
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Objectives: To advance our knowledge of disease mechanisms and therapeutic options, understanding cell cycle regulation is critical. Recent research has highlighted the importance of reactive oxygen species (ROS) in cell cycle regulation. Although excessive ROS levels can lead to age-related pathologies, ROS also play an essential role in normal cellular functions. Many cell cycle regulatory proteins are affected by their redox status, but the precise mechanisms and conditions under which ROS promote or inhibit cell proliferation are not fully understood.
Methods: This review presents data from the scientific literature and publicly available databases on changes in redox state during the cell cycle and their effects on key regulatory proteins.
Results: We identified redox-sensitive targets within the cell cycle machinery and analysed different effects of ROS (type, concentration, duration of exposure) on cell cycle phases. For example, moderate levels of ROS can promote cell proliferation by activating signalling pathways involved in cell cycle progression, whereas excessive ROS levels can induce DNA damage and trigger cell cycle arrest or cell death.
Discussion: Our findings encourage future research focused on identifying redox-sensitive targets in the cell cycle machinery, potentially leading to new treatments for diseases with dysregulated cell proliferation.
Introduction
Reactive oxygen species (ROS) are chemically reactive compounds generated as by-products of cellular metabolism and oxygen consumption. Far from being expendable metabolic derivatives, they are significant signaling molecules in various physiological and pathological processes. Cells produce ROS in response to various stimuli, including growth factors, cytokines, and stressors. ROS comprise free radicals such as superoxide anion () and hydroxyl radical (
), as well as non-radical species like hydrogen peroxide (H2O2) and singlet oxygen (1O2). The capacity of cellular oxidants to function as signaling molecules can be influenced by their potency. For instance, potent oxidants like ozone and nitrogen dioxide can non-selectively oxidize molecules, leading to irreversible pathophysiological reactions and loss of protein function [Citation1]. Conversely, physiological endogenously produced oxidants, such as controlled amounts of H2O2, often serve as specific and reversible signal transducers, providing additional regulatory mechanisms [Citation1].
Determining the precise threshold of exogenous oxidants applied to cells that leads to proliferation, oxidative stress, or apoptosis currently presents a significant challenge. Experimental investigations utilizing H2O2 often yield divergent outcomes with similar concentrations or analogous outcomes with different concentrations (). For instance, studies with NIH3T3 fibroblasts demonstrate that treatment with low H2O2 concentrations (0.02–0.13 µM) induces cell proliferation, while higher concentrations (0.25–2 µM) lead to cell death [Citation2,Citation3]. The oxidative damage and slower blastocyst formation have been detected in mouse zygotes at concentrations of 0.03 mM H2O2 [Citation4], and Martínez Munõz et al. [Citation5] even utilized concentrations as high as 3–5 mM H2O2 to induce oxidative stress without significant cell lysis in cell culture. Chang et al. [Citation6] synthesized findings from diverse studies, proposing that concentrations ranging from 1 to 15 µM signal proliferation, while the range of 100–200 µM halts the cell cycle, and concentrations spanning 0.5–5 mM induce apoptosis or necrosis [Citation6]. However, further research is still needed to accurately determine the most critical variables in the experimental application of H2O2 to induce and measure the effects of oxidative stress. The methodology of H2O2 dilution preparation may be one of the significant variables. Evidence suggests that dilution in the cell culture medium can cause H2O2 to interact with its components, influencing its final concentration [Citation5,Citation7]. Moreover, Ransy et al. [Citation8] pointed out that cellular catalase rapidly converts most of the applied H2O2 into oxygen within a few minutes [Citation8]. A high concentration of oxygen in the cell may increase the probability of the formation of singlet oxygen, contributing to oxidative damage [Citation8]. Therefore it is essential to better consider and understand the process preceding oxidative stress to prevent misidentification of molecular targets and mistakes in experimental interpretation. Other influential factors may include incubation time, cell line type, and the cell cycle phase during treatment.
Table 1. Summary of experimental outcomes and conditions used in H2O2 treatments across various cell lines and organisms.
The generation of ROS is primarily concentrated within specific cellular areas. Due to their short lifespan and heightened reactivity, endogenous ROS inflict the most damage on nearby cellular structures [Citation27]. Main intracellular ROS sources include mitochondria, cytochrome P450, endoplasmic reticulum, and peroxisomes. Within the mitochondria, approximately 1–5% of the oxygen involved in the electron transport chain undergoes reduction, resulting in the formation of [Citation28]. Complexes I and III within the respiratory chain are primary sites for the production of superoxide anions [Citation29]. Another major contributor to ROS generation is the family of NADPH oxidase enzymes (NOX), which facilitates the transfer of an electron from cytosolic NADPH to O2, resulting in the formation of
radicals. The impact of ROS on cellular structures depends on the specific ROS involved, their location, concentration, and the cell’s antioxidant defense system. Interestingly, it has been suggested that mitochondrial ROS are involved in cell death, while ROS produced by NOX are associated with promoting cell proliferation and migration [Citation30].
The cell cycle is a precisely regulated process of cell growth and division, influenced by both internal and external signals. Divided into G1, S, G2, and M phases, external signals, like growth factors and internal signals guide the cell through the resting phase (G0), differentiation, migration, or proliferation. Each phase depends on a complex network of proteins to ensure proper progression, with checkpoints at G1/S, G2/M, and M monitoring key events.
A key class of regulatory proteins in the cell cycle are cyclin-dependent kinases (CDKs) and their regulators, cyclins. CDKs are serine/threonine protein kinases responsible for phosphorylating substrates to regulate the timing and sequence of cell cycle events. They are activated by binding to specific cyclins at different stages of the cell cycle. CDKs form the catalytic component of heterodimeric kinases, with cyclins acting as the regulatory component responsible for substrate specificity and CDK activity. The accumulation and degradation of cyclins occur cyclically and determine when and where CDKs are activated, making cyclin levels a critical regulatory element of the cell cycle.
CDK activity is regulated by CDK inhibitors (CDKIs), which bind to the ATP-binding site of the CDK complex, inhibiting its ability to phosphorylate target proteins. This inhibition of CDK activity can induce cell cycle arrest, allowing cells to repair DNA damage or enter the G0 phase. Two families of CDKIs have been identified based on their structural characteristics: the INK4 proteins and the Cip/Kip family [Citation31]. The INK4 family, comprising p16INK4a, p15INK4b, and p19INK4d, specifically targets CDK4 and CDK6, with no binding to cyclins. On the other hand, the Cip/Kip family, including p21Cip1, p27Kip1 and p57Kip2, inhibits CDK activity by binding to both cyclins and CDKs [Citation31]. Dysregulation of CDKI expression or function is often associated with various diseases, including cancer and neurological disorders. Given the observed overactivity of CDKs in many cancer cells, CDK inhibitors are actively being explored as potential treatments for cancer [Citation32].
The impact of redox changes on the overall state of the cell in individual phases of the cell cycle is well-established, but the potential redox regulatory changes of individual proteins and their subsequent effects on major cellular pathways involved in cell cycle regulation remain unclear and need to be further explored, especially under pathophysiological conditions such as oxidative stress. This review will primarily examine the documented effects of ROS on specific proteins involved in cell cycle regulation. Several academic databases were consulted to complete this review, including PubMed, Google Scholar, Web of Science and Scopus. Combination of keywords such as ‘oxidation’, ‘redox regulation’, ‘reactive cysteines’, ‘oxidative stress’, ‘redox-sensitive proteins’, ‘ROS’, ‘cell cycle’, ‘cell cycle control’ and ‘cell cycle proteins’ were used to search for relevant studies. We excluded studies that showed poor statistical power to reduce the risk of bias and studies that did not report relevant redox-sensitive protein modifications and their effects on cell cycle regulation. Preferred were original experimental studies. Non-peer-reviewed publications were excluded.
ROS-mediated modulation of protein function
The oxidation of redox-sensitive cysteine and/or tyrosine residues, particularly in or near the active site of a protein, is a prevalent mechanism of redox signaling. This process critically influences protein structure and function, as the redox state of cysteine residues is a critical factor for proper protein functioning. Protein cysteines can exist as free thiols or participate in structural disulfides within the protein molecule. The reversible oxidation of thiol groups results in the formation of products such as sulfenic acid, which can further react with other thiol groups to form intraprotein or interprotein disulfide bonds or create disulfides with glutathione (GSH) [Citation33] (). Irreversible oxidation of cysteine residues to sulfinic or sulfonic acid can lead to loss of protein function and damage cellular processes [Citation33]. The reversibility of these oxidative modifications, alongside the potential for irreversible thiol oxidations, serves as an important cellular mechanism for assessing and addressing the extent of oxidative stress-induced damage [Citation33,Citation34]. Irreversible thiol oxidations can signal high oxidative damage and subsequently trigger cell death [Citation34].
Figure 1. Possible oxidative modifications of redox-sensitive cysteine.
The reversible oxidation of thiol groups can result in the formation of sulfenic acid, which can be further oxidized and create intra- or interprotein disulfide bonds or disulfides with glutathione. Irreversible oxidation to sulfinic or sulfonic acid can lead to protein dysfunction and cellular damage.
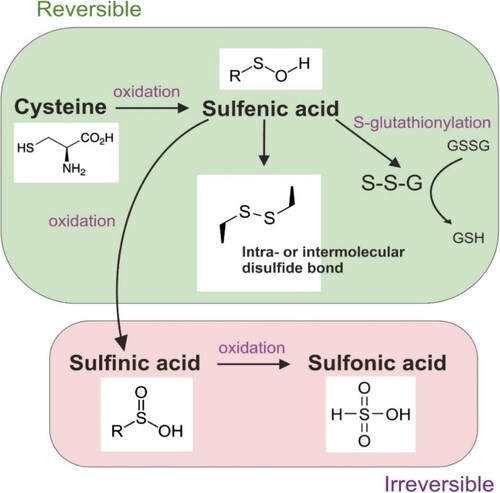
Reactive cysteine residues within antioxidant proteins undergo specific and regulated redox changes in their catalytic cysteines [Citation29]. This mechanism plays a role not only in reducing oxidized target proteins, protecting them from further oxidative damage, but also acts as a detector of harmful oxidative levels within cells [Citation29]. Moreover, the oxidation of cysteine residues may potentially displace specific metal cofactors, such as zinc ions within zinc finger motifs, disrupting the function of proteins featuring this motif, including certain transcription factors, kinases, and phosphatases where zinc or iron serves as a critical cofactor [Citation3,Citation34–37]. In addition to cysteine, protein tyrosine residues can undergo oxidation, resulting in the formation of tyrosyl radicals that initiate free radical chain reactions [Citation38]. The reversible oxidation of tyrosine residues to form sulfides can serve as a redox switch, regulating protein activity and signaling pathways.
The sensitivity of cysteine residues to redox changes is influenced by their steric accessibility and the pKa value of their thiol groups. Typically, the pKa value of free cysteine is approximately 8.2 [Citation39]. However, when cysteine residues are located near positively charged residues, their pKa drops to less than 6.5, making them more susceptible to oxidation [Citation34,Citation39]. This shift in pKa is associated with transforming redox-sensitive thiols into potent nucleophiles in the presence of basic residues [Citation29,Citation40]. Additionally, the local pH can influence the pKa value of cysteine residues [Citation41].
Antioxidant defense
The balance between ROS production and antioxidant defense is vital for maintaining cellular homeostasis. When the cellular redox system is overwhelmed with oxidized proteins and lacks sufficient reduced counterparts for their reduction, oxidative stress arises. In cases of temporary stress, proteins may undergo reversible oxidation, but in instances of substantial or persistent imbalance, irreversible oxidative changes can take place. Therefore, cells have developed a sophisticated antioxidant defense system to prevent excessive oxidative damage and maintain favorable redox homeostasis.
