
ABSTRACT
Introduction: The threat of chemical, biological, radiological, and nuclear (CBRN) warfare has been addressed as the uppermost risk to national security since the terrorist attacks on 11 September 2001. Despite significant scientific advances over the past several decades toward the development of safe, non-toxic and effective countermeasures to combat CBRN threats, relatively few countermeasures have been approved by the US Food and Drug Administration (US FDA). Therefore, countermeasures capable of protecting the population from the effects of CBRN attack remain a significant unmet medical need. Chemical and biological (CB) threat agents can be particularly hazardous due to their effectiveness in small quantities and ease of distribution.
Area covered: This article reviews the development of countermeasures for CB threats and highlights specific threats for which at least one countermeasure has been approved following the FDA Animal Rule. Patents of CB countermeasures since 2010 have been included.
Expert opinion: Nine CB countermeasures have received FDA approval for use in humans following the Animal Rule, and a number of promising CB countermeasures are currently under development. In the next few years, we should expect to have multiple countermeasures approved by the FDA for each indication allowing for more flexible and effective treatment options.
1. Introduction
A weapon of mass destruction is defined as a chemical, biological, radiological, and nuclear (CBRN) or other weapon capable of generating significant harm to a large number of humans [Citation1,Citation2]. Despite considerable investments prompted by the US 9/11 terrorist attacks in 2001, much work remains to prepare nations against CBRN threats. Based on the frequency of credible reports by journalists, threats of CBRN weapons being deployed by terrorist groups or by rogue nations clearly seem to be on the rise globally [Citation3]. Unfortunately, nations are currently limited by both the number of the currently approved medical countermeasures, as well as by the appropriateness of defense systems needed to guard against potential CBRN exposures. These medical countermeasures include not only drugs and vaccines, but also medical devices (including diagnostic tests, equipment, and supplies) crucial in responding to public health emergencies [Citation4]. Challenges of developing these medical countermeasures include the requirement of extensive government resources, the collaboration amongst public and private sector institutions, as well as the navigation and the compliance with the Animal Rule.
The development and regulatory approval of medical countermeasures for CBRN events remain a high priority of the US Department of Defense (DoD) and the Department of Health and Human Services (DHHS) which includes the FDA, the principal regulatory agency in this undertaking. The FDA is currently using a three-pillar approach to address key challenges of the regulatory review process of medical countermeasures; these involve but are not limited to gaps in regulatory science for medial countermeasure development and evaluation, as well as the legal regulatory and policy framework for an effective public health response [Citation5]. The three-pillar approach launched in 2010 with the Medical Countermeasure Initiative consists of (1) improving the medical countermeasure regulatory review process; (2) moving regulatory science for medical countermeasure development and evaluation forward; and (3) updating the legal, regulatory, and policy framework for a successful public health response. The aim behind such an approach was to improve capability to respond faster and more effectively to CBRN and emerging infectious disease threats. Filling in the gaps of the medical countermeasure enterprise is a major national undertaking and as such requires the collaborative cooperation of all stakeholders, including federal, corporate, and research organizations [Citation6,Citation7].
The FDA’s Animal Rule only permits the approval of drugs that have proven efficacy to counter CBRN health effects in animal models, proper safety profiles, and sufficient evidence that these drugs will yield a reasonable health benefit in humans [Citation8–Citation10]. Furthermore, there needs to be an understanding of the mechanisms of injury, drug efficacy, and the utilization of efficacy biomarkers [Citation11]. The Animal Rule can be applied to cases of infections or exposure to select types of agents with low natural incidences or exposure frequencies (anthrax or mustard gases, respectively) but would be unethical to expose human subjects to for the sake of efficacy testing. It is highly likely that additional new countermeasures will be approved by a combination of the Animal Rule and traditional approval processes. The Project BioShield Act specifically encourages and appropriates funding for the development of countermeasures against agents that may be used in terrorist attacks [Citation12]; it also created the Strategic National Stockpile (SNS) to store considerable quantities of drugs and medical supplies to protect the US population in case of a public health crisis [Citation13,Citation14]. The 2006 US Pandemic and All-Hazards Preparedness Act built upon the 2004 Project BioShield Act and offered the DHHS additional authority to fund the development and procurement of medical countermeasures against CBRN threats [Citation15].
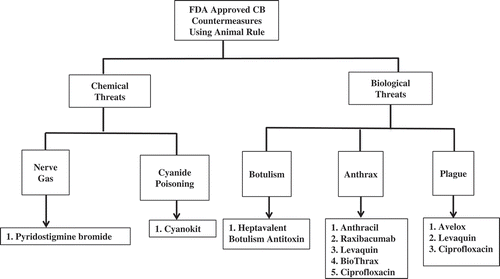
This is the first part of a two article series; here, we communicate countermeasures for CB threats, while in the second article, we discuss countermeasures for RN threats. Nine medical countermeasures have been approved by the FDA following the Animal Rule for various CB indications ( and ). In this report, we have highlighted only those CB threats/agents and recent patents for which at least one countermeasure has been approved by the FDA under the agency’s Animal Rule. These CB threats/agents include organophosphorus nerve agents, cyanide, mustard gas, bacteria-induced botulism, anthrax, and plague [Citation8,Citation9]. We have reviewed patents for CB countermeasures from 2010 to 2015. All of these patents are summarized in –.
Table 1. Patents of countermeasures for several indications.
Table 2. Patents of countermeasures for nerve gas.
Table 3. Patents of countermeasures for botulism.
Table 4. Patents of countermeasures for anthrax.
Table 5. Patents for plague countermeasures (protectors, mitigators, and therapeutics/treatments).
