
ABSTRACT
Mitochondria are the main source of reactive oxygen species (ROS) in cells. Early studies have shown that mitochondrial reactive oxygen species (mROS) are related to the occurrence and adverse outcomes of many diseases, and are thus regarded as an important risk factor that threaten human health. Recently, increasing evidence has shown that mROS are very important for an organism’s homeostasis. mROS can regulate a variety of signaling pathways and activate the adaptation and protection behaviors of an organism under stress. In addition, mROS also regulate important physiological processes, such as cell proliferation, differentiation, aging, and apoptosis. Herein, we review the mechanisms of production, transformation, and clearance of mROS and their biological roles in different physiological processes.
Introduction
Reactive oxygen species (ROS) are by-products of cell aerobic respiration. There are many kinds of ROS, including superoxide anion (O2-), hydrogen peroxide (H2O2), hydroxyl radical (HO·) and nitric oxide (NO), et al. NO also belongs to the RNS category. At present, the researches mainly focus on H2O2 and O2-. However, HO· produced by H2O2 and Fe2+ through the Fenton reaction is also an important type of ROS. ROS are generally regarded as toxic metabolites [Citation1], and one of the driving factors of cancer [Citation2–5], diabetes [Citation6–8], and cardiovascular diseases [Citation9,Citation10]. However, ROS has been proposed as an active factor regulating many kinds of life activities since the 1990s. For example, cytokines, insulin, growth factors, AP-1, and NF-KB signals all require H2O2 [Citation11]. Subsequent studies have shown that ROS play an important role in a wide range of physiological processes such as cell proliferation and differentiation, gene expression, post-translational protein modification, homeostasis and hypoxia adaptation [Citation12–18]. Therefore, in the normal physiological state, cells maintain a certain level of ROS to ensure homeostasis [Citation19] .
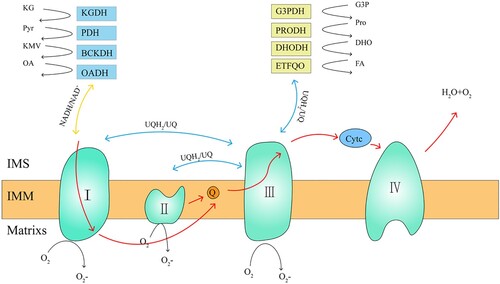
Figure 1. mROS production sites and mitochondrial electron transfer process. The mROS generation sites can be divided into two categories, namely NADH/NAD+ equipotential group (yellow) and the UQH2/UQ equipotential group (blue). The NADH/NAD+ group consists of KGDH, PDH, BCKDH, OADH, and complex I. The UQH2/UQ isopotential group is made up of complex II, PRODH, DHODH, ETFQO, and complex III. Complex I uses two equipotential groups to form reactive oxygen species. The red line indicates the electron transfer process of the mitochondria. mROS, mitochondrial reactive oxygen species; UQH2, ubisemiquinone; UQ, ubiquinone; KGDH, α-ketoglutarate dehydrogenase; PDH, pyruvate dehydrogenase; BCKDH, branched chain keto acid dehydrogenase; OADH, 2-oxoadipate dehydrogenase; PRODH, proline dehydrogenase; DHODH, dihydroorotate dehydrogenase; and ETFQO, electron transferring flavoprotein ubiquinone oxidoreductase.
Mitochondria are the main location of aerobic respiration to supply energy and are the main source of ROS in cells [Citation20–22]. The initial conception that mitochondrial ROS are essentially undesirable metabolites generated by the cellular respiratory chain has changed. A large body of experimental evidence indicates that mitochondrial reactive oxygen species (mROS) production is a continuous and tightly regulated process required for the regulation of many life activities [Citation23]. mROS have been proven to be involved in the regulation of many physiological processes, such as cell differentiation, senescence, signal transduction, hypoxic adaptation [Citation13–18,Citation24–26]. For example, Liu et al. [Citation27] showed that mROS can regulate the release of cytochrome c and thus mediate apoptosis. Chandel et al. [Citation24] revealed that in the hypoxic environment, mROS can promote the production of hypoxia inducible factors (HIFs) by regulating the expression of multiple functional genes, to initiate a protective mechanism of cells against hypoxia, thus playing an important role in the regulation of hypoxia homeostasis. Subsequently, it was found that H2O2 released by mitochondria can activate signal factors such as c-Jun amino-terminal kinase 1 (JNK1), p53, and nuclear factor kappa B (NF- κB) [Citation25,Citation26].
The production of mROS
In the 1970s, researchers first found that isolated mitochondria could produce superoxide and H2O2 [Citation28–32]. Since then, the production of mROS and its role in diseases have been studied in detail. The generation of mROS is mainly caused by the leakage of electrons in the electron transport chain (ETC) [Citation33,Citation34]. Under physiological conditions, 0.2–2% of the electrons in the ETC cannot be transferred normally, but leak out from ETC and interact with oxygen to form superoxide or H2O2 [Citation28–32]. The ETC is composed of transmembrane protein complex (I–IV) in the mitochondrial crest, the free-moving electron transfer vector ubiquinone (UQ) and cytochrome c [Citation34]. Together with F1F0 ATP synthetase (complex V), they become the basis of ATP production in oxidative phosphorylation (OXPHOS) [Citation35,Citation36].