Detoxification of ROS is achieved through a complex interplay of enzymatic and non-enzymatic molecules, including GSH, NADPH, thioredoxin (Trx), superoxide dismutase (SOD), glutathione peroxidase (GPx), glutathione reductase (GR), peroxiredoxin, and catalase (CAT). Superoxide radicals are converted into H2O2 by various isoforms of SOD (Cu/Zn-SOD, Mn-SOD, extracellular SOD), with catalase further converting H2O2 into water and oxygen [Citation42]. SOD isoforms are compartmentalised within specific cellular regions; for instance, cytoplasmic Cu/Zn-SOD is the main contributor to overall SOD activities, while Mn-SOD is the main mitochondrial antioxidant enzyme [Citation43]. Notably, elevated Cu/Zn-SOD expression has been associated with increased proliferation and invasiveness in tumor cells [Citation44]. Inhibition of Cu/Zn-SOD has been shown to induce cell cycle arrest in G1 and apoptosis [Citation43]. Cu/Zn-SOD overexpression has been linked to the positive regulation of Carnitine Palmitoyltransferase 1A (CPT1A), which may explain its role in promoting cancer cell growth [Citation44]. CPT1A is involved in cellular lipid metabolism, facilitating the entry of fatty acids into mitochondria and contributing to increased energy production.
Heme Oxygenase-1 (HO-1) is an enzyme that degrades heme, producing biliverdin, carbon monoxide (CO), and free iron. Biliverdin and CO contribute to the antioxidant effects of HO-1, protecting cells against oxidative stress and inflammation. However, elevated levels of HO-1 can have a pro-oxidant effect due to the release of free iron and consequent Fenton reaction [Citation45]. Intriguingly, HO-1 also promotes ferritin expression, essential for sequestering and storing free iron, mitigating its potential pro-oxidant effects. The role of HO-1 in cancer cell proliferation is gaining attention, with implications in various pathways such as BCR/ABL, c-Met–Ras, and EGFR–Src–NF-κB signaling to promote cell proliferation [Citation45–48].
GPx plays an important role in targeting and reducing lipid hydroperoxides and H2O2, safeguarding cellular membranes. The catalytic mechanism of GPx involves the use of GSH, and the resulting transformed form, glutathione disulfide (GSSG), is enzymatically converted back to its original state by glutathione reductase [Citation42]. Thioredoxin, a redox-active protein, contributes to antioxidant defense by reducing disulfide bonds in oxidized proteins. It has a role in cellular signaling and gene expression regulation, and the reduction of its oxidized form is catalyzed by thioredoxin reductase (TR), utilizing NADPH as an essential cofactor in the redox reaction [Citation35]. Additionally, peroxiredoxins often utilize thioredoxin as an electron donor during the reduction of peroxides [Citation49].
In response to oxidative stress, cells undergo distinct metabolic adaptations, altering their energy production and utilization. This includes a preference for glycolysis even when oxygen is available, known as the Warburg effect. Activating the pentose phosphate pathway (PPP) is crucial as it generates NADPH for antioxidant defenses and facilitates the recycling of key antioxidants, such as glutathione and thioredoxin. This metabolic shift also supports lipid production, essential for repairing and maintaining cell membranes [Citation50].
The antioxidant response can be indirectly triggered by oxidative stress. Inhibitors of the antioxidant response's effectors often contain a reactive cysteine residue, leading to the dissociation of inhibitor-target complexes upon cysteine oxidation [Citation34]. For example, the NF-E2-related factor 2 (Nrf2) family of transcription factors serves as the first-line defense activated by low increases in ROS levels [Citation51]. Nrf2 belongs to the cap'n'collar family of basic leucine zipper transcription factors. Under normal conditions, Nrf2 remains inactive in the cytoplasm, repressed by Kelch-like ECH-associated protein 1 (Keap1) [Citation52]. During oxidative stress, KEAP-1 forms a disulfide dimer at Cys151, losing its ability to bind Nrf2 [Citation53]. Activated Nrf2 then orchestrates the upregulation of antioxidant genes like SOD1, CAT, GPx, Trx, and HO-1, enhancing cellular resilience [Citation45,Citation54].
Another example of redox-regulated transcription factor is nuclear factor-κB (NF-κB). NF-κB is a family of transcription factors including p50, p52, p65, RelB, and c-Rel, regulated by inhibitory proteins (IκBs) in the cytoplasm [Citation55]. Upon activation triggered by stimuli like cytokines, pathogens, or stress, IκB undergoes phosphorylation by IKK (IκB kinase) and degradation in the proteasome, releasing NF-κB. Interestingly, IKK can be directly S-glutathionylated by H2O2, inhibiting its function [Citation20]. Similarly, the oxidation of native p50 leads to its glutathionylation, partially inhibiting its DNA binding ability [Citation56]. Once freed, NF-κB translocates to the nucleus, regulating gene transcription in response to changes in cell homeostasis. NF-κB is directly influenced by the TRX system, whereas it does not respond directly to the GSH system [Citation57]. In the cytoplasm, increased TRX levels inhibit the degradation of IκBα [Citation57]. Within the nucleus, TRX directly reduces cysteine modifications on NF-κB, enabling NF-κB-dependent gene expression. NF-κB plays a dual role in cellular redox balance, protecting against ROS by inducing the expression of antioxidant proteins like MnSOD, HO-1, or thioredoxin, while also assuming a pro-oxidant role by promoting the expression of genes such as the NOX2 subunit of NADPH oxidase [Citation55,Citation58,Citation59]. Prolonged oxidative stress has been observed to inhibit NF-κB activation by inactivating the proteasome and impeding I-κB degradation [Citation13,Citation55]. Furthermore, the complex interplay between Nrf2 and NF-κB shapes cellular responses to oxidative stress, with Nrf2 knockout enhancing NF-κB activity [Citation60]. Induced by Nrf2, HO-1 inhibits NF-κB [Citation61], while NF-κB suppresses Nrf2 by competing for the CBP–p300 complex [Citation62].
Metallothionein (MT) is a family of small, cysteine-rich proteins that bind heavy metal ions like zinc, copper, and cadmium. This family plays a key role in maintaining cellular metal homeostasis, detoxification, and shielding against metal toxicity, contributing to the cellular response to oxidative stress [Citation63]. A high GSH/GSSG ratio inhibits the release of zinc from the MT molecule [Citation64]. The zinc stored within MT molecules remains unavailable for the activation of specific target proteins that rely on zinc for their active conformation. This includes transcription factors with a zinc motif or p53, which utilizes zinc to stabilize its binding to DNA [Citation65–67]. Moreover, MT engages in antiapoptotic mechanisms by interacting with the p50 subunit of NF-κB, leading to the transactivation of the antiapoptotic NF-κB pathway [Citation67]. During oxidative stress, the depletion of free GSH coincides with the oxidation of thiol groups in cysteines within the MT molecule, resulting in the release of free zinc (II) [Citation63]. Released zinc (II) subsequently regulates various target proteins, initiating a positive feedback loop where it supports the expression of MT and activates Nrf2 [Citation68]. Nrf2, in turn, triggers the transcription of antioxidant enzymes such as HO-1 and CAT [Citation68]. MT concentration peaks in the G1 and G1/S phases of the cell cycle [Citation69]. In tumor cells, MT typically plays a protective role and contributes to cell proliferation [Citation70], although exceptions exist [Citation67]. For instance, phosphorylation induced by MT2A overexpression may inhibit cell proliferation through the inhibition of the Hippo signaling pathway [Citation63].
Redox status of the cell
Redox potential serves as a measurable indicator, reflecting a system's readiness for redox reactions by indicating its ability to donate or accept electrons. This property significantly influences the dynamic equilibrium of redox couples including NADH/NAD+, NADPH/NADP+, and GSH/GSSG. The GSH/GSSG ratio serves as a valuable indicator, often used to assess the cell's overall redox condition [Citation35]. GSH concentration is evenly distributed in the cytoplasm and nucleus, it peaks during the S and G2/M phases, and its depletion leads to reduced proliferation and apoptosis [Citation35].
The intracellular redox potential typically ranges from −160 to −260 mV, with cell death often occurring when it rises above −160 mV [Citation71,Citation72]. In noncancerous cells, a redox potential of approximately −207 mV has been identified as a threshold significant for pRB phosphorylation above which the cell cycle stops [Citation71,Citation72]. Intervening with antioxidants during the G1 phase induces a shift towards a more reducing redox state, resulting in increased hypophosphorylated pRB and subsequent cell cycle arrest [Citation73]. As cells progress to the S phase, their redox potential decreases, falling below the threshold for pRB dephosphorylation [Citation72]. Notably, the impact of oxidative stress on cells is closely tied to specific phases within the cell cycle; fibroblasts exposed to H2O2 undergo apoptosis in the S phase, while in G1 or G2/M phases, they stop proliferating [Citation11]. The cell's redox state emerges as a promising modulator of cell cycle progression, and antioxidant intervention holds the potential to regulate cell proliferation under pathophysiological conditions. However, pinning a definitive value to the cell's instantaneous overall redox potential remains challenging due to varying redox potentials among different cellular components [Citation74].
ROS impact on cell proliferation and progression through G1 phase
In the G1 phase of the cell cycle, cells decide whether to continue dividing or enter a state of rest known as quiescence or the G0 phase. In G0, cells focus on essential activities, and to re-enter the cell cycle, external cues like growth factors, hormones, and cytokines are needed.
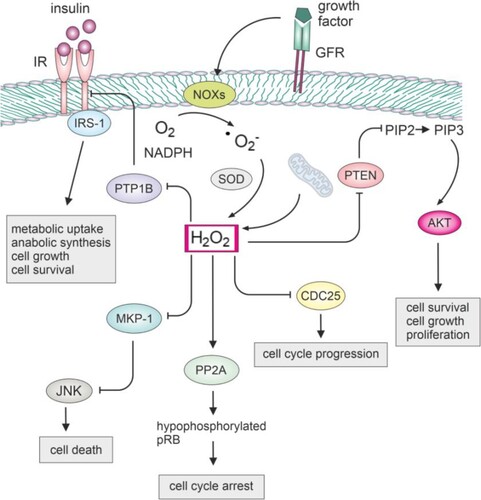
Ligand binding to growth factor receptors initiates a complex network of downstream signaling pathways. This process can trigger the production of H2O2 in nanomolar concentrations through the activity of NADPH oxidases like Nox1 and Nox4 [Citation75,Citation76]. Generated H2O2 can play a critical role in oxidizing and inactivating key phosphatases, that normally act to suppress proliferative pathways, as well as protein kinases. Specifically, redox-sensitive phosphatases, including protein tyrosine phosphatase 1B (PTP1B), phosphatase and tensin homolog (PTEN), protein phosphatase 2A (PP2A), Cdc25, and MAP kinase phosphatases (MKPs), are susceptible to oxidation due to the presence of cysteine with a low pKa in their catalytic sites () [Citation77–80]. Their inactivation can result in the physiological activation and amplification of proliferation signaling, but it may also contribute to the dysregulation of cell signaling and promote unchecked proliferative pathways. By oxidizing and inactivating JNK phosphatases within the MKP family in NF-κB deficient cells, H2O2 induced prolonged activation of JNK, leading to cell death [Citation14]. Oxidation of PTEN induces the formation of a disulfide bond, resulting in the inactivation of its phosphatase activity [Citation81]. Consequently, PTEN loses its ability to dephosphorylate PIP3, leading to the sustained activation of PI3 K/Akt signaling [Citation15,Citation82].
Figure 2. Impact of hydrogen peroxide on the function of key cellular phosphatases.
NOX produces superoxide, which can be converted to H2O2 by SOD. The generated H2O2 can then modulate redox-sensitive phosphatases. Inhibiting PTP1B may enhance insulin signaling, potentially improving glucose homeostasis. This can positively impact cell growth and metabolism. Inhibiting MKP-1 may lead to prolonged JNK activation, affecting stress signaling, apoptosis, or inflammatory processes. Inhibiting CDC25 could disrupt cell cycle progression, inducing cell cycle arrest. Inhibiting PTEN may lead to increased Akt signaling, promoting cell survival and growth. Dephosphorylation of pRB by PP2A results in the inhibition of E2F transcription factors, preventing the progression of the cell cycle from the G1 to the S phase.
Protein kinases, including members of the Src and fibroblast growth factor receptor (FGFR) families, are no exception from the adverse effect of oxidants (). Specifically, protein tyrosine kinases (PTKs) within these families can be directly affected by oxidation [Citation83,Citation84]. In the case of the Src family, Cys277 in the catalytic domain undergoes oxidation, resulting in the homodimerization of Src molecules connected by a disulfide bridge [Citation84]. This selective oxidative inactivation is prominent in crucial members of the Src family and all kinases within the FGFR family [Citation84]. Similarly, cAMP-dependent protein kinase (PKA) is also explored as a target for oxidative regulation, since it is susceptible to reversible inactivation by glutathionylation [Citation83].
Figure 3. Oxidative regulation of key protein kinases and cellular outcome.