Figure 1. Possible CBRN threats. Several CB threats have been identified for which there is need to develop medical countermeasures.
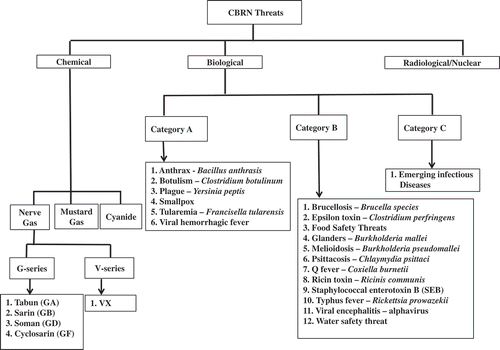
Figure 2. FDA-approved countermeasures for CB threats following the Animal Rule. In 2002, the FDA issued the Animal Rule to expedite the development of medical countermeasures against CBRN threats. In the past fourteen years, nine countermeasures have been approved by FDA following Animal Rule.
2. Countermeasures for chemical weapons
Chemical weapons use the toxic properties of their components to produce external or internal injuries. Some chemically based threats arise from nerve agents and sulfur mustard, which typically are liquids at ambient temperature. Nerve agents inhibit acetylcholinesterase, an essential molecule for signal transmission of neuronal tissues. This inhibition leads to a buildup of acetylcholine at neuronal synapses, producing an overstimulation of cholinergic receptors, ultimately leading to paralysis and often death. The organophosphorus nerve agents, tabun, sarin, soman, and cyclosarin, are among the most toxic chemical warfare agents known and grouped as G-series nerve agents [Citation16]. At room temperature, G-series nerve agents are volatile liquids, making them serious exposure risks from inhalation. V-series agents, including O-ethyl-S-(2-diisopropylaminoethyl) methylphosphonothioate (also known as VX), are among the most toxic chemical warfare nerve agents. The V agents are more poisonous than sarin but have low volatility and are primarily detrimental after skin exposure [Citation17].
G-series nerve agents display high volatility, which increases with rising temperatures; release of liquid G-type nerve agents in an enclosed environment may rapidly create lethal concentrations [Citation6]. In a military or terrorist scenario, exposure to chemical G-series nerve agents will happen mainly via inhalation; nevertheless, percutaneous absorption is also feasible if there is exposure to the skin or to the eyes with the less volatile (than the G-series agents) V-series nerve agents.
2.1. Nerve gas
Nerve agents are considered to be the deadliest of the common chemical warfare agents. By binding to or inactivating acetylcholinesterase, nerve agents alter the cholinergic transmission which leads to a rapidly increasing cholinergic crisis [Citation17]. This results in an uncontrolled and overwhelming stimulation of both muscarinic and nicotinic receptors leading to a wide variety of clinical symptoms, including death resulting from respiratory failure. Antidotal therapy largely consists of atropine, pralidoxime, and benzodiazepines, all of which must be administered immediately to limit the impact of nerve agents binding to the enzyme and to avoid the worsening of symptoms [Citation18].
Patents for agents under development to treat nerve gas exposure are presented in and . These recent patents include several enzymes such as hyperthermophilic phosphotriesterases, butyrylcholinesterase, paraoxonase, and novel peptides. Pyridostigmine bromide has been approved by the FDA for pretreatment against the nerve agent soman. Apart from medical pretreatment, if cholinergic crisis is detected, pralidoxime (2-PAM) and atropine must be administered as soon as possible; the additional use of benzodiazepines (diazepam) is suggested [Citation6]. Diazepam Chempack is currently stockpiled within the SNS, but will soon be substituted by the improved formulation, Midazolam, which is an intramuscular (im) injectable that acts faster than diazepam [Citation19]. Atropine and 2-PAM are also available in SNS. Some new medical countermeasures utilize nerve agent scavengers; human butyrylcholinesterase is the principal stoichiometric bioscavenger and human paraoxonase 1 is the leading catalytic bioscavenger. The proof of concept for bioscavenger gene-delivery has already been established [Citation20].
2.1.1. Pyridostigmine bromide
Soman triggers loss of muscle control and respiratory failure, leading to death. Results from studies in nonhuman primates (NHPs) and guinea pigs have provided support that pretreatment with pyridostigmine bromide, together with atropine and pralidoxime provided after exposure to soman, boosts rates of survival [Citation21,Citation22]. NHP-provided with mestinon (pyridostigmine) syrup-impregnated diet biscuits (40 mg/animal) displayed a reproducible inhibition of 40–50% whole blood cholinesterase activity for a period of 1–6 h. Pyridostigmine pretreatment was complemented by two doses of an antidotal combination (0.05 mg/kg atropine, 2.24 mg/kg trimedoxime bromide-4, and 0.4 mg/kg benactyzine) which ensured the survival of 5 of 6 animals following 3 separate exposures to soman concentrations of 10 LD50. The protective period of oral pyridostigmine supported by antidotal combination therapy was between 30 min and 8 h prior to soman exposure. Oral pyridostigmine pretreatment in combination with atropine therapy (3 doses of 0.07 or 1.00 mg/kg, im) also protected NHPs exposed to 10 LD50 soman. Oral pyridostigmine pretreatment was not able to protect NHPs from soman exposure, without the antidotal combination or atropine therapy [Citation22]. Based on these results, the FDA approved pyridostigmine bromide on 5 February 2003 to enhance survival after exposure to soman. Pyridostigmine bromide was the first drug approved by the FDA under the Animal Rule [Citation23]. The FDA approved pyridostigmine bromide specifically to be used prior to soman exposure and in combination with atropine and pralidoxime after exposure.