Up to now, 11 mROS production sites have been found in mammalian mitochondria, which are related to substrate catabolism, electron transport, and OXPHOS. Six sites work at the redox potential of the NADH/NAD+ isopotential pool; and five sites work at the redox potential of the UQH2/UQ isopotential pool () [Citation16,Citation37]. The former is composed of a flavin-dependent dehydrogenase that reduces or oxidizes nicotinamide, mainly including α-ketoglutarate dehydrogenase (KGDH), pyruvate dehydrogenase (PDH), branched chain keto acid dehydrogenase (BCKDH), and 2-oxoadipate dehydrogenase (OADH), as well as the flavin mononucleotide group of complex I. The latter is composed of enzymes that directly oxidize or reduce mitochondrial UQ to produce ROS, including complex I, complex II, complex III, sn-glyceral-3-phosphate dehydrogenase (G3PDH), proline dehydrogenase (PRODH), dihydroorotate dehydrogenase (DHODH), and electron transferring flavoprotein ubiquinone oxidoreductase (ETFQO) [Citation38].
Regulation of mROS
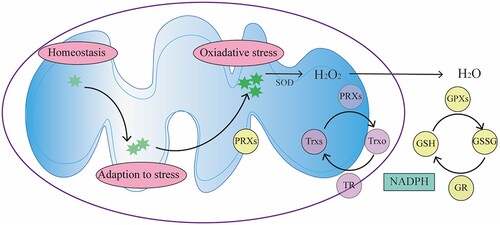
The in vivo level of mROS is related closely to their characteristics and physiological functions (); therefore, there is a precise control system for mROS to maintain the balance between its production and elimination. mROS scavenging system can reduce oxidative stress damage [Citation39]. This system mainly includes enzymatic and non-enzymatic parts. The non-enzymatic part of the system mainly consists of hydrophilic and lipophilic antioxidants, such as tocopherol, ascorbic acid, reducing coenzyme Q10 and glutathione. The antioxidant enzymes in cells mainly include superoxide dismutase (SOD), glutathione peroxidases (GPXs), peroxiredoxins (PRXs) and catalase (CAT). ROS, including mROS, can be efficiently eliminated, collectively referred to as the protective enzyme system. First, when the intracellular ROS concentration is high, the antioxidant defense system equipped by mitochondria can reduce the cytotoxicity caused by ROS. For example, O2- can be efficiently dismutated to H2O2 by Mn2+-dependent superoxide dismutase (Mn-SOD) [Citation40]. Then H2O2 is removed by PRXs and GPXs [Citation28–32,Citation41], which can be regenerated by glutathione (GSH) and thioredoxin (TRX) [Citation42,Citation43] (). Mammals express six PRX isoforms, including PRX3 and PRX5 in mitochondria [Citation41]. H2O2 oxidizes the active cysteine residue of PRXs and itself is reduced to H2O. The oxidized active cysteine residue can be reduced by TRX, thioredoxin reductase (TR), and NADPH for recycling; therefore, it can significantly reduce peroxide levels. GPXs exist widely in vivo and can catalyze the reaction between peroxide and glutathione (GSH), which protects the structure and function of cells from the interference and damage of oxides. CAT mainly exists in the peroxisome of cells, and its main role is to promote the decomposition of H2O2 into O2 and H2O. These antioxidant enzymes all play a key role in the biological defense system, but there are some differences in their functional processes. These differences come from the reaction rate (rate constant) at which they react with the substrate, as well as the concentration of substrate and enzyme [Citation44]. In general, PRXs have a high rate constant and high abundance and therefore can eliminate nanomolar ROS related to signal transmission. GPXs have a similar rate constant to PRXs, but are less abundant, so can only act at higher H2O2 concentration, such as hypoxia, hunger and other stressful environment [Citation44]. Thus, PRXs might be the key to turning off ROS signals, while GPXs might be the key to buffering high levels of ROS. The combined effect of the two enzymes can regulate the level of ROS precisely to ensure that cells can not only escape damage, but also initiate a signal stress response, which is crucial to maintain the homeostasis [Citation41]. In structurally and functionally intact mitochondria, the antioxidant capacity can maintain the balance of mROS, and the reduced antioxidant capacity is one of the reasons for the increase of mROS levels and the occurrence of oxidative stress. Notably, in addition to scavenging excess ROS, the protein repair and degradation are both important defense mechanisms against ROS induced damages.
Figure 2. Regulation of mROS. The level of mROS determines mitochondrial function and physiological outcomes. The organism needs low levels of mROS to maintain homeostasis, and when mROS rise to higher levels, cells can adapt to stress in a variety of ways. When the mROS level accumulates to a very high level, oxidative stress and damage occurs. SOD can convert O2- into H2O2. The generated H2O2 can be further converted into H2O by PRXs and GPXs. mROS, mitochondrial reactive oxygen species; SOD, superoxide dismutase; PRX, peroxiredoxin; GPX, glutathione peroxidase; TRXr/o, reduced/oxidized thioredoxin; GSH/GSSG, reduced/oxidized glutathione; TR, thioredoxin reductase; GR, glutathione reductase.