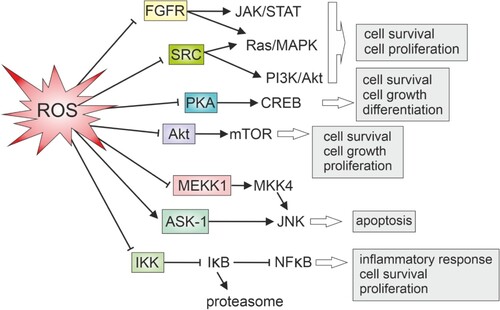
Protein kinase B, also known as Akt, is activated during growth factor signaling through sequential phosphorylation at Ser473 by mTORC2 and Thr308 by 3-phosphoinositide-dependent kinase 1 (PDK1) [Citation85]. Low levels of H2O2 promote Akt activation, possibly by oxidizing Cys310 to sulfenic acid near the catalytic loop. This oxidation may enhance Akt binding to PDK1, favoring Thr308 phosphorylation. Conversely, moderate to high H2O2 concentrations induce the oxidation of Akt1 Cys60 in the PH domain and Cys310 to sulfonic acid [Citation85]. In this oxidative environment, Akt and PDK1 binding is disrupted, preventing the second phosphorylation of Akt at Thr308 [Citation85]. Consequently, monophosphorylated Akt accumulates in mitochondria, leading to increased GSK-3α/β activity, reduced pro-survival signaling, and cytochrome c release, ultimately inducing apoptosis [Citation85].
MAPKs, including extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs), and p38 MAPK, contain cysteine residues sensitive to oxidation [Citation86–88]. The oxidation of these cysteine residues can induce conformational changes, influencing both kinase activity and downstream signaling.
According to one established model, Ask-1, a member of the MAP3K superfamily, interacts with reduced thioredoxin which acts as its inhibitor [Citation89]. Upon exposure to oxidants, thioredoxin undergoes oxidation, leading to its dissociation from the Ask-1 complex. Subsequently, Ask-1 can oligomerize, initiating downstream signaling for apoptosis [Citation89]. However, a more recent perspective suggests that H2O2 treatment oxidizes Ask1 directly and induces the formation of disulfide bonds between Ask1 molecules [Citation87]. Both phosphorylation and oxidation of Ask1, occurring independently, are considered crucial for competent signaling to MKK4/7, JNK, and apoptosis. While phosphorylation alone is adequate for Ask1 activation, oxidation plays a significant role in the subsequent phosphorylation of MKK4/7 and JNK. Notably, Trx1 transiently associates with Ask1 in this model, exerting a negative regulatory role by reducing Ask1 multimers [Citation87].
Similarly, within the MEKK1 (Mitogen-Activated Protein Kinase Kinase 1) domain, glutathionylation of cysteine residues induced by elevated oxidant levels results in the dysfunction of its kinase activity [Citation90]. MEKK1 is linked to stress responses and inflammation signaling, impacting downstream proteins like p38 MAPK or JNK in stress response signaling.
Furthermore, H2O2 is essential in autophagy by oxidizing cysteine residues in the structure of Atg4, a cysteine protease involved in autophagy. The oxidation inhibits the function of Atg4 and facilitates the required lipidation of Atg8, contributing to the autophagic process [Citation91]. Autophagy can be linked with cell cycle arrest and senescence, as reviewed elsewhere [Citation92]. The interplay between autophagy and cell cycle progression is evident from various signaling pathways, such as the involvement of AMBRA1 in both autophagy promotion and inhibition of cell proliferation via c-Myc regulation [Citation92]. Furthermore, CDKIs like p16, p21, and p27 are known to induce autophagy, suggesting coordinated stress responses connecting autophagy induction and cell cycle arrest [Citation92]. Additionally, autophagy proteins like Atg5 and Atg7 contribute to establishing senescence [Citation92].
The cyclin D-CDK4/6 complex has a main role in directing the cell cycle progression through the G1 phase, with its activity finely tuned by signaling events triggered by external cues for proliferation and growth. It was recently found that the oxidation of CDK4 triggers the formation of a reversible intermolecular disulfide bond between Cys135 of CDK4 and Cys7/8 of cyclin D, which inhibits cyclin D-CDK4 activity, consequently reducing cell proliferation in a mouse model of pulmonary hypertension [Citation93]. The activation of cyclin D promoters involves the integration of various pathways, including Ras-Raf-MEK-ERK, PI3K-Akt, and Wnt signaling [Citation94]. These pathways converge to activate specific transcription factors, such as AP-1, STAT, TCF, and c-myc, which bind to the consensus sequence within the cyclin D promoter and initiate its transcription [Citation94]. For example, in cardiac cell proliferation, Nox4-generated ROS triggers ERK1/2 activation, leading to c-myc phosphorylation and an increase in cyclin D2 expression [Citation76].
The AP-1 (Activator Protein 1) transcription factor plays a crucial role in initiating cyclin D transcription. It consists of various Jun family proteins (c-Jun, JunB, JunD) and Fos family proteins (c-Fos, FosB, Fra-1, Fra-2) [Citation95]. Specifically, c-Jun, Fra-1, and Fra-2 act as activators in cyclin D1 transcription. In contrast, c-Fos has a dual role, functioning as both a depressor and activator of the cyclin D1 promoter at different stages [Citation96]. Meanwhile, JunB consistently acts as a repressor in regulating the cyclin D1 promoter [Citation96]. Upon growth factor stimulation in quiescent cells, c-Fos is synthesized and translocated to the nucleus. Subsequently, its ERK-mediated phosphorylation and degradation occur, facilitating Fra1 protein access to chromatin and initiation of cyclin D1 expression [Citation97]. However, heightened H2O2 levels cause hyperactivation of ERK1/2, hindering cell cycle progression by stabilizing c-Fos binding to DNA and impeding Fra-1 access to chromatin, resulting in blocked cyclin D1 expression [Citation97,Citation98]. In contrast, Munõz [Citation5] observed that millimolar concentrations of H2O2 maintained cyclin D levels at a consistent level in Her14 fibroblasts. This discrepancy in findings might be attributed to the timing of hydrogen peroxide application, potentially occurring after cyclin D was already expressed within the cell. Notably, the observation that inhibiting cellular protein synthesis had no impact on cyclin D levels further supports this possibility. The other potential mechanism underlying the observed effect on cyclin D expression is that H2O2 interferes with the ubiquitin-proteasome degradation of cyclin D [Citation5], which is further supported by another study where sustained oxidative stress led to proteasome inhibition [Citation13].
Additionally, Menon et al. [Citation99] conducted a study involving the treatment of mouse fibroblasts with antioxidant N-acetylcysteine (NAC), resulting in an increased proportion of cells arrested in the G1 phase accompanied by a notable decrease in cyclin D1 levels. This effect was coupled with later elevation in MnSOD levels. Interestingly, after NAC application, the level of superoxide rose, particularly in cells with lower MnSOD levels, leading to a more pronounced G1 arrest. MnSOD's role in converting superoxide to H2O2 supports the possibility of a rapid rise in superoxide radicals after treatment with NAC. These findings collectively suggest that NAC may have an immediate prooxidant effect, contrasting its longer-term antioxidant capabilities. Notably, the expression of D cyclins is transient, with rapid degradation upon withdrawal of mitogenic signals. Cyclin D possesses two phosphorylation sites at T286 and T288, and withdrawal of growth factors activates glycogen synthase kinase-3 Beta (GSK-3Beta), marking D cyclins for ubiquitination and eventual degradation. The study suggests redox-sensitive regulation of T286 phosphorylation in cyclin D degradation, as nondegradable T286A mutants’ levels of cyclin D didn't change after NAC treatment [Citation99].
The signal transducer and activator of transcription 3 (Stat3) is known to play a role in promoting cell cycle progression, particularly from the G1 phase to the S phase. It regulates key proteins like cyclin D and CDKs. Stat3 possesses redox-sensitive cysteines, and their inactivation through mutation resulted in increased cell proliferation [Citation100]. Genes linked to immune function and cell adhesion were observed to be indirectly upregulated via Stat3 in response to oxidative stress, while genes displaying reduced activity were associated with developmental processes and cell death [Citation101]. Additionally, a synergistic interplay between Stat3 and Hif-1α in response to oxidative stress was described [Citation101]. In a study conducted by Xie et al. [Citation102], mild oxidizing conditions induced S-glutathionylation of Stat3 in HepG2 hepatocarcinoma cells. This post-translational modification adversely affected Stat3's functional state, influencing nuclear translocation, phosphorylation status, and, consequently, its ability to execute transcriptional functions. Furthermore, the altered Stat3 exhibited reduced favorability as a substrate for other signaling proteins, including JAK2 [Citation102]. In contrast, a different study demonstrated that H2O2, in conjunction with vanadate, induced tyrosine phosphorylation of Stat3, leading to its subsequent translocation into the nucleus in peripheral human lymphocytes [Citation7]. The authors hypothesized that the stimulating effects of oxidants, specifically H2O2, might arise from the inhibition of intracellular PTPases. They also suggested that intracellular Fe2+/Cu2+ ions could potentially serve as intermediates under conditions of H2O2-induced oxidative stress, enhancing the nuclear translocation of Stat3 [Citation7].
The contrasting effects of oxidants, not only on Stat3 function, observed in various studies may be attributed to variations in experimental design. Notably, different cell lines were employed, potentially yielding distinct responses to similar stimuli. The choice between cancerous and normal cell lines further introduces variability in the redox environment and cellular responses. Additionally, variations in the treatments, encompassing differences in chemical agents, duration, and other conditions, could contribute to the divergent outcomes. This heterogeneity in experimental parameters emphasizes the importance of considering diverse factors when interpreting the redox regulation of proteins and its implications for cell cycle dynamics.
The cyclin D-CDK4/6 complex phosphorylates the tumor suppressor pRB (retinoblastoma), a key regulatory event that tightly governs the progression of the cell cycle. The RB protein is hypophosphorylated upon entry into the G1 phase of the cell cycle and loses its inhibitory function. Monophosphorylation of the RB protein by the cyclin D-CDK4/6 complexes creates a transition between entry into the G0 phase and commitment to continue the cell cycle. The hyperphosphorylated RB protein is inactivated and releases E2F transcription factors, which can activate transcription in the nucleus, leading the cell to enter the next phase of the cycle. E2F-activated genes are responsible for cell cycle progression, DNA replication, genome protection, and cell growth. Transcription of E cyclins by E2F causes activation of CDK2, and this complex further phosphorylates pRB, leading to its complete inactivation and transition from G1 to S phase [Citation3]. A study by Cicchillitti et al. [Citation103] investigated the impact of H2O2 on retinoblastoma family proteins (pRb, p107, and p130) in endothelial cells. H2O2 induced rapid hypophosphorylation of these proteins, mediated by PP2A. Inhibiting PP2A prevented hypophosphorylation, and its activity positively correlated with H2O2 treatment [Citation103]. Moreover, PP2A inhibition prevented H2O2-induced DNA synthesis inhibition, suggesting a role for PP2A-mediated dephosphorylation of pRB in the cellular response to oxidative stress [Citation103].
DNA damage and S phase
During the S phase, also known as the synthesis phase, the cell undergoes genetic material duplication in preparation for subsequent cell division. This phase involves critical processes, including the proofreading and repair of DNA errors to ensure accurate genetic duplication.
CDK2 plays an important role during the transition from the G1 to the S phase. It transitions from binding cyclin E to cyclin A. During the transition phase, the negative regulator Kinase Associated Phosphatase (KAP) gains access to the T-loop of CDK2. This interaction between CDK2 and KAP is tightly regulated by the oxidation of CDK2 to sulfenic acid on Cys177, which is physiologically caused by mitochondrial ROS. This oxidation ensures the phosphorylation of CDK2 in the T-loop, a modification that obstructs KAP binding and facilitates the subsequent activation of CDK2 [Citation104]. The study by Deshpande et al. [Citation19] demonstrated that H2O2 had a dual impact on cell cycle regulators. Surprisingly, H2O2 initially increased CDK4 activity, potentially propelling cells from quiescence to mid-late G1. However, this was followed by a subsequent inhibition of cyclin A–CDK2 activity, causing cell cycle arrest at the G1/S interface. In response to serum, H2O2 completely suppressed cyclin A mRNA, essential for G1/S and S phase progression, while cyclin D1 mRNA remained unaffected [Citation19]. Additionally, H2O2 induced a significant increase in the expression of the cell cycle inhibitor p21, with no observed change in p27 protein levels. The heightened levels of p21 protein highlight its potential role as a major effector in H2O2-induced growth arrest [Citation19].