2.2. Cyanide poisoning
Hydrogen cyanide is one of the two cyanide chemical warfare agents, the other is cyanogen chloride. Cyanide is a fast acting, lethal agent when used in enclosed spaces where high concentrations can be attained easily. Because of the widespread use of cyanide in US industrial processes, this agent presents a credible threat for terrorist use. Cyanide poisoning uncouples mitochondrial oxidative phosphorylation and inhibits essential and life-sustaining cellular respiration; cyanide poisoning can be fatal despite the extent of oxygen available to the body, and without treatment, exposure to a high dose of cyanide may result in death within minutes [Citation24]. There are now two types of cyanide antidotes available, Cyanide Antidote kit and Cyanokit. Cyanide Antidote kit was the first available and contains inhalant amyl nitrite and injectable sodium nitrite and sodium thiosulfate. There is another version of this kit, Nithiodote, which contains injectable sodium nitrite and sodium thiosulfate. The second antidote, cyanide countermeasure (Cyanokit) is discussed below.
2.2.1. Cyanokit
Cyanokit is a cyanide antidote that includes hydroxocobalamin, a form of vitamin B12 which was approved by FDA on 15 December 2006 following the Animal Rule [Citation25]. Hydroxocobalamin binds to cyanide, allowing critical cellular respiration to resume. The ability of hydroxocobalamin for acute cyanide poisoning was compared with that of saline vehicle in canines [Citation26]. Adult canines were injected with potassium cyanide (0.4 mg/kg/min, intravenously [iv]) and then treated after 3 min following the onset of apnea. The treatments, consisting of hydroxocobalamin (75 or 150 mg/kg, iv) or the saline vehicle (as a control),were then infused over a 7.5-min period while animals were ventilated with 100% oxygen for 5 min. Cyanide exposure reduced vital physiological functions that ended in a moribund state requiring euthanasia (within 4 h) of 59% of vehicle-treated animals; cyanide exposure also caused neurological deficits which required euthanasia in an additional 23% of vehicle-treated animals (within 2–4 days postexposure), resulting in an overall mortality rate of 82%. By contrast, those subjects administered with 75-mg/kg hydroxocobalamin had a mortality rate of 21% at 14 days postexposure, whereas those administered 150-mg/kg hydroxocobalamin had a 0% mortality (all animals survived), suggesting that hydroxocobalamin reversed cyanide toxicity and reduced mortality in the canine model [Citation26].
2.3. Other important chemical threat
Sulfur mustard agents constitute another group of chemical threats and are often called blister agents since their injuries usually look like burns or blisters. Several cellular level mechanisms of action have been suggested for sulfur mustard [Citation27,Citation28]. Current therapy for mustard gas consists of treating the inflammation and where necessary, removing the dead eschar to expedite healing. Therapy for sulfur mustard poisoning is still a challenge although several promising methods are under investigation. Special attention is being paid to ameliorating the damage using low molecular weight antioxidants. Melatonin, epigallocatechin gallate, and flavone derivatives, in particular, have been studied recently [Citation29].
3. Countermeasures for biological threats – bioterrorism
At large, there are a select group of biological agents (infectious and/or toxic constituent) which can be weaponized using relatively easy means (i.e. inexpensive production and storage) by individuals with criminal intents. Aerosolization is believed to be the most likely, successful dispersal method of bioweapons [Citation30]. Furthermore, the injury produced by these weapons can spread well outside the initial delivery area, leading to dreadful results. These characteristics instill fear and incite public panic.
The Centers for Disease Control (CDC) and Prevention categorizes bioweapons into Categories A, B, and C, based on their infectious nature, mortality rate, public health impact, and ability to incite panic, and social disruption [Citation31]. These threat agents are as follows: category A consists of: anthrax – Bacillus anthracis, botulism – Clostridium botulinum toxin, plague – Yersinia pestis, smallpox – variola major, tularemia – Francisella tularensis, viral hemorrhagic fevers – filoviruses – Ebola and Marburg, arenaviruses – Lassa and Machupo; category B: brucellosis – Brucella species, epsilon toxin – Clostridium perfringens, food safety threats, glanders – Burkholderia mallei, melioidosis – Burkholderia pseudomallei, psittacosis – Chlamydia psittaci, Q fever – Coxiella burnetii, ricin toxin – Ricinus communis, staphylococcal enterotoxin B (SEB), typhus fever – Rickettsia prowazekii, viral encephalitis – alphaviruses, and water safety threats; and category C emerging infectious diseases such as nipah virus and hantavirus. There are large numbers of agents identified as countermeasures for biological threats (, –); a few of these have obtained FDA approval as a result of the Animal Rule and are discussed below. The foremost therapeutic developments in countering exposures to given biologic agents focus on vaccine development. In addition to antibiotics, a few countering biologics have been approved; several of such agents are under development [Citation32].
3.1. Botulism
Botulinum toxin is an exotoxin produced by the gram-positive bacteria Clostridium botulinum. Botulinum toxin-producing bacteria are divided into six groups: C. botulinum groups I–IV, as well as some strains of Clostridium baratii and Clostridium butyricum [Citation33]. There are seven well-known serotypes of the botulism toxin, labeled A through G; almost all human cases of botulism are caused by types A, B, and E, with type E being the most prevalent [Citation34]. It is the most powerful toxin known and has been projected (per unit mass) to be ~100,000 times more lethal than sarin gas [Citation35]. The three main clinical presentations of botulism are as follows: foodborne botulism, infant botulism, and wound botulism; the main symptom of exposure is flat muscle paralysis. Though botulism is rare, it yields a life-threatening neuroparalytic syndrome. Patents for agents to treat botulism have been listed in and . The majority of listed patented countering agents are antimicrobial by nature and do not directly neutralize the botulism toxin. The only therapeutic countermeasure for the exotoxicemic effects of botulism, namely heptavalent botulinum antitoxin (HBAT), has been approved by the FDA using the Animal Rule and is discussed below.