The cells also regulate the production of mROS to control their levels. However, the current understanding of this aspect is only based on experimental studies of mitochondria and cells in vitro, and the mechanism controlling the production of mROS in vivo has not yet been determined [Citation45]. According to existing results, there are two determining factors that affect the production of mROS. One is the redox state of the ETC. Inhibition of ETC electronic carriers will increase the possibility of superoxide production. The second is the proton motive force (PMF). When the PMF increases, enhanced mROS production is observed [Citation46]. Another study showed that the number of mitochondria also affects the production of mROS, but because of the presence of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α), which regulates mitochondrial production and thus inhibits the production of mROS, the number of mitochondria is not necessarily proportional to the amount of mROS [Citation47].
In addition, the signaling capacity of mROS might be altered by the mitochondria’s location. ROS are generally short-lived molecules; therefore, the coexistence of their production sites and signal functional sites may increase their efficiency.
Physiological roles of mROS
mROS and signal transduction
To date, many studies have shown that ROS can activate signal transduction. One of the mechanisms is ROS-mediated oxidation of amino acid residues on a target protein. These amino acid residues mainly include cysteine residues and methionine residues [Citation48]. Phosphatase-containing active cysteine residues are one of the most studied oxidative modification sites. ROS have been proven to inhibit the activities of many phosphatases, such as angiotensin homologous enzyme and mitogen activated protein kinase. The kelch-like ECH-associated protein 1 (Keap1)/nuclear factor E2-related factor 2 (Nrf2) signaling system is one of the main signaling pathways activated by ROS via the above mechanisms. The destruction of Keap1 by ROS leads to the dissociation of the Nrf2-Keap1 complex and the activation of Nrf2.
Low doses of H2O2 can induce calcium signaling [Citation49–51]. For example, mROS plays an important role in central nervous signal transaction. H2O2 produced by monoamine oxidase (MAO), an effective pharmacological target of the central nervous system, can stimulate lipid peroxidation, activate phospholipase C (PLC), and induce inositol 1,4,5-trisphosphate (IP3)-dependent calcium signaling. The calcium signal produced by dopamine in astrocytes is also induced by H2O2 produced by MAO [Citation52]. It can be inferred that the activation of signal transduction by this mechanism might be a common phenomenon in nerve cells; however, further studies are needed to prove this hypothesis. ROS play an important role in the regulation of various ion channels. On the one hand, the redox environment in the cell can regulate the gated characteristics of ion channels and their activities. On the other hand, mROS can regulate the activity of amino acid residues of different channels or receptor proteins [Citation53–55], thus mROS can control a variety of signaling pathways through the regulation of ion channels.
mROS and aging
One of the most popular theories of aging is the ‘free radical theory’ of aging proposed by Denham Harman in the 1950s, which suggests that aging of organisms is caused by the accumulation of free radicals in cells. Free radicals, as by-products of oxidative metabolism, can cause damage to cellular proteins, lipids and DNA, resulting in the loss of the overall ability to adapt over time. Experimental phenomena such as free radical inhibitors and antioxidants can prolong the lifespan of animals or cells, and species with low free radical production have longer lifespans have confirmed this conception. However, several recent studies have shown the opposite effect, for example, several in vivo studies have shown that increasing the antioxidant capacity doesn’t increase lifespan. For example, it has been reported that the lifespan of C. elegans mutants lacking mitochondrial SOD was not severely affected, while SOD2 single mutants lived even longer than wild-type. After adding additional oxidative stress (caused by paraquat), SOD mutants have a shortened lifespan even much faster than killing the wild type [Citation56]. A reasonable explanation for these interesting observations is that, in the absence of additional oxidative stress, moderate oxidative stress can induce sufficient adaptation to protect these mutants from permanent damage caused by endogenous ROS. Chen and Andziak et al. [Citation57,Citation58] showed that the ROS expressed by long-lived species and their accompanying oxidative damage were not always at a low level. For example, ROS prolongs the lifespan of worms [Citation59,Citation60]. In yeast, inhibition of target of rapamycin (TOR) increased intracellular mROS and prolonged chromosome lifespan [Citation61]; Caloric restriction (CR) prolonged the life span of yeast by promoting the production of H2O2 [Citation62,Citation63]. In addition to the model organisms mentioned above, ROS has been shown to prolong the life span of human fibroblasts under hypoxia. These phenomena add more support to the above argument, but do not mean that the ‘radical theory’ is wrong, they only suggest to us that any theory claims ROS is the sole cause of biological aging is discredited. A plausible concept is to explore the protective effects of ROS on the aging of organisms [Citation64].
Mitochondria are the main production sites of intracellular ROS, and changes in mitochondria and their functions have been regarded as the driving factors of aging. mROS can induce permanent cell cycle arrest and play a very important role in initiating and maintaining cell senescence [Citation65,Citation66]. However, some studies have also shown that mROS and mROS-induced mitochondrial damage can initiate signals that activate multiple pathways that protect mitochondria from stress, delay aging, and inhibit cell death. Therefore, increasing mROS levels within a certain range can prolong lifespan [Citation67–69]. For example, Stefanatos et al. showed that an increase of mROS in the brain of mice prolonged their lifespan, indicating that mROS may also protect brain signal pathways [Citation70]. In addition, some scholars believe that although high levels of mROS play an important role in the occurrence of neurodegenerative diseases, such as Parkinson's disease, they are not a direct cause of aging. The relationship between mROS and aging requires further extensive verification.