A recent study conducted by Kirova et al. [Citation104] has presented compelling evidence that showcases the essential role of oxidants in managing cell cycle progression during the S-phase. The study findings reveal that antioxidant treatment leads to a dose-dependent reduction in proliferation. The study effectively reduced mitochondrial ROS production by limiting the metabolite supply to the TCA cycle in mitochondria. Consequently, this alteration led to a slower proliferation rate and an extended duration of the S phase. The reduced formation of mitochondrial ROS was also associated with a delayed onset of DNA replication in the S phase, suggesting a direct involvement of ROS in promoting DNA replication [Citation104]. However, this observation may be attributable to the disruption of mitochondrial metabolism, whereby metabolites essential for DNA synthesis might become unavailable. The observed natural increase in ROS levels during the S and G2/M phases could be a consequence of heightened mitochondrial activity, including the shift from glycolysis to oxidative phosphorylation and an increase in mitochondrial mass [Citation104].
Before DNA replication, the cell evaluates nutrient, energy, and growth factor availability, and checks for DNA damage. The Intra-S phase checkpoint maintains replication fork integrity, ensuring accurate DNA replication, and enables a seamless transition into the mitotic phase. Activated in response to various forms of damage, including oxidative stress, this checkpoint suppresses the activation of late replication origins [Citation105]. Oxidative stress-induced DNA damage manifests in a variety of deleterious effects on genetic material, such as the conversion of guanine to 8-oxoguanine (8-oxoG), alterations in base pairs, the formation of apurinic/apyrimidinic (AP) sites, single-strand breaks (SSBs), double-strand breaks (DSBs), mutations, changes in gene expression and increased susceptibility to cancer [Citation88]. The MRN (MRE11-RAD50-NBS1) complex recognizes DNA damage and activates the damage sensor and signal transducer protein kinase ataxia-telangiectasia mutated (ATM), which, in turn, phosphorylates numerous substrates. Among these substrates, Chk1/2 and p53 are considered the most crucial players in the cellular response to DNA damage. Elevated levels of ROS activate ATM, which in turn can lead to cell cycle arrest through the ATM-Chk2 pathway [Citation106]. Inhibiting ATM under elevated levels of oxidants did not activate p53 and Chk2, indicating that oxidative stress primarily activates p53 through ATM [Citation107].
In DNA repair, apurinic/apyrimidinic endonuclease-1 (APE1) is needed for the base excision repair (BER) pathway, addressing damage to single-stranded DNA (ssDNA) and AP sites from ROS or alkylating agents [Citation108,Citation109]. The protein's expression peaks during the S phase of the cell cycle, signifying an increased risk of DNA damage due to heightened DNA replication activity and ROS production [Citation108]. H2O2-induced APE1 expression enhances overall cellular resistance to oxidative stress [Citation108]. Moreover, APE1 enhances AP-1 binding to DNA by reducing the conserved Cys272 residue in the DNA-binding domain of c-Jun, and the oxidation of APE1 significantly impairs this ability which can lead to a decrease in cyclin D expression [Citation108,Citation110,Citation111]. APE1 possesses seven conserved cysteines implicated in redox regulation, but their accessibility varies. While some studies point to Cys65 as an active redox site, Bhakat's protein model suggests its limited accessibility for oxidation [Citation108,Citation112–114]. Conversely, Howpay Manage et al. [Citation109] detected oxidation of five out of seven cysteines in APE1, including Cys65, when exposed to CO3•– [Citation109].
P53, a critical tumor suppressor, plays a key role in the cellular response to DNA damage, capable of inducing cell cycle arrest, DNA repair, or apoptosis upon activation [Citation115]. The multifaceted and context-dependent nature of oxidation in p53 function is a subject of ongoing research. In response to oxidative stress, p53 activates the transcription of genes such as p21 and redox enzymes like Gpx1 and MnSOD, forming a protective mechanism against the damaging effects of ROS [Citation71,Citation88,Citation116,Citation117]. Accordingly, reduced p53 activity contributes to ROS accumulation, further supporting its antioxidant function [Citation71,Citation117,Citation118]. However, some studies propose a potential pro-oxidant role for p53, linking its activation to increased ROS levels and apoptosis induction, while also functioning as a transactivator for prooxidative genes like PIG3 and PIG6 [Citation88,Citation119,Citation120]. It was shown that p53 counteracts Nrf2-induced transcription of certain antioxidant genes, suggesting a negative control mechanism to prevent the generation of a robust antioxidant environment that might impede the induction of apoptosis [Citation120].
The dual nature of p53's effects on ROS and apoptosis depends on cellular levels of oxidants and their signaling duration. Transient oxidation tends to induce temporal cell cycle arrest and antioxidant function of p53, while prolonged induction or high oxidative stress are associated with apoptosis and pro-oxidative effects of p53 signaling [Citation71,Citation120,Citation121]. Oxidation of specific cysteine residues in p53's DNA-binding domain can impact the protein's ability to bind to specific DNA sequences. P53 has ten cysteine residues in the DNA-binding domain [Citation88]. Cys182 has been identified as sensitive to oxidation, potentially leading to structural damage in the p53 protein [Citation122]. On the other hand, oxidation of Cys277 may contribute to the recognition of specific p53 response elements in genes, enhancing their activation in response to redox regulation [Citation88,Citation123]. In conclusion, the role of oxidation in p53 function is complex and dynamic, involving a delicate balance between pro-oxidant and antioxidant effects. The context-dependent nature of p53's response to oxidative stress underscores the need for further research to identify the molecular mechanisms underlying its dual role in maintaining cellular redox homeostasis.
G2, M phase, and ROS influence
In the G2 phase of the cell cycle, DNA replication is finalized, and the cell readies itself for mitosis. This phase also acts as a checkpoint to detect any DNA damage or incomplete replication that could potentially cause problems. The CDK2/cyclin A complex is involved in the control of DNA replication during the S phase and is still active in the G2 phase, contributing to the regulation of the G2/M transition [Citation124]. CDK1 (or CDC2) and cyclin B complex have a main role in the progression from G2 to mitosis – the levels of cyclin B rise during the G2 phase and peak at the G2/M transition. As A/B-CDK1 activity surpasses a threshold, the prophase of mitosis is initiated. This elevation in A/B-CDK1 activity leads to the phosphorylation of numerous CDK1 substrates, including mitotic kinases. It induces cellular rounding, chromatin condensation within the nucleus, nuclear envelope disassembly, and permeabilization [Citation32].
Cdc25 (Cell Division Cycle 25) phosphatase enzymes belong to PTPs and play a key role in cell cycle progression by activating CDKs through dephosphorylation. Specifically, Cdc25B and Cdc25C are crucial in regulating G2/M transitions through dephosphorylating Cdk1 [Citation125,Citation126]. The activation of Cdc25C in the G2/M transition involves its dissociation from 14-3-3 proteins [Citation126] (p201). The activation of Cdc25B in the G2/M transition involves its phosphorylation by PLK1, PKA, and Aurora-A kinase [Citation126]. The phosphorylated form of Cdc25B concentrates at the centrosome, where it colocalizes with Aurora-A [Citation126,Citation127].
In response to H2O2 exposure, disulfide bonds form between Cys377 and Cys330 in the Ccd25C, leading to the inactivation of its function [Citation128,Citation129]. The active-site cysteine in Cdc25 can be shielded from rapid oxidation to irreversibly inactivated sulfinic acid by swiftly forming intramolecular disulfide bonds with adjacent (back-door) cysteines. This protective mechanism prevents the potential consequences of irreversible oxidation and is efficiently reduced by thioredoxin/thioredoxin reductase [Citation129].
The Anaphase-Promoting Complex/Cyclosome (APC/C) is a multi-subunit E3 ubiquitin ligase with the primary function of targeting specific proteins for ubiquitination and subsequent degradation by the proteasome. Cdh1, or Cdc20 Homolog 1, is an activator subunit of APC/C, particularly in late mitosis and the G1 phase. Its activation of APC/C allows it to recognize and ubiquitinate its substrates such as cyclin A, cyclin B, and securin, promoting the progression to anaphase, while securin degradation releases separase, which cleaves cohesin and separates the chromatids. It also initiates the formation of an actomyosin contractile ring that contracts during cytokinesis, dividing the cell into two. Cdh1 is phosphorylated by CDK1, leading to inhibition of its interaction with APC/C. With an increased level of endogenous H2O2, coordination cysteine is oxidized in the APC11 unit of the APC complex, causing the display of zinc from the molecule resulting in APC's inability to bind to the enzyme ubiquitin-conjugating enzyme E2 4 (Ubc4) [Citation6,Citation130]. Ubc4, as an E2 ubiquitin-conjugating enzyme, catalyzes the transfer of ubiquitin molecules to target proteins, marking them for degradation by the proteasome.
In prometaphase, the nuclear envelope disintegrates, allowing condensed chromosomes to interact with microtubules, thereby prompting the formation of the mitotic spindle [Citation32]. Kinases such as Plk, Aurora A, and Aurora B actively contribute to spindle assembly and connection. The activity of Aurora kinases is triggered by auto-phosphorylation at conserved threonine residues, mediated by co-factors [Citation131]. It was found that oxidative stress caused hyperphosphorylation of Aurora A at the beginning of mitosis, abnormal mitotic spindle formation, stabilized levels of cyclin B, and a significantly delayed mitosis [Citation22]. Moreover, other studies pointed out that increased phosphorylation of Aurora A at T288 by PP1[Citation132], PAK1[Citation133], or PP6[Citation134] can negatively affect its function [Citation22]. It was also discovered that the catalytic activity of the Ser/Thr kinase Aurora A is inhibited when the conserved cysteine residue (Cys290) adjacent to the critical phosphorylation site Thr288 undergoes oxidation [Citation24]. Similarly, a study by Chang et al. [Citation6] observed that H2O2 inhibits cyclin B degradation and delays mitosis; however, these results had been attributed to the oxidation of APC/C [Citation6].
Contrary to this, Lim et al. [Citation135] proposed a different perspective. They found that during the cell cycle, the centrosome is shielded from endogenous H2O2 influence by the H2O2-eliminating enzyme PrxI. At the beginning of mitosis, PrxI is inactivated by Cdk1, causing a temporary increase in H2O2 levels in the centrosome. This increase deactivates phosphatases that negatively regulate Cdk1, especially Cdc14B, near the centrosome, leading to further activation of Cdk1 [Citation135]. The experiment demonstrated that H2O2 is necessary during mitosis to enhance the full activation of Cdk1 [Citation135]. This finding could potentially explain why persistent H2O2 signaling results in increased hyperphosphorylation of Aurora A and halted degradation of cyclin B, with the cause potentially being sustained signaling of active Cdk1. It is noteworthy that full activation of Aurora during G2/M is downstream of Cdk1 [Citation136], and activated Cdk1 inhibits APC/C. This inhibition ensures the prevention of cyclin B degradation. Taken together, this suggests that the temporal rise of endogenous H2O2, produced under physiological conditions, may be crucial for mitotic transition, while unnatural levels and timing of its activity could lead to mitotic arrest, potentially caused by sustained CDK1 signaling. However, the specific mechanisms of this outcome and the role of prolonged CDK1 activation require further exploration.
Therapeutic strategies
ROS are critical regulators of the cell cycle, exerting both beneficial and detrimental effects depending on their concentration, duration of action, and the cellular context. At physiological levels, ROS act as signaling molecules that promote normal cell cycle progression and maintain cellular homeostasis. ROS regulate key physiological processes, including cell proliferation, differentiation, and survival through the activation of signaling pathways such as MAPK, PI3 K/Akt, and NF-κB, which are critical for cell cycle progression, immune response, and homeostasis. ROS also modulate the activity of cyclins and CDKs, which are key regulators of the cell cycle. In 2003, Nick Lane introduced a theory of ageing and disease termed the double-agent theory [Citation137]. Central to this theory is the role of the transcription factor NF-κB in ageing and age-related diseases. While NF-κB is crucial for cellular survival during acute infections, a shift in the intracellular redox state during ageing could cause chronic activation of NF-κB, leading to prolonged inflammatory responses that contribute to many age-related chronic diseases. Thus, oxidative stress and NF-κB activation could be beneficial when transiently activated during infections but harmful when persistently elevated. Exploring the similarities and differences between acute and prolonged oxidative stress can offer valuable insights into the pathogenesis of many age-related diseases. Excessive ROS can cause oxidative damage, leading to cell cycle arrest, apoptosis, and various pathological conditions, including cancer and neurodegenerative diseases. Understanding the dual roles of ROS in cell cycle regulation is crucial for developing therapeutic strategies aimed at modulating ROS levels and mitigating their harmful effects.