3.1.1. HBAT
The C. botulinum toxin blocks acetylcholine released from motor neurons in a dose-dependent manner, which results in acute symmetric diplopia, dysphagia, dysarthria, dysphonia, and possible neurologic sequelae. Botulism is characterized by symmetrical descending paralysis that may develop to respiratory arrest. HBAT contains equine-derived, heptavalent antibody to the seven known botulinum toxin types (A–G) and is comprised of <2% intact immunoglobulin G (IgG) and >90% Fab and F(ab′)2 immunoglobulin fragments; these fragments are created by the enzymatic cleavage and removal of Fc components [Citation36]. In March 2010, the US CDC released HBAT as an Investigational New Drug (IND), for use as the preferred treatment for food-borne botulism, including type E, which had not been covered by the previously offered bivalent antitoxin. On 22 March 2013, the FDA approved the botulism antitoxin for use in neutralizing all seven known botulinum nerve toxin serotypes [Citation37]. This product’s efficacy was studied utilizing guinea pig and NHP animal models under Good Laboratory Practice. In the guinea pig model, treatment with the vaccine resulted in a statistically significant improvement in the survival rate of animals across all serotypes tested at a dose of 1.5 LD50 dose for guinea pigs injected im into their right hind limbs. In a controlled therapeutic NHP efficacy study, treatment with the antitoxin resulted in a statistical improvement in survival in animals challenged with 1.7 LD50 dose of the toxin. These results provided substantial evidence that the antitoxin is likely to help humans with botulism, allowing for the FDA’s approval.
3.2. Anthrax
Since the anthrax (B. anthracis) spore incidents in the fall of 2001 involving its use in the US mail, the threat of weaponized biological agents being used by terrorists has been of the paramount concern to national security [Citation30]. Anthrax is one of the seven globally ignored zoonotic and endemic diseases, according to the World Health Organization [Citation38]. Anthrax is the potentially fatal disease triggered by infection of gram-positive B. anthracis; this disease can present in one of four distinct clinical patterns depending on the route of infection (cutaneous, gastrointestinal, pneumonic, or injectional). Once the bacterial spore gains entry to a mammalian host, it is taken up by phagocytic cells and transported to the draining lymph nodes; the host cells undergo apoptosis and the bacteria germinate into vegetative cells able to produce the tri-partite complex of exotoxins. Pulmonary anthrax arises after the inhalation of small, ~2–5 µm, aerosolized spores; spores of this size are able to bypass the mucociliary system in the sinonasal tract and reach deep within the lung into the alveolar spaces [Citation39,Citation40]. The incubation time can be up to 100 days in the mediastinal lymph nodes, eventually leading to toxin-mediated edema, hemorrhage, and necrosis [Citation41,Citation42]. Inhalational symptoms include fevers, chills, sore throat, malaise and fatigue, nausea and vomiting, nonproductive cough, and respiratory distress. Ongoing attempts to produce more effective countermeasures involve three basic approaches: (1) improving the quality of existing vaccines as well as developing new and improved vaccines; (2) developing and testing more efficient antimicrobials; and (3) developing arrays of antitoxins. The process of producing higher quality and defined therapeutics is based on a growing understanding of the complex pathogenesis of anthrax infection; it is insufficient to simply reduce the bacteremia with antibiotic therapy, since beyond a specific point, the toxemia associated with anthrax infection is often fatal. Five agents have received FDA approval for human use following the Animal Rule to counteract exposure to anthrax; several more countermeasures have been identified and patented ( and ). Comparatively, more countermeasures for anthrax have been developed as an indirect result of the public concern and pressure following the 2001 anthrax incident that resulted directly in more scientific interest, coupled to increased government funding. In aggregate, these listed patents clearly cover all three of the R&D approaches mentioned earlier; however, the vast majority of them (~75%) represent new types/approaches for antimicrobial therapies, while the remaining patents are for new types of vaccines and antitoxin therapies. Useful prophylactic strategies as well as postexposure therapies remain under development [Citation43]. The agents that have been approved by the FDA under the Animal Rule are specifically identified and discussed below.
3.2.1. Anthrasil
Anthrasil (or more commonly referred to as Anthrax ImmunoGlobulin) is a preparation of purified polyclonal human IgG for iv administration, with specificity to the anthrax toxin; these antibodies are present in the plasma of individuals vaccinated against anthrax. It is recommended for the treatment of inhalation anthrax in adult and pediatric patients in conjunction with antibacterial drugs. Anthrasil efficacy studies were accomplished in rabbit and NHP models [Citation44]. In one study, NHPs were exposed to lethal doses of aerosolized B. anthracis spores, treated with Anthrasil or a placebo, and then assessed for survival. The survival of NHPs treated with Anthrasil (7.5, 15, or 30 mg/kg, started after animal became toxemic) ranged from 36% to 70% compared to 0% in the control group; survival rates enhanced as the treatment doses of Anthrasil were increased. Rabbits treated with Anthrasil after infection exhibited 26% survival compared to 2% survival in the control group. Another rabbit study demonstrated that a combination of Anthrasil and antibiotics (one of the fluoroquilones) resulted in 71% survival compared to 25% survival of those animals treated with antibiotics alone [Citation44]. Simultaneous administration of Anthrasil with levofloxacin or ciprofloxacin in anthrax-exposed rabbits and anthrax-exposed NHPs, respectively, did not decrease the effectiveness of antibacterial therapy. This report also supported the concept that Anthrasil essentially alleviates the toxemic effects initiated by anthrax infection when administered in combination with antibiotics. These results offered sufficient support that Anthrasil is expected to benefit humans with inhalation anthrax. The safety of Anthrasil was examined in 74 healthy human volunteers [Citation45]; the most frequently observed side effects were back pain, headache, nausea, and infusion site pain and swelling [Citation44]. On 24 March 2015, Anthrasil was approved by the FDA following the Animal Rule [Citation45].
