancreatic cancer cell growth and invasion. Oncotarget. 2017;8(12):20165–20178.
- Abdo S, Lo CS, Chenier I, et al. Heterogeneous nuclear ribonucleoproteins F and K mediate insulin inhibition of renal angiotensinogen gene expression and prevention of hypertension and kidney injury in diabetic mice. Diabetologia. 2013 Jul;56(7):1649–1660. doi:10.1007/s00125-013-2910-4. PubMed PMID: 23609310.
- Lichtnekert J, Kulkarni OP, Mulay SR, et al. Anti-GBM glomerulonephritis involves IL-1 but is independent of NLRP3/ASC inflammasome-mediated activation of caspase-1. PloS one. 2011;6(10):e26778.
- Perlman H, Pagliari LJ, Liu H, et al. Rheumatoid arthritis synovial macrophages express the Fas-associated death domain-like interleukin-1beta-converting enzyme-inhibitory protein and are refractory to Fas-mediated apoptosis. Arthriti Rheum. 2001;44(1):21–30.
- Huang QQ, Birket R, Doyle RE, et al. Increased F4/80(hi) macrophages is associated with suppression of serum transfer induced arthritis in mice with flip reduced in myeloid cells. Arthriti Rheum. 2017.
- Chen L-C, Chung I-C, Hsueh C, et al. The antiapoptotic protein, FLIP, is regulated by heterogeneous nuclear ribonucleoprotein K and correlates with poor overall survival of nasopharyngeal carcinoma patients. Cell Death Differ. 2010;17(9):1463–1473.
- Tan RZ, Liu J, Zhang YY, et al. Curcumin relieved cisplatin-induced kidney inflammation through inhibiting Mincle-maintained M1 macrophage phenotype. Phytomedicine: Int J Phytother Phytopharmacol. 2019 Jan;52:284–294. doi:10.1016/j.phymed.2018.09.210. PubMed PMID: 30599909; eng.
- Chenxu G, Minxuan X, Yuting Q, et al. Irhom2 loss alleviates renal injury in long-term PM2.5-exposed mice by suppression of inflammation and oxidative stress. Redox Biol. 2018 Oct;19:147–157. doi:10.1016/j.redox.2018.08.009. PubMed PMID: 30165303; PubMed Central PMCID: PMCPMC6118040. eng.
- Hudalla H, Karenberg K, Kuon RJ, et al. LPS-induced maternal inflammation promotes fetal leukocyte recruitment and prenatal organ infiltration in mice. Pediatr Res. 2018 Nov;84(5):757–764. doi:10.1038/s41390-018-0030-z. PubMed PMID: 30135596; eng.
- Ontawong A, Srimaroeng C, Boonphang O, et al. Spirogyra neglecta aqueous extract attenuates LPS-induced renal inflammation. Biol Pharm Bull. 2019;42(11):1814–1822. doi:10.1248/bpb.b19-00199. PubMed PMID: 31685765; eng.
- Liang S, Yao Q, Wei D, et al. KDM6B promotes ovarian cancer cell migration and invasion by induced transforming growth factor-beta1 expression. J Cell Biochem. 2019 Jan;120(1):493–506. doi:10.1002/jcb.27405. PubMed PMID: 30277596.
- Yang Y, Yang F, Yu X, et al. miR-16 inhibits NLRP3 inflammasome activation by directly targeting TLR4 in acute lung injury. Biomed Pharmacother. 2019 Feb 19;112:108664. doi:10.1016/j.biopha.2019.108664. PubMed PMID: 30784935.
- Pan Y, Shen B, Gao Q, et al. Caspase-1 inhibition attenuates activation of BV2 microglia induced by LPS-treated RAW264.7 macrophages. J Biomed Res. 2016 May;30(3):225–233. doi:10.7555/JBR.30.20150141. PubMed PMID: 27533933; PubMed Central PMCID: PMC4885171.
- Chen H, Zhu J, Liu Y, et al. Lipopolysaccharide induces chronic kidney injury and fibrosis through activation of mTOR signaling in macrophages. Am J Nephrol. 2015;42(4):305–317. doi:10.1159/000441506. PubMed PMID: 26517816.
- Hwang I, Uddin MJ, Lee G, et al. Peroxiredoxin 3 deficiency accelerates chronic kidney injury in mice through interactions between macrophages and tubular epithelial cells. Free Radic Biol Med. 2019 Feb 1;131:162–172. doi:10.1016/j.freeradbiomed.2018.12.002. PubMed PMID: 30529270; eng.
- Nagase M, Kurihara H, Aiba A, et al. Deletion of Rac1GTPase in the myeloid lineage protects against inflammation-mediated kidney injury in mice. PloS one. 2016;11(3):e0150886. doi:10.1371/journal.pone.0150886. PubMed PMID: 26939003; PubMed Central PMCID: PMCPMC4777421. eng.
- Luo YP, Jiang L, Kang K, et al. Hemin inhibits NLRP3 inflammasome activation in sepsis-induced acute lung injury, involving heme oxygenase-1. Int Immunopharmacol. 2014 May;20(1):24–32. doi:10.1016/j.intimp.2014.02.017. PubMed PMID: 24583148.
- Jang SE, Jeong JJ, Hyam SR, et al. Ursolic acid isolated from the seed of Cornus officinalis ameliorates colitis in mice by inhibiting the binding of lipopolysaccharide to toll-like receptor 4 on macrophages. J Agric Food Chem. 2014 Oct 8;62(40):9711–9721. doi:10.1021/jf501487v. PubMed PMID: 25213465.
- Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014 May 22;157(5):1013–1022. doi:10.1016/j.cell.2014.04.007. PubMed PMID: 24855941.
- Vilaysane A, Chun J, Seamone ME, et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol – JASN. 2010 Oct;21(10):1732–1744. doi:10.1681/ASN.2010020143. PubMed PMID: 20688930; PubMed Central PMCID: PMC3013544.
- Xu Y, Li R, Zhang K, et al. The multifunctional RNA-binding protein hnRNPK is critical for the proliferation and differentiation of myoblasts. BMB Rep. 2018;51(7):350–355.
- Liu XH, Ma J, Feng JX, et al. Regulation and related mechanism of GSN mRNA level by hnRNPK in lung adenocarcinoma cells. Biol Chem. 2018 Dec 1. doi:10.1515/hsz-2018-0417. PubMed PMID: 30771276.
- Li Z, Liu X, Ma J, et al. hnRNPK modulates selective quality-control autophagy by downregulating the expression of HDAC6 in 293 cells. Int J Oncol. 2018 Nov;53(5):2200–2212. doi:10.3892/ijo.2018.4517. PubMed PMID: 30106132.
- Barboro P, Ferrari N, Balbi C. Emerging roles of heterogeneous nuclear ribonucleoprotein K (hnRNP K) in cancer progression. Cancer Lett. 2014 Oct 1;352(2):152–159. doi:10.1016/j.canlet.2014.06.019. PubMed PMID: 25016060.
- Hai-Xia XU, Yong-Jie XU, Liang XJ, et al. The biological function of heterogeneous nuclear ribonucleoprotein K(hnRNP K) and Its Roles in Spermatogenisis. J Agr Biotechnol. 2015;23(5):661–670.