Therapeutic strategies targeting oxidative stress include antioxidants, redox-active compounds, and modulators of redox-sensitive signaling pathways to mitigate damage and improve patient outcomes. One therapeutic strategy involves the direct exogenous intake of antioxidants. However, due to the rapid reactivity of oxidants, external scavengers often fall short compared to the numerous endogenous molecules already present in biological systems [Citation138]. Enzymatic antioxidants like SOD and GPx are more efficient at removing ROS compared to small molecule antioxidants. SOD mimetics and GPx mimics, which replicate these enzymes’ activities, have been extensively studied for their potential to mitigate oxidative stress [Citation138]. SOD mimetics, synthetic compounds replicating SOD activity, show significant promise in therapy. For example, manganese porphyrins shield healthy cells from radiation-induced damage in mouse models [Citation139]. Administration of GC4419 reduced radiation-induced oral mucositis in head and neck cancer patients [Citation140], while salen-Mn complexes have shown neuroprotective effects in neurodegenerative disease models [Citation141]. GPx mimetics, like Ebselen and ALT-2074, reduce hydrogen peroxide and lipid peroxides by utilizing glutathione as a substrate. Ebselen has shown efficacy in reducing ischemia-reperfusion injury in stroke and myocardial infarction [Citation142,Citation143] and holds promise for neuroprotection and anti-inflammatory effects in neurodegenerative diseases [Citation144].
Levels of GSH, a critical intracellular antioxidant, can be enhanced using GSH esters (monomethyl, monoethyl, diethyl, and isopropyl) to improve cellular uptake or by supplementing NAC, a GSH precursor used in treating acetaminophen overdose, chronic bronchitis, and some psychiatric disorders [Citation138]. NOX inhibitors (diphenyleneiodonium, Ebselen, CYR5099, Apocynin, GKT137831) reduce ROS production and protect tissues, showing potential in cardiovascular diseases, neurodegenerative disorders, and cancer [Citation138,Citation145,Citation146].
ROS modulation in cancer therapy
In cancer treatment, ROS modulation is a double-edged sword. While high levels of ROS can induce apoptosis in cancer cells, excessive oxidative stress can also contribute to cancer progression and resistance to therapy. Pro-oxidant cancer therapy aims to selectively increase ROS levels in cancer cells, exploiting their already elevated oxidative stress. Chemotherapeutic agents such as doxorubicin and cisplatin generate ROS as part of their cytotoxic mechanism, leading to DNA damage, cell cycle dysregulation, and apoptosis in cancer cells. Combining ROS-inducing agents with other therapies can improve outcomes. This approach is particularly effective in tumors resistant to conventional therapies [Citation147]. For example, apigenin selectively induces cell cycle arrest in cancer cells, inhibits the upregulation of thymidylate synthase, enhancing the efficacy of 5-FU in colorectal cancer cells [Citation148]. It also improves the effectiveness of paclitaxel in HeLa cells through induction of ROS production [Citation149]. Similarly, quercetin and cucurbitacin B also increase ROS production, enhancing the efficacy of paclitaxel in prostate cancer treatment and cisplatin in ovarian cancer therapy [Citation150,Citation151]. Interestingly, quercetin exhibits dose-dependent effects: at low doses, it acts as an antioxidant, reducing oxidative stress and offering protective benefits in conditions like diabetes and neurodegenerative diseases [Citation152,Citation153].
Moreover, compounds like β-lapachone and elesclomol, specifically increase ROS production and induce cell cycle arrest and apoptosis in cancer cells in preclinical models by targeting metabolic pathways unique to these cells [Citation154,Citation155]. However, out of 20 examined clinical trials (clinicaltrials.gov, selected filters: Intervention/treatment: elesclomol or β-lapachone), only one with elesclomol has posted results, which were evaluated as too insignificant for further investigation [Citation156]. Additionally, elesclomol's effectiveness as an anticancer agent relies on oxygen and OXPHOS, making it potentially ineffective in glycolytic tumors [Citation157,Citation158].
Therapeutic applications of antioxidants
The therapeutic potential of antioxidants has been explored in various contexts, particularly in cancer treatment. Several well-known antioxidants, such as L-ascorbic acid (vitamin C) and α-tocopherol (vitamin E), along with essential enzyme cofactors like selenium and riboflavin, play crucial roles in maintaining redox balance, and their potential therapeutic benefits are explored through various ongoing clinical studies and trials.
Vitamin C, a potent reducing agent, participates in neutralizing free radicals but has been shown to exhibit pro-oxidant effects under certain conditions, particularly at high doses [Citation159]. This dual function is exploited in cancer therapy, where high-dose intravenous vitamin C, combined with chemotherapy, has demonstrated enhanced tumor shrinkage in mouse models and reduced side effects in ovarian cancer patients [Citation160]. A combination of vitamin C and chemotherapy or radiotherapy is actively investigated in several clinical trials (NCT03508726, NCT04634227, NCT01852890, NCT03541486, NCT02905578, NCT06083454). Its pro-oxidant mechanism involves the disruption of intracellular iron metabolism and sensitization of cancer cells to oxidative damage [Citation160,Citation161]. Similarly, vitamin E, particularly in its delta-tocotrienol form, has shown promise in inducing apoptosis in neoplastic cells and enhancing chemopreventive outcomes in pancreatic cancer patients [Citation162]. Astaxanthin, a carotenoid with strong antioxidant properties, has shown beneficial effects in Polycystic Ovary Syndrome (PCOS) by enhancing serum Total Antioxidant Capacity and activating the Nrf2 pathway in granulosa cells [Citation163].
The meta-analyses consistently show that green tea consumption is associated with a reduced risk of several types of cancers, particularly those related to reproductive organs, the lung, non-Hodgkin's lymphoma, and oral cancer [Citation164]. Some studies, such as those assessing green tea's impact on cardiovascular risk factors and glycemic control, show both significant and non-significant outcomes, reflecting its complex role in health [Citation165,Citation166]. In the context of HPV-related cervical disease, green tea extract did not significantly impact disease progression [Citation167]. Despite mixed results, green tea is being extensively investigated as evidenced by a search on ClinicalTrials.gov for active studies using ‘green tea’ as an intervention/treatment, which yielded 132 results as of May 2024.
Melatonin, another potent antioxidant, has shown mixed results, with significant benefits observed in advanced cancer patients but not in early-stage disease [Citation168]. Curcumin, a compound found in turmeric, shows significant therapeutic potential due to its anti-inflammatory, antioxidant, and anticancer properties [Citation169]. Studies have demonstrated its ability to modulate inflammatory pathways and enhance the efficacy of cancer treatments. For instance, oral curcumin reduced the severity of radiation dermatitis in breast cancer patients [Citation170]. However, its clinical application is hampered by poor bioavailability, which results in low plasma and tissue levels, thereby limiting its efficacy [Citation171]. Despite promising findings, some clinical trials report mixed results; for example, adding curcumin to chemoradiotherapy did not improve complete response rates in patients with metastatic prostate cancer or locally advanced rectal cancer [Citation172,Citation173]. These inconsistent outcomes highlight the need for improved curcumin formulations with better bioavailability and further research to determine optimal dosages that ensure safety and efficacy.
Cancer cells often have upregulated antioxidant systems to manage their higher ROS levels. Inhibiting these antioxidant defenses can disrupt the redox balance, leading to oxidative stress cell cycle arrest and cell death. This strategy targets the cancer cells’ reliance on antioxidant systems to survive in a high-ROS environment. Agents like buthionine sulfoximine, which depletes GSH, enhance the efficacy of ROS-inducing therapies by preventing cancer cells from neutralizing ROS [Citation147]. Inhibitors of enzymes such as SOD and catalase can also reduce the capacity of cancer cells to neutralize ROS.
Given the mixed outcomes and the potential for antioxidants to both aid and hinder therapy, their use must be tailored to individual patient profiles and specific treatment contexts. Advances in personalized medicine, including the identification of biomarkers for oxidative stress and antioxidant capacity, will be crucial in optimizing antioxidant use in therapy. Large-scale, well-designed clinical trials are needed to provide definitive evidence of redox treatment efficacy and safety.
Conclusion
Maintaining redox homeostasis within the cell is critical, as its disturbance can lead to dysregulated proliferation or cell death. Experimental studies demonstrate the possibility for exogenous control of this homeostasis, offering potential therapeutic avenues for diseases associated with heightened oxidative stress. However, leveraging this knowledge in therapy requires a thorough understanding and prediction of the cell's response to oxidative stress. The precise role of ROS in cell cycle regulation remains uncertain. While some studies highlight the necessity of protein oxidation for its progression, others emphasize undesirable inhibitory effects (). Redox regulation of proteins involved in the cell cycle, as detailed in the , shows how reactive cysteines can influence their function. For instance, modifications such as disulfide bonds and sulfenic acid formation can activate or inactivate key proteins, impacting cell cycle progression. It is evident that the concentration of oxidants within the cell has a key role, but determining what concentrations are considered favorable or damaging for cell signaling remains unclear. To employ redox signaling in clinical research, it is imperative to conduct further research that accurately describes the effects of ROS based on the concentration, cell phase, and cell type. Only through comprehensive investigations can we gather the necessary insights to influence cell fate through redox signaling.
Figure 4. Redox-mediated regulation of cell cycle phases.
Increased ROS during the transition from G0 to G1 signaling can cause continuous ERK signaling and halt c-fos degradation, subsequently preventing the access of the transcription factor Fra-1 to chromatin, which is necessary for the transcription of cyclin D. In the G1 phase, ROS can cause the cell to halt the cycle due to the inhibition of pRB hyperphosphorylation, which, in this state, will not allow the release of E2F transcription factors necessary for progression to the S phase. ROS can affect the signaling function of STAT3 and inhibit cycle progression. ROS was suggested to prevent the breakdown of cyclin D by inhibiting the proteasome. However, positive effects have also been described, where ROS mediate c-myc activation through ERK signaling and upregulation of cyclin D. In the S phase, ROS can contribute to cellular resistance to oxidative stress through APE-1 and enhancing the repair capacity of the cell, while also mediating passage through the S phase by inhibiting KAP, thus enabling phosphorylation of Cdk2. During the S phase, ROS can also contribute to the cell's decision to stop or exit the cell cycle by damaging cell structures and activating the checkpoint through ATM and p53, or they can activate p21, which subsequently inhibits the activity of CDK2. In the G2/M phase, ROS exert an inhibitory effect on CDC25 and APC/C, preventing mitosis progression. Still, it is assumed that a lower amount of ROS is necessary for the inhibition of CDC14B, which subsequently leads to the activation of APC. Pathways indicated in red color show the inhibitory effect of ROS on cell cycle progression, while green lines represent pathways supporting its progression.
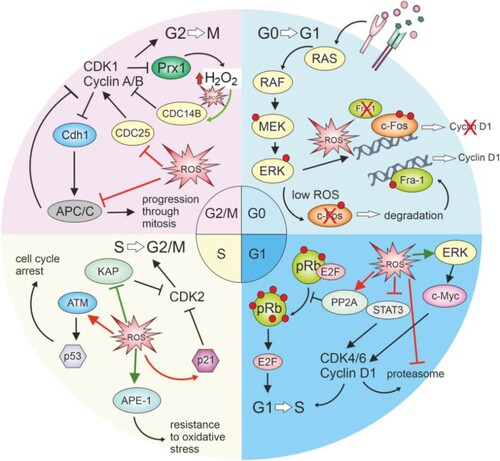
Table 2. Identified reactive cysteine modifications in cell cycle regulators.
Author statement
Viktoria Mackova participated in writing of manuscript, design of figures and table preparations.
Martina Raudenska participated in writing and supervision of manuscript and created all figures in graphical software. Hana Holcova Polanska and Milan Jakubek participated in writing and supervision of manuscript. Michal Masarik design concept of manuscript and participated in manuscript supervision.