3.2.2. Raxibacumab
Raxibacumab (ABthrax) is an anthrax-specific antitoxin, composed of human IgG1λ monoclonal antibody directed against the B. anthracis Protective Antigen. Therapy with Raxibacumab prevents the progression of the disease by binding the protective antigen and eventually inhibiting both, lethal factor and edema factor from entering the host cells [Citation46]. Raxibacumab received fast track designation from the FDA and then approval for its indication on 14 December 2012 [Citation47]; it was the first biologic product to be approved for adult and pediatric patients through the FDA Animal Rule [Citation48].
A number of animal models have been used to determine Raxibacumab efficacy in both preexposure and postexposure situations [Citation47]. Due to the unpredictable nature of potential anthrax releases, Raxibacumab may need to be administered to individuals in high-risk groups even before exposure events; therefore, the efficacy of prophylactic therapy is significant. Postexposure studies were vital in determining the therapeutic window in which the drug could be administered and still remain effective.
Raxibacumab’s efficacy for inhalation anthrax was shown in NHP and rabbit models [Citation49,Citation50]. All animals were administered aerosolized B. anthracis spores and received different doses of Raxibacumab, placebo, or antibiotics normally used to treat anthrax. In experiments treating anthrax-exposed animals (185 LD50 spore dose for NHP and 202.5 LD50 for rabbit) with a treatment dose of 40 mg/kg Raxibacumab, 64% of the NHPs and 44% of the rabbits survived, compared to 0% survival in the control groups of NHPs and rabbits; all surviving animals produced toxin-neutralizing antibodies. Another study with rabbits demonstrated that 82% of animals treated with a combination of antibiotics and Raxibacumab survived exposure compared with 65% of animals receiving antibiotic treatment alone [Citation46,Citation47]. To investigate the therapeutic window of treatment with Raxibacumab, rabbits were exposed to 100 LD50 of B. anthracis spores and administered Raxibacumab at a single dose of 40 mg/kg at the time of exposure, 12, 24, or 36 h after exposure. Survival was 100% in animals treated at the time of exposure or 12 h after exposure, but decreased to 50% and 42% in animals treated at 24 and 36 h after exposure, respectively [Citation51]. Raxibacumab has undergone human safety trials (326 healthy volunteers); common side effects included rash, extremity pain, itching, and drowsiness [Citation46,Citation52].
3.2.3. Levaquin
Levaquin (levofloxacin), a constituent of the fluoroquinolone family, is a synthetic broad-spectrum antibacterial agent for oral and iv administration. It is the l-isomer that provides the primary antibacterial activity of racemic ofloxacin, a quinolone antimicrobial agent [Citation53]. Levofloxacin and other fluoroquinolone antimicrobials inhibit bacterial topoisomerase IV and DNA gyrase, enzymes needed for DNA replication, transcription, repair, and recombination [Citation54].
Levaquin is indicated for inhalation anthrax to inhibit the occurrence or advancement of disease following exposure to aerosolized B. anthracis spores [Citation55]. An animal study using NHPs exposed to an inhaled average dose of 49 LD50 (~2.7 × 106 spores) of B. anthracis was conducted; NHPs were administered a 30-day regimen of oral Levaquin beginning 24-h postexposure. The minimal inhibitory concentration of Levaquin for the anthrax strain used in this study was 0.125 μg/ml. Mortality of animals that received Levaquin (1/10) was significantly lower, compared to the control group (9/10). The one levofloxacin-treated animal that died of anthrax did so following the 30-day drug administration period.
The estimated effective human dose of Levaquin is based on the mean plasma concentrations of Levaquin combined with a statistically significant enhancement in survival over control in the NHP model of inhalation anthrax. Plasma concentrations serve as a surrogate end point that has a realistic likelihood of predicting clinical advantage. Levaquin plasma concentrations reach or exceed those determined in NHP experiments in adult and pediatric patients receiving the recommended oral and iv dosage regimens [Citation56]. Levaquin received FDA approval for this indication following the Animal Rule on the 24 November 2004 [Citation56].
3.2.4. BioThrax
Anthrax Vaccine Adsorbed was originally licensed on 10 November 1970 and re-licensed to Emergent as BioThrax on 12 November 1998. On 23 November 2015, the FDA approved the supplement requesting a new indication for BioThrax: postexposure prophylaxis of suspected or confirmed Bacillus anthracis exposure, when combined with the recommended course of antimicrobial therapy. This was the first approval of a vaccine for any indication based on the Animal Rule [Citation57]. BioThrax produces antibodies against protective antigen, ultimately neutralizing the activities of the cytotoxic lethal toxin and edema toxin of B. anthracis. Although other B. anthracis proteins besides those against protective antigen may be present in BioThrax, their role in protection against anthrax exposure has not been established.
Pivotal animal efficacy studies were conducted in rabbits and NHPs; animals received two im vaccinations 4 weeks apart with serial dilutions of BioThrax and were subjected to lethal quantities of aerosolized B. anthracis spores on study day 70 [Citation58]. Logistic regression analysis established that an antibody 50% neutralization factor level of 0.56 in rabbits and 0.29 in NHPs was linked with 70% probability of survival.