mROS and stem cell differentiation
The abundance of mitochondria in stem cells is very low; therefore, these cells rely mainly on glycolysis to obtain energy. However, the importance of mROS in regulating stem cell activity and differentiation is often overlooked [Citation71]. In recent years, research on whether mROS is required for stem cell differentiation has been carried out. Owusu-Ansah et al. found that the elimination of ROS by enhancing the expression of GTPx-1 could delay the differentiation of Drosophila multipotent hematopoietic progenitor cells. Increasing ROS by inducing the deletion of mitochondrial complex I protein ND75 or SOD2 could promote differentiation [Citation72]. Tormos et al. showed that the differentiation of human mesenchymal stem cells (MSCs) into adipocytes could be inhibited by either knocking out the complex III protein complex III protein Rieske Iron-Sulfur Protein or using mitochondria targeted antioxidants to reduce mROS levels [Citation73]. In addition, a study by Hamanaka et al. confirmed that mROS are important regulators of epidermal differentiation [Citation74]. Bigarella et al. showed that mROS are also important for the differentiation of neural stem cells (NSCs) [Citation75].
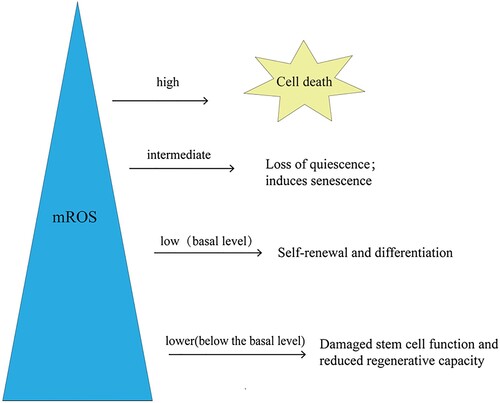
Mitochondrial activity significantly affects the function and differentiation of stem cells. Stem cells initially prioritize glycolysis over oxidative phosphorylation; therefore, they exhibit lower mitochondrial activity, which limits electron flux, inhibits mROS production, and maintains their regenerative potential. When metabolism is converted to oxidative phosphorylation, mitochondrial activity increases, which promotes the production of mROS and reduces stem cells’ regenerative potential [Citation76,Citation77]. These results indicate that the regenerative potential of stem cells is closely related to the level of mROS and the redox status in vivo. Lower mROS levels can maintain the balance between cell quiescent differentiation and self-renewal [Citation78–80]. However, when mROS are lower than the basal level, a significant decrease in regeneration ability will occur (). For example, when the levels of ROS in NSCs and hematopoietic stem cells (HSCs) were lower than the basal level, their proliferation, differentiation, and self-renewal abilities were decreased [Citation78,Citation81,Citation82]. In contrast, excess ROS is associated with reduced stem cell function and regenerative potential [Citation48,Citation78,Citation83–90]. For example, the long-term accumulation of ROS in HSCs severely impaired their reconstitution potential, and the stem cell pool was depleted [Citation78,Citation86,Citation87]. Notably, further increases in ROS lead to cell death [Citation91,Citation92]. Therefore, physiological levels of mROS are necessary for stem cells to perform their normal physiological functions.
Figure 3. mROS levels affect stem cell function and fate. The level of mROS is closely related to the fate and function of stem cells. Stem cells maintain basic ROS levels to balance self-renewal and differentiation. When mROS levels are below the baseline, stem cell function is impaired and metabolic capacity is reduced. When mROS accumulate to an intermediate level, loss of immobility and the induction of senescence occur. Further accumulation of mROS to a high level leads to cell death. mROS, mitochondrial reactive oxygen species.
mROS and hypoxic adaptation
When the organisms face a hypoxic environment, it will reduce oxygen consumption in various ways, and increase the supply of oxygen at the same time. Ironically, most of the adaptation pathways to hypoxia are mediated by mROS.
When cells are exposed to a low oxygen concentration for a short period of time, adaptive responses can be induced through the protein kinase pathway, which is activated by adenosine monophosphate (AMP) to promote glycolysis and increase the energy supply under hypoxic conditions. However, when the cells is subjected to chronic hypoxia, this protective measure is no longer effective, and instead, it forms an adaptation to hypoxia by stimulating HIF [Citation93,Citation94]. HIF is a heterodimeric transcription factor composed of an oxygen-sensitive α subunit and a constitutively expressed β subunit, and is a determinant of cell adaptation to hypoxia [Citation95]. Under normal oxygen conditions, HIF-α is rapidly hydroxylated by prolyl hydroxylase 2 (PHD2), and the hydroxylated HIF-α subunit is degraded by the Von Hippel-Lindau tumor suppressor (VHL) [Citation96]. In hypoxia, PHD2 is inhibited by mROS and HIF-α does not undergo hydroxylation, thus maintaining HIF homeostasis [Citation97,Citation98]. To date, many studies have confirmed that inhibition of PHD2 is regulated by mROS. Chandel et al. showed that the cells without mitochondrial DNA could not maintain the stability of HIF, and could not initiate a variety of transcription pathways involving HIF. Therefore, it was speculated that the stability of HIF was related to the mitochondrial electron transport chain. Subsequent studies found that the cells expressing mutant cytochrome subunits could not consume oxygen for oxidative phosphorylation, but could produce ROS and inhibit HIF degradation, which further confirmed that mROS were the main factors that stabilize HIF [Citation99].