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Janssen-Heininger YMW, Mossman BT, Heintz NH, et al. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med. 2008;45(1):1–17. doi:10.1016/j.freeradbiomed.2008.03.011
- Laurent A, Nicco C, Chéreau C, et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005;65(3):948–956. doi:10.1158/0008-5472.948.65.3
- Sarsour EH, Kumar MG, Chaudhuri L, et al. Redox control of the cell cycle in health and disease. Antioxid Redox Signal. 2009;11(12):2985–3011. doi:10.1089/ars.2009.2513
- Zhang Y, Qian D, Li Z, et al. Oxidative stress-induced DNA damage of mouse zygotes triggers G2/M checkpoint and phosphorylates Cdc25 and Cdc2. Cell Stress Chaperones. 2016;21(4):687–696. doi:10.1007/s12192-016-0693-5
- Martínez Munõz C, Post JA, Verkleij AJ, et al. The effect of hydrogen peroxide on the cyclin D expression in fiborblasts. Cell Mol Life Sci CMLS. 2001;58(7):990–996. doi:10.1007/PL00013204
- Chang TS, Jeong W, Lee DY, et al. The RING-H2–finger protein APC11 as a target of hydrogen peroxide. Free Radic Biol Med. 2004;37(4):521–530. doi:10.1016/j.freeradbiomed.2004.05.006
- Carballo M, Conde M, El Bekay R, et al. Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J Biol Chem. 1999;274(25):17580–17586. doi:10.1074/jbc.274.25.17580
- Ransy C, Vaz C, Lombès A, et al. Use of H2O2 to cause oxidative stress, the catalase issue. Int J Mol Sci. 2020;21(23):9149. doi:10.3390/ijms21239149
- Lin KY, Chung CH, Ciou JS, et al. Molecular damage and responses of oral keratinocyte to hydrogen peroxide. BMC Oral Health. 2019;19(1):10. doi:10.1186/s12903-018-0694-0
- Tan J, Li P, Xue H, et al. Cyanidin-3-glucoside prevents hydrogen peroxide (H2O2)-induced oxidative damage in HepG2 cells. Biotechnol Lett. 2020;42(11):2453–2466. doi:10.1007/s10529-020-02982-2
- Chen QM, Liu J, Merrett JB. Apoptosis or senescence-like growth arrest: influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem J. 2000;347(Pt 2):543–551. doi:10.1042/bj3470543
- Wang F, Nguyen M, Qin FXF, et al. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6(4):505–514. doi:10.1111/j.1474-9726.2007.00304.x
- Wu M, Bian Q, Liu Y, et al. Sustained oxidative stress inhibits NF-kappaB activation partially via inactivating the proteasome. Free Radic Biol Med. 2009;46(1):62–69. doi:10.1016/j.freeradbiomed.2008.09.021
- Kamata H, Honda SI, Maeda S, et al. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120(5):649–661. doi:10.1016/j.cell.2004.12.041
- Le Belle JE, Orozco NM, Paucar AA, et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3 K/Akt-dependant manner. Cell Stem Cell. 2011;8(1):59–71. doi:10.1016/j.stem.2010.11.028
- Kundu N, Zhang S, Fulton AM. Sublethal oxidative stress inhibits tumor cell adhesion and enhances experimental metastasis of murine mammary carcinoma. Clin Exp Metastasis. 1995;13(1):16–22. doi:10.1007/BF00144014
- Coyle CH, Martinez LJ, Coleman MC, et al. Mechanisms of H2O2-induced oxidative stress in endothelial cells. Free Radic Biol Med. 2006;40(12):2206–2213. doi:10.1016/j.freeradbiomed.2006.02.017
- Zhang X, Liang S, Gao X, et al. Protective effect of chitosan oligosaccharide against hydrogen peroxide-mediated oxidative damage and cell apoptosis via activating Nrf2/ARE signaling pathway. Neurotox Res. 2021;39(6):1708–1720. doi:10.1007/s12640-021-00419-w
- Deshpande NN, Sorescu D, Seshiah P, et al. Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid Redox Signal. 2002;4(5):845–854. doi:10.1089/152308602760599007
- Reynaert NL, van der Vliet A, Guala AS, et al. Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase β. Proc Natl Acad Sci. 2006;103(35):13086–13091. doi:10.1073/pnas.0603290103
- Guo S, Fei HD, Chen JF, et al. Activation of Nrf2 by MIND4-17 protects osteoblasts from hydrogen peroxide-induced oxidative stress. Oncotarget. 2017;8(62):105662–105672. doi:10.18632/oncotarget.22360
- Wang GF, Dong Q, Bai Y, et al. Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free Radic Biol Med. 2017;103:177–187. doi:10.1016/j.freeradbiomed.2016.12.031
- Cosar MY, Erdogan MA, Yilmaz O. Epigallocatechin-3-gallate and resveratrol attenuate hydrogen peroxide induced damage in neuronal cells. Bratisl Lek Listy. 2023;124(3):205–211. doi:10.4149/BLL_2023_033
- Byrne DP, Shrestha S, Galler M, et al. Aurora A regulation by reversible cysteine oxidation reveals evolutionarily conserved redox control of Ser/Thr protein kinase activity. Sci Signal. 2020;13(639):eaax2713. doi:10.1126/scisignal.aax2713
- Pyo CW, Choi JH, Oh SM, et al. Oxidative stress-induced cyclin D1 depletion and its role in cell cycle processing. Biochim Biophys Acta BBA – Gen Subj. 2013;1830(11):5316–5325. doi:10.1016/j.bbagen.2013.07.030
- Saijo H, Hirohashi Y, Torigoe T, et al. Plasticity of lung cancer stem-like cells is regulated by the transcription factor HOXA5 that is induced by oxidative stress. Oncotarget. 2016;7(31):50043–50056. doi:10.18632/oncotarget.10571
- Giannoni E, Taddei ML, Chiarugi P. Src redox regulation: again in the front line. Free Radic Biol Med. 2010;49(4):516–527. doi:10.1016/j.freeradbiomed.2010.04.025
- Loschen G, Azzi A, Richter C, et al. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974;42(1):68–72. doi:10.1016/0014-5793(74)80281-4
- Radzinski M, Oppenheim T, Metanis N, et al. The cys sense: thiol redox switches mediate life cycles of cellular proteins. Biomolecules. 2021;11(3):469. doi:10.3390/biom11030469
- Perillo B, Di Donato M, Pezone A, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52(2):192–203. doi:10.1038/s12276-020-0384-2
- Denicourt C, Dowdy SF. Cip/Kip proteins: more than just CDKs inhibitors. Genes Dev. 2004;18(8):851–855. doi:10.1101/gad.1205304
- Matthews HK, Bertoli C, de Bruin RAM. Cell cycle control in cancer. Nat Rev Mol Cell Biol. 2022;23(1):74–88. doi:10.1038/s41580-021-00404-3
- Cumming RC, Andon NL, Haynes PA, et al. Protein disulfide bond formation in the cytoplasm during oxidative stress*. J Biol Chem. 2004;279(21):21749–21758. doi:10.1074/jbc.M312267200
- Chiu J, Dawes IW. Redox control of cell proliferation. Trends Cell Biol. 2012;22(11):592–601. doi:10.1016/j.tcb.2012.08.002
- Lukosz M, Jakob S, Büchner N, et al. Nuclear redox signaling. Antioxid Redox Signal. 2010;12(6):713–742. doi:10.1089/ars.2009.2609
- Webster KA, Prentice H, Bishopric NH. Oxidation of zinc finger transcription factors: physiological consequences. Antioxid Redox Signal. 2001;3(4):535–548. doi:10.1089/15230860152542916
- Whittal RM, Benz CC, Scott G, et al. Preferential oxidation of zinc finger 2 in estrogen receptor DNA-binding domain prevents dimerization and, hence, DNA binding. Biochemistry. 2000;39(29):8406–8417. doi:10.1021/bi000282f
- Houée-Lévin C, Bobrowski K, Horakova L, et al. Exploring oxidative modifications of tyrosine: an update on mechanisms of formation, advances in analysis and biological consequences. Free Radic Res. 2015;49(4):347–373. doi:10.3109/10715762.2015.1007968
- Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45(5):549–561. doi:10.1016/j.freeradbiomed.2008.05.004
- Poole LB. The basics of thiols and cysteines in redox biology and chemistry. Free Radic Biol Med. 2015;80:148–157. doi:10.1016/j.freeradbiomed.2014.11.013
- Marino SM, Gladyshev VN. Analysis and functional prediction of reactive cysteine residues. J Biol Chem. 2012;287(7):4419–4425. doi:10.1074/jbc.R111.275578
- Reuter S, Gupta SC, Chaturvedi MM, et al. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49(11):1603–1616. doi:10.1016/j.freeradbiomed.2010.09.006
- Liu S, Li B, Xu J, et al. SOD1 promotes cell proliferation and metastasis in non-small cell lung cancer via an miR-409-3p/SOD1/SETDB1 epigenetic regulatory feedforward loop. Front Cell Dev Biol. 2020:8 [cited 2023 Dec 28]. Available from: https://www.frontiersin.org/articles/10.3389fcell.2020.00213
- Li S, Fu L, Tian T, et al. Disrupting SOD1 activity inhibits cell growth and enhances lipid accumulation in nasopharyngeal carcinoma. Cell Commun Signal. 2018;16(1):28. doi:10.1186/s12964-018-0240-3
- Chiang SK, Chen SE, Chang LC. The role of HO-1 and its crosstalk with oxidative stress in cancer cell survival. Cells. 2021;10(9):2401. doi:10.3390/cells10092401
- Balan M, Chakraborty S, Flynn E, et al. Honokiol inhibits c-Met-HO-1 tumor-promoting pathway and its cross-talk with calcineurin inhibitor-mediated renal cancer growth. Sci Rep. 2017;7(1):5900. doi:10.1038/s41598-017-05455-1
- Lien GS, Wu MS, Bien MY, et al. Epidermal growth factor stimulates nuclear factor-κB activation and heme oxygenase-1 expression via c-Src, NADPH oxidase, PI3 K, and Akt in human colon cancer cells. PLoS One. 2014;9(8):e104891. doi:10.1371/journal.pone.0104891
- Mayerhofer M, Florian S, Krauth MT, et al. Identification of heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in chronic myeloid leukemia. Cancer Res. 2004;64(9):3148–3154. doi:10.1158/0008-5472.CAN-03-1200
- Stancill JS, Corbett JA. The role of thioredoxin/peroxiredoxin in the β-cell defense against oxidative damage. Front Endocrinol. 2021:12 [cited 2024 Jan 9]. Available from: https://www.frontiersin.org/articles/10.3389fendo.2021.718235
- Hönigova K, Navratil J, Peltanova B, et al. Metabolic tricks of cancer cells. Biochim Biophys Acta BBA – Rev Cancer. 2022;1877(3):188705. doi:10.1016/j.bbcan.2022.188705
- Xiao GG, Wang M, Li N, et al. Use of proteomics to demonstrate a hierarchical oxidative stress response to diesel exhaust particle chemicals in a macrophage cell line. J Biol Chem. 2003;278(50):50781–50790. doi:10.1074/jbc.M306423200
- Surh YJ, Kundu JK, Na HK, et al. Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J Nutr. 2005;135(12 Suppl):2993S–3001S. doi:10.1093/jn/135.12.2993S
- Fourquet S, Guerois R, Biard D, et al. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J Biol Chem. 2010;285(11):8463–8471. doi:10.1074/jbc.M109.051714
- He F, Ru X, Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci. 2020;21(13):4777. doi:10.3390/ijms21134777
- Lingappan K. NF-κB in oxidative stress. Curr Opin Toxicol. 2018;7:81–86. doi:10.1016/j.cotox.2017.11.002
- Pineda-Molina E, Klatt P, Vázquez J, et al. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40(47):14134–14142. doi:10.1021/bi011459o
- Hirota K, Murata M, Sachi Y, et al. Distinct roles of thioredoxin in the cytoplasm and in the nucleus: a two-step mechanism of redox regulation of transcription factor NF-κB *. J Biol Chem. 1999;274(39):27891–27897. doi:10.1074/jbc.274.39.27891
- Anrather J, Racchumi G, Iadecola C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281(9):5657–5667. doi:10.1074/jbc.M506172200
- Djavaheri-Mergny M, Javelaud D, Wietzerbin J, et al. NF-kappaB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFalpha-treated Ewing sarcoma cells. FEBS Lett. 2004;578(1-2):111–115. doi:10.1016/j.febslet.2004.10.082
- Pan H, Wang H, Wang X, et al. The absence of Nrf2 enhances NF-κB-dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediators Inflamm. 2012;2012:217580. doi:10.1155/2012/217580
- Seldon MP, Silva G, Pejanovic N, et al. Heme oxygenase-1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of NF-kappaB RelA phosphorylation at serine 276. J Immunol Baltim Md 1950. 2007;179(11):7840–7851. doi:10.4049/jimmunol.179.11.7840
- Liu GH, Qu J, Shen X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta. 2008;1783(5):713–727. doi:10.1016/j.bbamcr.2008.01.002
- Liu X, Quan J, Shen Z, et al. Metallothionein 2A (MT2A) controls cell proliferation and liver metastasis by controlling the MST1/LATS2/YAP1 signaling pathway in colorectal cancer. Cancer Cell Int. 2022;22(1):205. doi:10.1186/s12935-022-02623-w
- Jiang LJ, Maret W, Vallee BL. The glutathione redox couple modulates zinc transfer from metallothionein to zinc-depleted sorbitol dehydrogenase. Proc Natl Acad Sci U S A. 1998;95(7):3483–3488. doi:10.1073/pnas.95.7.3483
- Méplan C, Richard MJ, Hainaut P. Metalloregulation of the tumor suppressor protein p53: zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells. Oncogene. 2000;19(46):5227–5236. doi:10.1038/sj.onc.1203907
- Méplan C, Verhaegh G, Richard MJ, et al. Metal ions as regulators of the conformation and function of the tumour suppressor protein p53: implications for carcinogenesis. Proc Nutr Soc. 1999;58(3):565–571. doi:10.1017/S0029665199000749
- Ruttkay-Nedecky B, Nejdl L, Gumulec J, et al. The role of metallothionein in oxidative stress. Int J Mol Sci. 2013;14(3):6044–6066. doi:10.3390/ijms14036044
- Arisumi S, Fujiwara T, Yasumoto K, et al. Metallothionein 3 promotes osteoclast differentiation and survival by regulating the intracellular Zn2 + concentration and NRF2 pathway. Cell Death Discov. 2023;9(1):1–13. doi:10.1038/s41420-023-01729-y
- Nagel WW, Vallee BL. Cell cycle regulation of metallothionein in human colonic cancer cells. Proc Natl Acad Sci USA. 1995;92(2):579–583. doi:10.1073/pnas.92.2.579
- Kondo Y, Rusnak JM, Hoyt DG, et al. Enhanced apoptosis in metallothionein null cells. Mol Pharmacol. 1997;52(2):195–201. doi:10.1124/mol.52.2.195
- Burhans WC, Heintz NH. The cell cycle is a redox cycle: linking phase-specific targets to cell fate. Free Radic Biol Med. 2009;47(9):1282–1293. doi:10.1016/j.freeradbiomed.2009.05.026
- Hoffman A, Spetner LM, Burke M. Ramifications of a redox switch within a normal cell: its absence in a cancer cell. Free Radic Biol Med. 2008;45(3):265–268. doi:10.1016/j.freeradbiomed.2008.03.025
- Menon SG, Sarsour EH, Spitz DR, et al. Redox regulation of the G1 to S phase transition in the mouse embryo fibroblast cell Cycle1. Cancer Res. 2003;63(9):2109–2117.
- Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780(11):1273–1290. doi:10.1016/j.bbagen.2008.01.011
- Gough DR, Cotter TG. Hydrogen peroxide: a jekyll and hyde signalling molecule. Cell Death Dis. 2011;2(10):e213–e213. doi:10.1038/cddis.2011.96
- Murray TVA, Smyrnias I, Schnelle M, et al. Redox regulation of cardiomyocyte cell cycling via an ERK1/2 and c-Myc-dependent activation of cyclin D2 transcription. J Mol Cell Cardiol. 2015;79:54–68. doi:10.1016/j.yjmcc.2014.10.017
- Lee SR, Kwon KS, Kim SR, et al. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273(25):15366–15372. doi:10.1074/jbc.273.25.15366
- Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018;80:50–64. doi:10.1016/j.semcdb.2017.05.023
- Östman A, Frijhoff J, Sandin Å, et al. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem (Tokyo). 2011;150(4):345–356. doi:10.1093/jb/mvr104
- Raman D, Pervaiz S. Redox inhibition of protein phosphatase PP2A: potential implications in oncogenesis and its progression. Redox Biol. 2019;27:101105. doi:10.1016/j.redox.2019.101105
- Kwon J, Lee SR, Yang KS, et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101(47):16419–16424. doi:10.1073/pnas.0407396101
- Mahadev K, Zilbering A, Zhu L, et al. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem. 2001;276(24):21938–21942. doi:10.1074/jbc.C100109200
- Humphries KM, Juliano C, Taylor SS. Regulation of cAMP-dependent protein kinase activity by glutathionylation. J Biol Chem. 2002;277(45):43505–43511. doi:10.1074/jbc.M207088200
- Kemble DJ, Sun G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc Natl Acad Sci U S A. 2009;106(13):5070–5075. doi:10.1073/pnas.0806117106
- Arciuch VGA, Galli S, Franco MC, et al. Akt1 intramitochondrial cycling is a crucial step in the redox modulation of cell cycle progression. PLoS One. 2009;4(10):e7523. doi:10.1371/journal.pone.0007523
- Keyes JD, Parsonage D, Yammani RD, et al. Endogenous, regulatory cysteine sulfenylation of ERK kinases in response to proliferative signals. Free Radic Biol Med. 2017;112:534–543. doi:10.1016/j.freeradbiomed.2017.08.018
- Nadeau PJ, Charette SJ, Toledano MB, et al. Disulfide bond-mediated multimerization of Ask1 and its reduction by thioredoxin-1 regulate H2O2-induced c-Jun NH2-terminal kinase activation and apoptosis. Mol Biol Cell. 2007;18(10):3903–3913. doi:10.1091/mbc.e07-05-0491
- Shi T, Dansen TB. Reactive oxygen species induced p53 activation: DNA damage, redox signaling, or both? Antioxid Redox Signal. 2020;33(12):839–859. doi:10.1089/ars.2020.8074
- Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17(9):2596–2606. doi:10.1093/emboj/17.9.2596
- Cross JV, Templeton DJ. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem J. 2004;381(Pt 3):675–683. doi:10.1042/BJ20040591
- Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26(7):1749–1760. doi:10.1038/sj.emboj.7601623
- Mathiassen SG, De Zio D, Cecconi F. Autophagy and the cell cycle: a complex landscape. Front Oncol. 2017:7. doi:10.3389/fonc.2017.00051
- Knight H, Abis G, Kaur M, et al. Cyclin D-CDK4 disulfide bond attenuates pulmonary vascular cell proliferation. Circ Res. 2023;133(12):966–988. doi:10.1161/CIRCRESAHA.122.321836
- Pawlonka J, Rak B, Ambroziak U. The regulation of cyclin D promoters – review. Cancer Treat Res Commun. 2021;27:100338. doi:10.1016/j.ctarc.2021.100338
- Jiang X, Xie H, Dou Y, et al. Expression and function of FRA1 protein in tumors. Mol Biol Rep. 2020;47(1):737–752. doi:10.1007/s11033-019-05123-9
- Guo ZY, Hao X-h, Tan FF, et al. The elements of human cyclin D1 promoter and regulation involved. Clin Epigenetics. 2011;2(2):63–76. doi:10.1007/s13148-010-0018-y
- Burch PM, Yuan Z, Loonen A, et al. An extracellular signal-regulated kinase 1- and 2-dependent program of chromatin trafficking of c-Fos and Fra-1 Is required for cyclin D1 expression during cell cycle reentry. Mol Cell Biol. 2004;24(11):4696–4709. doi:10.1128/MCB.24.11.4696-4709.2004
- Phalen TJ, Weirather K, Deming PB, et al. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J Cell Biol. 2006;175(5):779–789. doi:10.1083/jcb.200606005
- Menon SG, Sarsour EH, Kalen AL, et al. Superoxide signaling mediates N-acetyl-L-cysteine-induced G1 arrest: regulatory role of cyclin D1 and manganese superoxide dismutase. Cancer Res. 2007;67(13):6392–6399. doi:10.1158/0008-5472.CAN-07-0225
- Li L, Cheung S-h, Evans EL, et al. Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Res. 2010;70(20):8222–8232. doi:10.1158/0008-5472.CAN-10-0894
- Grillo M, Palmer C, Holmes N, et al. Stat3 oxidation-dependent regulation of gene expression impacts on developmental processes and involves cooperation with Hif-1α. PLoS One. 2020;15(12):e0244255. doi:10.1371/journal.pone.0244255
- Xie Y, Kole S, Precht P, et al. S-Glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology. 2009;150(3):1122–1131. doi:10.1210/en.2008-1241
- Cicchillitti L, Fasanaro P, Biglioli P, et al. Oxidative stress induces protein phosphatase 2A-dependent dephosphorylation of the pocket proteins pRb, p107, and p130*. J Biol Chem. 2003;278(21):19509–19517. doi:10.1074/jbc.M300511200
- Kirova DG, Judasova K, Vorhauser J, et al. A ROS-dependent mechanism to drive progression through S phase. Published online 2022 Mar 31. doi:10.1101/2022.03.31.486607
- Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, et al. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi:10.1146/annurev.biochem.73.011303.073723
- He L, Nan MH, Oh HC, et al. Asperlin induces G2/M arrest through ROS generation and ATM pathway in human cervical carcinoma cells. Biochem Biophys Res Commun. 2011;409(3):489–493. doi:10.1016/j.bbrc.2011.05.032
- Guo Z, Kozlov S, Lavin MF, et al. ATM activation by oxidative stress. Science. 2010;330(6003):517–521. doi:10.1126/science.1192912
- Bhakat KK, Mantha AK, Mitra S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid Redox Signal. 2009;11(3):621. doi:10.1089/ars.2008.2198
- Howpay Manage SA, Fleming AM, Chen HN, et al. Cysteine oxidation to sulfenic acid in APE1 aids G-quadruplex binding while compromising DNA repair. ACS Chem Biol. 2022;17(9):2583–2594. doi:10.1021/acschembio.2c00511
- Xanthoudakis S, Miao G, Wang F, et al. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11(9):3323–3335. doi:10.1002/j.1460-2075.1992.tb05411.x
- Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11(2):653–665. doi:10.1002/j.1460-2075.1992.tb05097.x
- Fritz G, Grösch S, Tomicic M, et al. APE/Ref-1 and the mammalian response to genotoxic stress. Toxicology. 2003;193(1):67–78. doi:10.1016/S0300-483X(03)00290-7
- Seo YR, Kelley MR, Smith ML. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc Natl Acad Sci U S A. 2002;99(22):14548–14553. doi:10.1073/pnas.212319799
- Walker LJ, Robson CN, Black E, et al. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol Cell Biol. 1993;13(9):5370–5376. doi:10.1128/mcb.13.9.5370-5376.1993
- Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6(3):a026104. doi:10.1101/cshperspect.a026104
- Tan M, Li S, Swaroop M, et al. Transcriptional activation of the human glutathione peroxidase promoter by p53. J Biol Chem. 1999;274(17):12061–12066. doi:10.1074/jbc.274.17.12061
- Sablina AA, Budanov AV, Ilyinskaya GV, et al. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11(12):1306–1313. doi:10.1038/nm1320
- Hussain SP, Amstad P, He P, et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004;64(7):2350–2356. doi:10.1158/0008-5472.CAN-2287-2
- Drane P, Bravard A, Bouvard V, et al. Reciprocal down-regulation of p53 and SOD2 gene expression-implication in p53 mediated apoptosis. Oncogene. 2001;20(4):430–439. doi:10.1038/sj.onc.1204101
- Faraonio R, Vergara P, Di Marzo D, et al. P53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J Biol Chem. 2006;281(52):39776–39784. doi:10.1074/jbc.M605707200
- Liu D, Xu Y. P53, oxidative stress, and aging. Antioxid Redox Signal. 2011;15(6):1669–1678. doi:10.1089/ars.2010.3644
- Scotcher J, Clarke DJ, Mackay CL, et al. Redox regulation of tumour suppressor protein p53: identification of the sites of hydrogen peroxide oxidation and glutathionylation. Chem Sci. 2013;4(3):1257–1269. doi:10.1039/c2sc21702c
- Buzek J, Latonen L, Kurki S, et al. Redox state of tumor suppressor p53 regulates its sequence-specific DNA binding in DNA-damaged cells by cysteine 277. Nucleic Acids Res. 2002;30(11):2340–2348. doi:10.1093/nar/30.11.2340
- Oakes V, Wang W, Harrington B, et al. Cyclin A/Cdk2 regulates Cdh1 and claspin during late S/G2 phase of the cell cycle. Cell Cycle. 2014;13(20):3302–3311. doi:10.4161/15384101.2014.949111
- Shen T, Huang S. The role of Cdc25A in the regulation of cell proliferation and apoptosis. Anticancer Agents Med Chem. 2012;12(6):631–639. doi:10.2174/187152012800617678
- Sur S, Agrawal DK. Phosphatases and kinases regulating CDC25 activity in the cell cycle: clinical implications of CDC25 overexpression and potential treatment strategies. Mol Cell Biochem. 2016;416(1-2):33–46. doi:10.1007/s11010-016-2693-2
- Dutertre S, Cazales M, Quaranta M, et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J Cell Sci. 2004;117(Pt 12):2523–2531. doi:10.1242/jcs.01108
- Savitsky PA, Finkel T. Redox regulation of CDC25C. J Biol Chem. 2002;277(23):20535–20540. doi:10.1074/jbc.M201589200