The ability of BioThrax to increase rates of survival when the treatments were supplemented by antimicrobial treatment was examined in two postexposure animal model studies. In these studies, rabbits were challenged with aerosolized B. anthracis spores and then treated with levofloxacin, administered via oral gavage once daily for 7 days starting at 6–12 h postexposure, with or without two im injections of BioThrax 1 week apart. Rates of survival among animals receiving both levofloxacin and BioThrax ranged from 70% to 100% and incrementally increased as dose of the vaccine increased. In contrast, only 44% and 23% survival were noted among animals that received only antimicrobial treatment [Citation58].
3.2.5. Ciprofloxacin hydrochloride
Ciprofloxacin hydrochloride (Cipro), a fluoroquinolone, is the monohydrochloride monohydrate salt of 1-cyclopropyl-6-fluoro-1, 4-dihydro-4-oxo-7-(1-piperazinyl)-3-quinolinecarboxylic acid. The FDA approved Cipro in 2015 to decrease the chance of developing anthrax infection following exposure to the anthrax spores [Citation59]. A study using NHPs exposed to an inhaled mean dose of 11 LD50 (~5.5 × 105 spores; range 5–30 LD50) of B. anthracis was conducted. The minimal inhibitory concentration of ciprofloxacin for the anthrax strain used in this study was 0.08 µg/ml [Citation60,Citation61]. NHPs received a 30-day regimen of oral ciprofloxacin, initiated 24 h postexposure; the mortality of ciprofloxacin-treated NHPs was significantly lower (1/9), compared to the control group (9/10) [Citation61]. The mean serum concentrations of ciprofloxacin associated with a statistically significant enhancement in survival in the NHP model of inhalation anthrax are attained or exceeded in adult and pediatric patients receiving oral and iv regimens [Citation62,Citation63].
3.3. Plague
Y. pestis is a gram-negative coccobacillus that is the causal agent of a rare but possibly fatal bacterial infection known as plague. It is classified by the CDC as a category A bioterrorism agent with the three most common forms being: bubonic plague (infection of the lymph nodes), septicemic plague (infection of the blood), and pneumonic plague (infection of the lungs) [Citation30]. Y. pestis has been used as a bio warfare weapon in the past [Citation64]. Agents that have been identified and patented for the treatment of plague are listed in and . The majority of these newly patented agents fall into three major groups: (1) vaccines against antigens that are predicted to confer protection to host; (2) new antimicrobials; and (3) new approaches to enhance specific immunity. Discussed below are three countermeasures with indications to treat plague that have been approved by the FDA utilizing the Animal Rule.
3.3.1. Avelox
Avelox (moxifloxacin hydrochloride) is a fluoroquinolone antibiotic, effective in treating experimental pneumonic plague in an animal infection model [Citation65]. The FDA approved Avelox on 8 May 2015 to treat patients with pneumonic and septicemic plague [Citation66]. In the NHP model of pneumonic plague, the mortality of Avelox-treated NHPs was significantly lower compared to the control group. Avelox’s safety was characterized in clinical studies and post-marketing information for the drug’s existing clinical uses. Common side effects are diarrhea, nausea, headache, and dizziness.
3.3.2. Levaquin
On 27 April 2012, the FDA approved Levaquin (levofloxacin) for the prophylactic use and treatment of patients with plague, including pneumonic and septicemic plague, caused by Y. pestis [Citation47]. Approval for this indication is based on an efficacy study conducted in NHPs exposed to an inhaled mean dose of 65 LD50 (range 3–145 LD50) of Y. pestis [Citation55]. The minimal inhibitory concentration of Levaquin for the Y. pestis strain used in this study was 0.03 μg/ml. Mean plasma concentrations of Levaquin attained at the end of a single 30-min iv infusion ranged from 2.84 to 3.50 μg/ml in NHPs. The mortality in the Levaquin-treated group was significantly lower (1/17) compared to the control group (7/7).
3.3.3. Cipro
The FDA approved Cipro on 2 February 2015 for treatment and prophylaxis of plague due to Y. pestis in adults and pediatric patients from birth to 17 years of age [Citation67]. Studies of Cipro for use in the treatment of plague were conducted in NHPs [Citation68]. Ciprofloxacin was significantly efficacious in the treatment of pneumonic plague in NHPs (10-day regimen, 15 mg/kg iv administered over 60 min twice a day, mimics the human regimen of 400 mg, iv twice a day).
3.4. Other biological agents
In addition to the infectious agents discussed above, there are several other infectious agents and toxins that pose significant health threats when weaponized. Some of these agents include tularemia, Q fever, smallpox, viral hemorrhagic fever, SEB (Staphylococcal aureus enterotoxin B), ricin, trichothecene mycotoxins including T-2 mycotoxin, saxotoxin, and tetrodotoxins [Citation69]. Tularemia is a zoonotic disease caused by infection with the gram-negative, facultative intracellular bacterium Francisella tularensis. A live, attenuated vaccine is currently available with IND and is effective against aerosol infection [Citation70]. Q fever is a zoonosis triggered by rickettsia-like organism, Coxiella burnetii, an obligate gram-negative intracellular bacterium. To treat Q fever, a 2-week course of doxycycline is recommended for adults and children aged ≥8 years. A Q fever vaccine is licensed in Australia but not in the United States [Citation71].