HIF activates the transcription of more than 70 genes in vivo [Citation1]. For example, when cells are exposed to hypoxia, HIF can regulate the oxygen supply in the blood by controlling the transcription of several functional genes [Citation100,Citation101], such as EPO [Citation102] encoding erythropoietin and VEGF [Citation100] encoding vascular endothelial growth factor. HIF-1 also inhibits the conversion of pyruvate by promoting the expression of 3-phosphoinositide-dependent protein kinase-1 (PDK1) and lactate dehydrogenase A (LDHA) in cells. At the same time, acetyl-Coenzyme A promotes the conversion of pyruvate to lactic acid. The produced lactic acid can regenerate coenzyme Ⅰ, so as to carry out continuous glycolysis to provide enough energy [Citation103–105]. HIF can also increase the concentration of glucose transporters and key enzymes in glycolysis to improve the rate of glycolysis and increase the production of anaerobic energy [Citation103]. In addition, HIF can also alleviate the damage caused by ROS. Studies have shown that HIF protein can promote ROS clearance and enhance the oxidative defense ability by upregulating the levels of SOD2 and GSH under hypoxic conditions [Citation106].
In addition to HIF-mediated transcriptional effects, there are other non-transcriptional effects, including increasing intracellular calcium storage, triggering the contraction of pulmonary vasculature to divert blood from hypoxic lung regions [Citation107,Citation108], the release of neurotransmitters by the carotid body to increase the respiration rate, and the reduction of ATP usage [Citation41].
Conclusion
mROS are produced in large quantities under stress conditions such as hypoxia, starvation, and pathogen infection, and are regarded as a marker for changes in the internal and external environment of the cell. The accumulation of mROS can cause damage to DNA, proteins, and lipids, and induces a variety of pathological processes. However, their effects will change with environmental alterations; therefore, antioxidants do not have a definite therapeutic effect. Physiological levels of mROS have dual functions of promoting cell damage and cell adaptation, making them a potential therapeutic target. However, due to their complex regulatory mechanisms and diverse biological functions in vivo, challenges remain regarding their practical.
Authors’ contributions
CYP, CF, YJY, CQY, XXW, CJG and ZLC provide ideas and directions for article writing. BYZ read literatures and write articles. All authors read and approved the inal manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Indo HP, Hawkins CL, Nakanishi I, et al. Role of mitochondrial reactive oxygen species in the activation of cellular signals, molecules, and function. Handb Exp Pharmacol. 2017;240:439–456. doi:10.1007/164_2016_117.
- Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010 May;44(5):479–496. doi:10.3109/10715761003667554.
- Tong L, Chuang CC, Wu S, et al. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015 Oct 10;367(1):18–25. doi:10.1016/j.canlet.2015.07.008.
- Stojnev S, Ristic-Petrovic A, Jankovic-Velickovic L. Reactive oxygen species, apoptosis and cancer. Vojnosanit Pregl. 2013 Jul;70(7):675–678. doi:10.2298/VSP1307675S.
- Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996 Jan 1;313(Pt 1):17–29. doi:10.1042/bj3130017.
- Gratas-Delamarche A, Derbre F, Vincent S, et al. Physical inactivity, insulin resistance, and the oxidative-inflammatory loop. Free Radic Res. 2014 Jan;48(1):93–108. doi:10.3109/10715762.2013.847528.
- Bashan N, Kovsan J, Kachko I, et al. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol Rev. 2009 Jan;89(1):27–71. doi:10.1152/physrev.00014.2008.
- Rains JL, Jain SK. Oxidative stress,: insulin signaling, and diabetes. Free Radic Biol Med. 2011 Mar 1;50(5):567–575. doi:10.1016/j.freeradbiomed.2010.12.006.
- Wang R, Ding G, Liang W, et al. Role of LOX-1 and ROS in oxidized low-density lipoprotein induced epithelial-mesenchymal transition of NRK52E. Lipids Health Dis. 2010 Oct 19;9:120.
- He F, Zuo L. Redox roles of reactive oxygen species in cardiovascular diseases. Int J Mol Sci. 2015 Nov 20;16(11):27770–27780. doi:10.3390/ijms161126059.
- Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998 Apr;10(2):248–253. doi:10.1016/S0955-0674(98)80147-6.
- Murphy MP, Holmgren A, Larsson NG, et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011 Apr 6;13(4):361–366. doi:10.1016/j.cmet.2011.03.010.
- Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012 May;24(5):981–990. doi:10.1016/j.cellsig.2012.01.008.
- Phull AR, Nasir B, Haq IU, et al. Oxidative stress, Consequences and ROS mediated cellular signaling in rheumatoid arthritis. Chem Biol Interact. 2018 Feb 1;281:121–136. doi:10.1016/j.cbi.2017.12.024.
- Newsholme P, Cruzat VF, Keane KN, et al. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem J. 2016 Dec 15;473(24):4527–4550. doi:10.1042/BCJ20160503C.
- Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med. 2016 Nov;100:14–31. doi:10.1016/j.freeradbiomed.2016.04.001.
- D'Autreaux B, Toledano MB. Ros as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007 Oct;8(10):813–824. doi:10.1038/nrm2256.
- De Giusti VC, Caldiz CI, Ennis IL, et al. Mitochondrial reactive oxygen species (ROS) as signaling molecules of intracellular pathways triggered by the cardiac renin-angiotensin II-aldosterone system (RAAS). Front Physiol. 2013;4:126.