- Sohn J, Rudolph J. Catalytic and chemical competence of regulation of CDC25 phosphatase by oxidation/reduction. Biochemistry. 2003;42(34):10060–10070. doi:10.1021/bi0345081
- Heo S, Kim S, Kang D. The role of hydrogen peroxide and peroxiredoxins throughout the cell cycle. Antioxidants. 2020;9(4):280. doi:10.3390/antiox9040280
- Willems E, Dedobbeleer M, Digregorio M, et al. The functional diversity of aurora kinases: a comprehensive review. Cell Div. 2018;13(1):7. doi:10.1186/s13008-018-0040-6
- Katayama H, Zhou H, Li Q, et al. Interaction and feedback regulation between STK15/BTAK/Aurora-A kinase and protein phosphatase 1 through mitotic cell division cycle*. J Biol Chem. 2001;276(49):46219–46224. doi:10.1074/jbc.M107540200
- Zhao Zs, Lim JP, Ng YW, et al. The GIT-associated kinase PAK targets to the centrosome and regulates Aurora-A. Mol Cell. 2005;20(2):237–249. doi:10.1016/j.molcel.2005.08.035
- Zeng K, Bastos RN, Barr FA, et al. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J Cell Biol. 2010;191(7):1315–1332. doi:10.1083/jcb.201008106
- Lim JM, Lee KS, Woo HA, et al. Control of the pericentrosomal H2O2 level by peroxiredoxin I is critical for mitotic progression. J Cell Biol. 2015;210(1):23–33. doi:10.1083/jcb.201412068
- Van Horn RD, Chu S, Fan L, et al. Cdk1 activity is required for mitotic activation of Aurora A during G2/M transition of human cells. J Biol Chem. 2010;285(28):21849–21857. doi:10.1074/jbc.M110.141010
- Lane N. A unifying view of ageing and disease: the double-agent theory. J Theor Biol. 2003;225(4):531–540. doi:10.1016/S0022-5193(03)00304-7
- Forman HJ, Zhang H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov. 2021;20(9):689–709. doi:10.1038/s41573-021-00233-1
- Chatterjee A, Zhu Y, Tong Q, et al. The addition of manganese porphyrins during radiation inhibits prostate cancer growth and simultaneously protects normal prostate tissue from radiation damage. Antioxid Basel Switz. 2018;7(1):21. doi:10.3390/antiox7010021
- Anderson CM, Lee CM, Saunders DP, et al. Phase IIb, randomized, double-blind trial of GC4419 versus placebo to reduce severe oral mucositis due to concurrent radiotherapy and cisplatin For head and neck cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2019;37(34):3256–3265. doi:10.1200/JCO.19.01507
- Chidambaram SB, Anand N, Varma SR, et al. Superoxide dismutase and neurological disorders. IBRO Neurosci Rep. 2024;16:373–394. doi:10.1016/j.ibneur.2023.11.007
- Cheng B, Zhong JP, Wu FX, et al. Ebselen protects rat hearts against myocardial ischemia-reperfusion injury. Exp Ther Med. 2019;17(2):1412–1419. doi:10.3892/etm.2018.7089
- Yamaguchi T, Sano K, Takakura K, et al. Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial. Ebselen Study Group. Stroke. 1998;29(1):12–17. doi:10.1161/01.str.29.1.12
- Landgraf AD, Alsegiani AS, Alaqel S, et al. Neuroprotective and anti-neuroinflammatory properties of ebselen derivatives and their potential to inhibit neurodegeneration. ACS Chem Neurosci. 2020;11(19):3008–3016. doi:10.1021/acschemneuro.0c00328
- Elbatreek MH, Mucke H, Schmidt HHHW. NOX inhibitors: from bench to naxibs to bedside. Handb Exp Pharmacol. 2021;264:145–168. doi:10.1007/164_2020_387
- Szekeres FLM, Walum E, Wikström P, et al. A small molecule inhibitor of NOX2 and NOX4 improves contractile function after ischemia–reperfusion in the mouse heart. Sci Rep. 2021;11(1):11970. doi:10.1038/s41598-021-91575-8
- Kim SJ, Kim HS, Seo YR. Understanding of ROS-inducing strategy in anticancer therapy. Oxid Med Cell Longev. 2019;2019:e5381692. doi:10.1155/2019/5381692
- Yang C, Song J, Hwang S, et al. Apigenin enhances apoptosis induction by 5-fluorouracil through regulation of thymidylate synthase in colorectal cancer cells. Redox Biol. 2021;47:102144. doi:10.1016/j.redox.2021.102144
- Xu D, Rovira II, Finkel T. Oxidants painting the cysteine chapel: redox regulation of PTPs. Dev Cell. 2002;2(3):251–252. doi:10.1016/S1534-5807(02)00132-6
- El-Senduny FF, Badria FA, El-Waseef AM, et al. Approach for chemosensitization of cisplatin-resistant ovarian cancer by cucurbitacin B. Tumour Biol J Int Soc Oncodevelopmental Biol Med. 2016;37(1):685–698. doi:10.1007/s13277-015-3773-8
- Zhang X, Huang J, Yu C, et al.Quercetin enhanced paclitaxel therapeutic effects towards PC-3 prostate cancer through ER stress induction and ROS production. OncoTargets Ther. 2020;13:513–523. doi:10.2147/OTT.S228453
- Aghababaei F, Hadidi M. Recent advances in potential health benefits of quercetin. Pharmaceuticals. 2023;16(7):1020. doi:10.3390/ph16071020
- Vieira-Frez FC, Sehaber-Sierakowski CC, Perles JVCM, et al. Anti- and pro-oxidant effects of quercetin stabilized by microencapsulation on interstitial cells of Cajal, nitrergic neurons and M2-like macrophages in the jejunum of diabetic rats. Neurotoxicology. 2020;77:193–204. doi:10.1016/j.neuro.2020.01.011
- Gomes CL, de Albuquerque Wanderley Sales V, Gomes de Melo C, et al. Beta-lapachone: natural occurrence, physicochemical properties, biological activities, toxicity and synthesis. Phytochemistry. 2021;186:112713. doi:10.1016/j.phytochem.2021.112713
- Zheng P, Zhou C, Lu L, et al. Elesclomol: a copper ionophore targeting mitochondrial metabolism for cancer therapy. J Exp Clin Cancer Res CR. 2022;41(1):271. doi:10.1186/s13046-022-02485-0
- Monk BJ, Kauderer JT, Moxley KM, et al. A phase II evaluation of elesclomol sodium and weekly paclitaxel in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube or primary peritoneal cancer: an NRG oncology/gynecologic oncology group study. Gynecol Oncol. 2018;151(3):422–427. doi:10.1016/j.ygyno.2018.10.001
- Gao J, Wu X, Huang S, et al. Novel insights into anticancer mechanisms of elesclomol: more than a prooxidant drug. Redox Biol. 2023;67:102891. doi:10.1016/j.redox.2023.102891
- O’Day SJ, Eggermont AMM, Chiarion-Sileni V, et al. Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31(9):1211–1218. doi:10.1200/JCO.2012.44.5585
- Kaźmierczak-Barańska J, Boguszewska K, Adamus-Grabicka A, et al. Two faces of vitamin C—antioxidative and pro-oxidative agent. Nutrients. 2020;12(5):1501. doi:10.3390/nu12051501
- Ma Y, Chapman J, Levine M, et al. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med. 2014;6(222):222ra18. doi:10.1126/scitranslmed.3007154
- Schoenfeld JD, Sibenaller ZA, Mapuskar KA, et al. O2⋅− and H2O2-mediated disruption of Fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell. 2017;31(4):487–500.e8. doi:10.1016/j.ccell.2017.02.018
- Springett GM, Husain K, Neuger A, et al. A phase I safety, pharmacokinetic, and pharmacodynamic presurgical trial of vitamin E δ-tocotrienol in patients with pancreatic ductal neoplasia. EBioMedicine. 2015;2(12):1987–1995. doi:10.1016/j.ebiom.2015.11.025
- Gharaei R, Alyasin A, Mahdavinezhad F, et al. Randomized controlled trial of astaxanthin impacts on antioxidant status and assisted reproductive technology outcomes in women with polycystic ovarian syndrome. J Assist Reprod Genet. 2022;39(4):995–1008. doi:10.1007/s10815-022-02432-0
- Abe SK, Inoue M. Green tea and cancer and cardiometabolic diseases: a review of the current epidemiological evidence. Eur J Clin Nutr. 2021;75:865–876. doi:10.1038/s41430-020-00710-7
- Gao N, Ni M, Song J, et al. Causal relationship between tea intake and cardiovascular diseases: A Mendelian randomization study. Front Nutr. 2022:9. doi:10.3389/fnut.2022.938201
- Martin BJ, Tan RB, Gillen JB, et al. No effect of short-term green tea extract supplementation on metabolism at rest or during exercise in the fed state. Int J Sport Nutr Exerc Metab. 2014;24(6):656–664. doi:10.1123/ijsnem.2013-0202
- Garcia FAR, Cornelison T, Nuño T, Greenspan DL, Byron JW, Hsu C-H, Alberts DS, Chow H-HS. Results of a phase II randomized, double-blind, placebo-controlled trial of Polyphenon E in women with persistent high-risk HPV infection and low-grade cervical intraepithelial neoplasia. Gynecol. Oncol. 2014;132:377–382. doi:10.1016/j.ygyno.2013.12.034
- Seely D, Legacy M, Auer RC, et al. Adjuvant melatonin for the prevention of recurrence and mortality following lung cancer resection (AMPLCaRe): a randomized placebo controlled clinical trial. EClinicalMedicine. 2021;33:100763. doi:10.1016/j.eclinm.2021.100763
- Kah G, Chandran R, Abrahamse H. Curcumin a natural phenol and its therapeutic role in cancer and photodynamic therapy: a review. Pharmaceutics. 2023;15(2):639. doi:10.3390/pharmaceutics15020639
- Ryan JL, Heckler CE, Ling M, et al. Curcumin for radiation dermatitis: a randomized, double-blind, placebo-controlled clinical trial of thirty breast cancer patients. Radiat Res. 2013;180(1):34–43. doi:10.1667/RR3255.1
- Hegde M, Girisa S, BharathwajChetty B, et al. Curcumin formulations for better bioavailability: what we learned from clinical trials thus far? ACS Omega. 2023;8(12):10713–10746. doi:10.1021/acsomega.2c07326
- Gunther JR, Chadha AS, Guha S, et al. A phase II randomized double blinded trial evaluating the efficacy of curcumin with pre-operative chemoradiation for rectal cancer. J Gastrointest Oncol. 2022;13(6):2938–2950. doi:10.21037/jgo-22-259
- Passildas-Jahanmohan J, Eymard J, Pouget M, et al. Multicenter randomized phase II study comparing docetaxel plus curcumin versus docetaxel plus placebo in first-line treatment of metastatic castration-resistant prostate cancer. Cancer Med. 2021;10(7):2332–2340. doi:10.1002/cam4.3806