Despite the eradication of naturally occurring smallpox and the availability of a vaccine, the potential weaponization of the variola virus continues to pose a significant health threat to the military and civilian populations, alike. A Vaccinia vaccine is an effective vaccine for preexposure prophylaxis against smallpox [Citation72]. IMVAMUNE (also known as IMVANEX), which is based on a live, attenuated, modified Vaccinia Ankara virus that lacks replicative capacity in human hosts, is being developed for the prevention of smallpox infection, particularly in patients contraindicated to the traditional smallpox vaccine, such as immunosuppressed individuals. This vaccine has been approved by European Medical Agency in 2013 and may get FDA approval in the near future [Citation73].
SEB is an exotoxin produced by Staphylococcus aureus. It is one of the toxins responsible for staphylococcal food poisoning in humans and has been produced previously as a biological weapon [Citation74]. SEB is a superantigen; it acts by stimulating cytokine release and inflammation [Citation75]. Ricin, a toxin derived from the castor bean, is a by-product of the process used to extract castor bean oil. Ricin acts against cells’ ribosomes, preventing the cells from producing proteins, thus leading to cell death, and possibly resulting in organ failure and death. Since there is no cure for ricin poisoning, treatment focuses on addressing the symptoms and, if possible, flushing the ricin out of the system [Citation74]. Trichothecene is a type of mycotoxin produced by several toxic fungi such as Stachybotrys chartarum. Trichothecene is one of the most notorious mycotoxins because it is extremely toxic and difficult to destroy. There are 60 known types of trichothecene mycotoxins, though T-2 trichothecene mycotoxins are the only ones which have been used in biological weapons [Citation74]. Saxitoxins and tetrodotoxins are a group of chemical agents produced by certain species of marine algae and fish. The lethal dose for a human is about 0.5–2.0 mg when entering the body orally (as in with food), and 0.05 mg if injected (as bite or sting) [Citation76]. Saxitoxin and tetrodotoxin poisoning produces neurological symptoms such as muscle weakness, headaches, dysphonia, floating feeling, paralysis of the cranial and peripheral nerves, astigmatism, as well as paresthesia around the lips, tongue, gums, and distal segments of the limbs. There is no specific antidote for saxitoxins and tetrodotoxins, but supportive therapies are recommended [Citation76].
3.5. Recent patents for CB threat countermeasures
A large number of agents have been patented as countermeasures for CB threats during the last few years. We have listed such agents in – and have discussed them below.
3.5.1. Chemical/Nerve gas
Improvements in the therapeutic efficacies of the presently available treatment options for unwanted exposures to highly toxic, often lethal effects of organophosphorous nerve agents are needed. In this regard, galantamine (GAL-Hbr) has been recognized to be beneficial, at least experimentally, improving survival following exposures to various nerve agents [Citation77,Citation78]. Galantamine is a unique, dual mode-of-action agent. It is a reversible and weak competitive inhibitor of acetylcholinesterase and is one of a few drugs with activity as an allosteric modulator of nicotinic acetylcholine receptors [Citation79]. Galantamine protects acetylcholinesterase from irreversible inhibition by organophosphate while preserving the scavenger capacity of plasma butyrylcholinesterase for organophosphates. Soman-induced inhibition of a major regulatory, neurotransmitter, GABA, was not observed in the hippocampus of galantamine-treated guinea pigs [Citation79]. When administered instantly after VX exposure and in combination with two other well-known antidotes, atropine and pyridine-2-aldoxoime methochloride, GAL-Hbr effectively reduces the extent of diaphragmatic paralysis; this improves survival of the nerve agent-exposed guinea pigs [Citation78,Citation80]. Atropine, a current therapy for organophosphate poisoning and a muscarinic receptor antagonist, is administered postexposure. Galantamine can be administered multiple times per day for a protracted period of time. Another agent often discussed alongside galantamine as a potentially useful antidote is huperzine (HUP), a potent, naturally occurring, reversible inhibitor of acetylcholinesterase that can be successfully administered prior to or immediately following exposure to nerve agent, e.g. soman [Citation81]. Despite HUP’s equivalent efficacy to that of physostigmine in mice (i.e. a pyridiostigmine-like related drug), the drug seems to be devoid of serious deleterious effects in healthy subjects [Citation82].
3.5.2. Botulism
Several patents have recently been accepted as effective treatments for botulism (all seven known types of botulinum serotypes). These agents, currently under research and development, fall into several categories including biologics and nutraceuticals. Only one of the agents presented in , an oligoclonal (2–10 specificities) antibody of botulinum, has been tested for efficacy specifically against botulism. With prophylactic administration, the oligoclonal botulinum antibody binds solubilized toxin, preventing the toxin’s ability to disrupt natural levels of acetylcholine at synaptic neuromuscular junctions [Citation83]. Experiments using an ex vivo mouse model suggest neuroparalysis can be significantly delayed by combining single chain Fv antibody fragments and either epitope 1 (S25) or 2 (C25) antibody fragments. This improved treatment modality may aid in improving the current treatment regime more effectively.
3.5.3. Anthrax
Patents for anthrax countermeasures have a wide range of drug-delivery schedules, including before or after exposure to B. anthracis. Activated C protein (Xigris) has shown efficacy in NHPs and mice when administered before exposure [Citation84]. Untreated, control mice presented anthrax dose-dependent lethality whereas Xigris-treated mice were protected. Angiopoietins, members of vascular growth factor family, were augmented during the first 48 h of exposure to the bacterium and declined after 48 h in the individuals that survived infection. Angiopoietins seem to be a useful biomarker for anthrax infection and survival prediction. Additionally, poly-γ-d-glutamic acid (γDPGA), which is present on the surface of B. anthracis, can be targeted by prophylactic treatments; monoclonal antibodies bound to this surface molecule can protect mice from anthrax infection [Citation85]. A treatment protocol for humans was developed utilizing these monoclonal antibodies and may prove suitable in the clinical management of victims following exposure. Treatment with a high-affinity anti-Protective Antigen (anti-PA) monoclonal antibody after exposure to B. anthracis may also be helpful [Citation86] as it has shown to provide exposure mitigation to NHPs. When combined with levofloxacin, NHPs and rabbits demonstrated greater protection than with antibiotic treatment alone. Anti-PA provided comparable pharmacokinetics and bioavailability when administered either through iv or im routes, with the im route being more suitable during a mass causality scenario.