- Poli G, Leonarduzzi G, Biasi F, et al. Oxidative stress and cell signalling. Curr Med Chem. 2004 May;11(9):1163–1182. doi:10.2174/0929867043365323.
- Chouchani ET, Pell VR, Gaude E, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014 Nov 20;515(7527):431–435.
- Sanz A, Pamplona R, Barja G. Is the mitochondrial free radical theory of aging intact? Antioxid Redox Signal. 2006 Mar-Apr;8(3-4):582–599.
- Scialo F, Sriram A, Fernandez-Ayala D, et al. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 2016 Apr 12;23(4):725–734. doi:10.1016/j.cmet.2016.03.009.
- Figueira TR, Barros MH, Camargo AA, et al. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid Redox Signal. 2013 Jun 1;18(16):2029–2074. doi:10.1089/ars.2012.4729.
- Chandel NS, Maltepe E, Goldwasser E, et al. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998 Sep 29;95(20):11715–11720. doi:10.1073/pnas.95.20.11715.
- Chandel NS, Vander Heiden MG, Thompson CB, et al. Redox regulation of p53 during hypoxia. Oncogene. 2000 Aug 10;19(34):3840–3848. doi:10.1038/sj.onc.1203727.
- Chandel NS, Trzyna WC, McClintock DS, et al. Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin. J Immunol. 2000 Jul 15;165(2):1013–1021. doi:10.4049/jimmunol.165.2.1013.
- Liu X, Kim CN, Yang J, et al. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996 Jul 12;86(1):147–157. doi:10.1016/S0092-8674(00)80085-9.
- Loschen G, Flohe L, Chance B. Respiratory chain linked H(2)O(2) production in pigeon heart mitochondria. FEBS Lett. 1971 Nov 1;18(2):261–264. doi:10.1016/0014-5793(71)80459-3.
- Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. general properties and effect of hyperbaric oxygen. Biochem J. 1973 Jul;134(3):707–716. doi:10.1042/bj1340707.
- Loschen G, Azzi A, Richter C, et al. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974 May 15;42(1):68–72. doi:10.1016/0014-5793(74)80281-4.
- Boveris A, Cadenas E. Mitochondrial production of superoxide anions and its relationship to the antimycin insensitive respiration. FEBS Lett. 1975 Jul 1;54(3):311–314. doi:10.1016/0014-5793(75)80928-8.
- Cadenas E, Boveris A, Ragan CI, et al. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys. 1977 Apr 30;180(2):248–257. doi:10.1016/0003-9861(77)90035-2.
- Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol. 2017 Apr;11:613–619. doi:10.1016/j.redox.2016.12.035.
- Zhao RZ, Jiang S, Zhang L, et al. Mitochondrial electron transport chain, ROS generation and uncoupling (review). Int J Mol Med. 2019 Jul;44(1):3–15.
- Guo R, Zong S, Wu M, et al. Architecture of human mitochondrial Respiratory megacomplex I2III2IV2. Cell. 2017 Sep 7;170(6):1247–1257.e12. doi:10.1016/j.cell.2017.07.050.
- Iwata S, Lee JW, Okada K, et al. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 1998 Jul 3;281(5373):64–71.
- Mailloux RJ. An update on mitochondrial reactive oxygen species production. Antioxid (Basel). 2020 Jun 2;9(6):1–14. doi:10.3390/antiox9060472.
- Mailloux RJ. Mitochondrial antioxidants and the maintenance of cellular hydrogen peroxide levels. Oxid Med Cell Longev. 2018;2018:7857251.
- Millare B, O'Rourke B, Trayanova N. Hydrogen peroxide diffusion and scavenging shapes mitochondrial network instability and failure by sensitizing ROS-induced ROS release. Sci Rep. 2020 Sep 25;10(1):15758), doi:10.1038/s41598-020-71308-z.
- Nickel A, Kohlhaas M, Maack C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol. 2014 Aug;73:26–33. doi:10.1016/j.yjmcc.2014.03.011.
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012 Oct 26;48(2):158–167. doi:10.1016/j.molcel.2012.09.025.
- Aon MA, Stanley BA, Sivakumaran V, et al. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J Gen Physiol. 2012 Jun;139(6):479–491. doi:10.1085/jgp.201210772.
- Ying W. Nad+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008 Feb;10(2):179–206. doi:10.1089/ars.2007.1672.
- Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008 Sep 1;45(5):549–561. doi:10.1016/j.freeradbiomed.2008.05.004.
- Munro D, Pamenter ME. Comparative studies of mitochondrial reactive oxygen species in animal longevity: technical pitfalls and possibilities. Aging Cell. 2019 Oct;18(5):e13009, doi:10.1111/acel.13009.
- Echtay KS, Murphy MP, Smith RA, et al. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. studies using targeted antioxidants. J Biol Chem. 2002 Dec 6;277(49):47129–47135. doi:10.1074/jbc.M208262200.
- St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006 Oct 20;127(2):397–408. doi:10.1016/j.cell.2006.09.024.
- Garcia-Prat L, Martinez-Vicente M, Perdiguero E, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016 Jan 7;529(7584):37–42.
- Domijan AM, Kovac S, Abramov AY. Lipid peroxidation is essential for phospholipase C activity and the inositol-trisphosphate-related Ca(2)(+) signal. J Cell Sci. 2014 Jan 1;127(Pt 1):21–26.
- Granados MP, Salido GM, Pariente JA, et al. Modulation of CCK-8-evoked intracellular Ca2 + waves by hydrogen peroxide in mouse pancreatic acinar cells. J Physiol Pharmacol. 2007 Sep;58(3):423–440.
- Gonzalez-Pacheco FR, Caramelo C, Castilla MA, et al. Mechanism of vascular smooth muscle cells activation by hydrogen peroxide: role of phospholipase C gamma. Nephrol Dial Transplant. 2002 Mar;17(3):392–398. doi:10.1093/ndt/17.3.392.
- Vaarmann A, Gandhi S, Abramov AY. Dopamine induces Ca2 + signaling in astrocytes through reactive oxygen species generated by monoamine oxidase. J Biol Chem. 2010 Aug 6;285(32):25018–25023. doi:10.1074/jbc.M110.111450.
- Tang XD, Garcia ML, Heinemann SH, et al. Reactive oxygen species impair Slo1 BK channel function by altering cysteine-mediated calcium sensing. Nat Struct Mol Biol. 2004 Feb;11(2):171–178. doi:10.1038/nsmb725.
- Ciorba MA, Heinemann SH, Weissbach H, et al. Modulation of potassium channel function by methionine oxidation and reduction. Proc Natl Acad Sci U S A. 1997 Sep 2;94(18):9932–9937. doi:10.1073/pnas.94.18.9932.
- Kolbe K, Schonherr R, Gessner G, et al. Cysteine 723 in the C-linker segment confers oxidative inhibition of hERG1 potassium channels. J Physiol. 2010 Aug 15;588(Pt 16):2999–3009. doi:10.1113/jphysiol.2010.192468.
- Van Raamsdonk JM, Hekimi S. Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci U S A. 2012 Apr 10;109(15):5785–5790. doi:10.1073/pnas.1116158109.
- Andziak B, O'Connor TP, Qi W, et al. High oxidative damage levels in the longest-living rodent, the naked mole-rat. Aging Cell. 2006 Dec;5(6):463–471. doi:10.1111/j.1474-9726.2006.00237.x.
- Chen JH, Hales CN, Ozanne SE. Dna damage: cellular senescence and organismal ageing: causal or correlative? Nucleic Acids Res. 2007;35(22):7417–7428. doi:10.1093/nar/gkm681.
- Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010 Dec 7;8(12):e1000556, doi:10.1371/journal.pbio.1000556.
- Zarse K, Schmeisser S, Groth M, et al. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012 Apr 4;15(4):451–465. doi:10.1016/j.cmet.2012.02.013.
- Pan Y, Schroeder EA, Ocampo A, et al. Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 2011 Jun 8;13(6):668–678. doi:10.1016/j.cmet.2011.03.018.
- Mesquita A, Weinberger M, Silva A, et al. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc Natl Acad Sci U S A. 2010 Aug 24;107(34):15123–15128. doi:10.1073/pnas.1004432107.
- Zuin A, Carmona M, Morales-Ivorra I, et al. Lifespan extension by calorie restriction relies on the Sty1 MAP kinase stress pathway. EMBO J. 2010 Mar 3;29(5):981–991. doi:10.1038/emboj.2009.407.
- Liochev SI. Reactive oxygen species and the free radical theory of aging. Free Radic Biol Med. 2013 Jul;60:1–4. doi:10.1016/j.freeradbiomed.2013.02.011.
- Correia-Melo C, Marques FD, Anderson R, et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016 Apr 1;35(7):724–742. doi:10.15252/embj.201592862.
- Passos JF, Nelson G, Wang C, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:347.
- Sanz A. Mitochondrial reactive oxygen species: Do they extend or shorten animal lifespan? Biochim Biophys Acta. 2016 Aug;1857(8):1116–1126. doi:10.1016/j.bbabio.2016.03.018.
- Desjardins D, Cacho-Valadez B, Liu JL, et al. Antioxidants reveal an inverted U-shaped dose-response relationship between reactive oxygen species levels and the rate of aging in Caenorhabditis elegans. Aging Cell. 2017 Feb;16(1):104–112. doi:10.1111/acel.12528.
- Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016 Mar 3;61(5):654–666. doi:10.1016/j.molcel.2016.01.028.
- Stefanatos R, Sanz A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018 Mar;592(5):743–758. doi:10.1002/1873-3468.12902.
- Rafalski VA, Mancini E, Brunet A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J Cell Sci. 2012 Dec 1;125(Pt 23):5597–5608. doi:10.1242/jcs.114827.
- Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009 Sep 24;461(7263):537–541.
- Tormos KV, Anso E, Hamanaka RB, et al. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011 Oct 5;14(4):537–544. doi:10.1016/j.cmet.2011.08.007.
- Hamanaka RB, Glasauer A, Hoover P, et al. Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci Signal. 2013 Feb 5;6(261):ra8, doi:10.1126/scisignal.2003638.
- Zhang H, Menzies KJ, Auwerx J. The role of mitochondria in stem cell fate and aging. Development. 2018 Apr 13;145(8), doi:10.1242/dev.143420.
- Sukumar M, Liu J, Mehta GU, et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016 Jan 12;23(1):63–76. doi:10.1016/j.cmet.2015.11.002.
- Vannini N, Girotra M, Naveiras O, et al. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat Commun. 2016 Oct 12;7:13125.
- Takubo K, Goda N, Yamada W, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010 Sep 3;7(3):391–402. doi:10.1016/j.stem.2010.06.020.
- Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011 Oct 4;9(4):298–310. doi:10.1016/j.stem.2011.09.010.
- Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007 Oct 15;110(8):3056–3063. doi:10.1182/blood-2007-05-087759.
- Juntilla MM, Patil VD, Calamito M, et al. Akt1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010 May 20;115(20):4030–4038. doi:10.1182/blood-2009-09-241000.
- Le Belle JE, Orozco NM, Paucar AA, et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3 K/Akt-dependant manner. Cell Stem Cell. 2011 Jan 7;8(1):59–71. doi:10.1016/j.stem.2010.11.028.
- Borodkina A, Shatrova A, Abushik P, et al. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging (Albany NY). 2014 Jun;6(6):481–495.
- Burova E, Borodkina A, Shatrova A, et al. Sublethal oxidative stress induces the premature senescence of human mesenchymal stem cells derived from endometrium. Oxid Med Cell Longev. 2013;2013:474931.
- Choo KB, Tai L, Hymavathee KS, et al. Oxidative stress-induced premature senescence in wharton's jelly-derived mesenchymal stem cells. Int J Med Sci. 2014;11(11):1201–1207. doi:10.7150/ijms.8356.
- Ito K, Hirao A, Arai F, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004 Oct 21;431(7011):997–1002.
- Ito K, Hirao A, Arai F, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006 Apr;12(4):446–451. doi:10.1038/nm1388.
- Khacho M, Clark A, Svoboda DS, et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. 2016 Aug 4;19(2):232–247. doi:10.1016/j.stem.2016.04.015.
- Paik JH, Ding Z, Narurkar R, et al. Foxos cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell. 2009 Nov 6;5(5):540–553. doi:10.1016/j.stem.2009.09.013.
- Pan H, Guan D, Liu X, et al. Sirt6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. 2016 Feb;26(2):190–205. doi:10.1038/cr.2016.4.
- Merchant AA, Singh A, Matsui W, et al. The redox-sensitive transcription factor Nrf2 regulates murine hematopoietic stem cell survival independently of ROS levels. Blood. 2011 Dec 15;118(25):6572–6579. doi:10.1182/blood-2011-05-355362.
- Cho J, Yusuf R, Kook S, et al. Purinergic P2Y(1)(4) receptor modulates stress-induced hematopoietic stem/progenitor cell senescence. J Clin Invest. 2014 Jul;124(7):3159–3171. doi:10.1172/JCI61636.
- Viollet B, Athea Y, Mounier R, et al. Ampk: lessons from transgenic and knockout animals. Front Biosci (Landmark Ed). 2009 Jan 1;14:19–44. doi:10.2741/3229.
- Gonzalez FJ, Xie C, Jiang C. The role of hypoxia-inducible factors in metabolic diseases. Nat Rev Endocrinol. 2018 Dec;15(1):21–32. doi:10.1038/s41574-018-0096-z.
- Fuhrmann DC, Brune B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017 Aug;12:208–215. doi:10.1016/j.redox.2017.02.012.
- Kaelin WG. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002 Sep;2(9):673–682. doi:10.1038/nrc885.
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003 Oct;3(10):721–732. doi:10.1038/nrc1187.
- Kaelin WG, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008 May 23;30(4):393–402. doi:10.1016/j.molcel.2008.04.009.
- Bell EL, Klimova TA, Eisenbart J, et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol. 2007 Jun 18;177(6):1029–1036. doi:10.1083/jcb.200609074.
- Forsythe JA, Jiang BH, Iyer NV, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996 Sep;16(9):4604–4613. doi:10.1128/MCB.16.9.4604.
- Elvidge GP, Glenny L, Appelhoff RJ, et al. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1alpha. HIF: 2a), lpha, and other pathways. J Biol Chem. 2006 Jun 2;281(22):15215–15226.
- Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992 Dec;12(12):5447–5454.
- Nagao A, Kobayashi M, Koyasu S, et al. HIF-1-Dependent reprogramming of glucose metabolic pathway of cancer cells and Its therapeutic significance. Int J Mol Sci. 2019 Jan 9;20(2), doi:10.3390/ijms20020238.
- Semba H, Takeda N, Isagawa T, et al. HIF-1alpha-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. 2016 May 18;7:11635.
- Papandreou I, Cairns RA, Fontana L, et al. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006 Mar;3(3):187–197. doi:10.1016/j.cmet.2006.01.012.
- Befani C, Mylonis I, Gkotinakou IM, et al. Cobalt stimulates HIF-1-dependent but inhibits HIF-2-dependent gene expression in liver cancer cells. Int J Biochem Cell Biol. 2013 Nov;45(11):2359–2368. doi:10.1016/j.biocel.2013.07.025.
- Waypa GB, Chandel NS, Schumacker PT. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res. 2001 Jun 22;88(12):1259–1266. doi:10.1161/hh1201.091960.
- Waypa GB, Guzy R, Mungai PT, et al. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ Res. 2006 Oct 27;99(9):970–978. doi:10.1161/01.RES.0000247068.75808.3f.