3.5.4. Plague
Elucidation of the pathogenic mechanisms of a given infectious microbe is generally an essential step for developing effective treatments. A recently published patent concerning a new type of Y pestis vaccine suggests that by selectively modifying Y. pestis’s lipopolysaccharide (LPS) by in vitro culturing at 37°C (typical body temperature), microbial infectivity becomes limited, and thus can serve as the basis of an effective, noninfectious, whole-organism vaccine. Limiting the infectivity of Y pestis and the potency of its cell wall-derived endotoxin might well serve in as a treating the causative root for plague symptoms [Citation87]. The LPS may be part of the therapy in some situations. Protollin, a proteasome-LPS adjuvant (combination of Neisseria meningitides and Shigella flexneri), significantly enhanced mouse survival when combined with Y. pestis antigens to provide protection from plague [Citation88,Citation89].
In two recently published patents, NHPs were used to study the efficacy of the therapies for Y. pestis infections. NHPs were protected from Y. pestis exposure after immunization with plant-produced (F1-LcrV-LicKM (thermostable enzyme lichenase from Clostridium thermocellum) double fusion protein) (novel expression system) antigens [Citation90]. No Y. pestis bacteria were evident in surviving NHP organs. A second biologic, flagellin, and F1/V antigen enhanced an appropriate antibody response in mice, suggesting that it may protect against exposure to Y. pestis [Citation91]. Mice treated with this biologic had a 93% survival rate compared to 7% in the control group after administration of 100 LD50 plague bacteria. The antibody efficacy is B-cell mediated, as further control tests revealed 100% mortality in both groups after exposure to 150 LD50 doses of Y pestis in a B-cell knock-out mouse strain. Similarly, using the host’s natural immune system may allow for better protection. A novel trivalent nanoemulsion-based vaccine administered in mice demonstrated an antibody pattern that mimics that of untreated individuals that survive the plague [Citation92]. Mice administered with this trivalent nanoemulsion vaccine demonstrated 90% protection when exposed to 150 LD50. This vaccine can be used effectively through intranasal or im routes.
4. Conclusion
The availability of CBRN countermeasures has become a critical US homeland security concern since the terrorist attacks in 2001. The challenges facing medical countermeasure development and availability are complex. Generally, medical countermeasure development and approval must follow the FDA’s arduous product review process. Yet, when human efficacy data are limited, the scientific and regulatory obstacles may be greater than the challenges faced during regular drug development. Furthermore, medical product development is very costly in terms of financial and research resources; it is roughly approximated that the cost to develop a new medical product spans from ~$1 million to well over ~$1 billion and the likelihood of failure is considerable (i.e. >85%) [Citation4,Citation93]. Although the 2004 Project BioShield Act funded a government market to procure additional medical countermeasures, the available funds are small compared to the possible monetary return made by developing and marketing a successful drug intended for other indications [Citation94]. These challenges and the absence of a commercial market for many CBRN countermeasures have left those successful drug companies most experienced at navigating the complex regulatory process hesitant to enter into the business of CBRN countermeasures development. Stepping up are smaller biotechnology companies that are often technically strong, but have little or no experience developing any drug through the FDA review and approval process. Therefore, it is important that the FDA provides more guidance to such companies throughout the development process. This approach is not uncommon to the FDA as there are a large number of countermeasures for various threats at different stages of development and several recent review articles cover this area of development [Citation95–Citation97].
The DHHS as well as the DoD are committed to develop medical countermeasures for CBRN threats. Interagency interaction, cooperation, and support are essential for long-term goals to be met successfully and efficiently within and across the civilian and military sectors. These efforts will be important to allocate and dispense medical countermeasures to those who will need them in order to prevent, mitigate, and/or treat the detrimental effects of CBRN injuries. A critical consideration for medical countermeasure development is the diversity of the civilian population. In contrast to the DoD programs, which largely focus on the needs of a young, healthy military population, the Biomedical Advanced Research and Development Authority’s (BARDA, DHHS) efforts account for the total civilian population, including infants, children, pregnant women, elderly, and immunocompromised individuals. The special medical needs of each of these subpopulations must be predicted and accounted. One of BARDA’s approaches is to repurpose drugs already approved by the FDA for other indications; this strategy decreases risk within the development pipeline because these drugs have already proven some measure of safety in these select populations and often have well-defined biochemical targets that are relevant to chemical threats.
5. Expert opinion
Several factors have contributed to the mounting threat of a major crisis which would result in enormous numbers of CB exposure-related casualties. Despite the robust International Health Regulations Treaty that gave exceptional authority to World Health Organization after the 2005 severe acute respiratory syndrome outbreak, cutbacks in funding rapidly reduced the preparedness capability of responding to such an incident, especially before the 2015 Ebola crisis. This situation emphasizes the need for additional funding so that multiple countermeasures against various threats can be developed, stockpiled, and efficiently deployed under such circumstances. The US government has taken appropriate steps to develop a procurement system for CBRN threat countermeasures to protect citizens in the event of a public health emergency. The government has created a market and encouraged developers of medical countermeasures to participate in programs such as Project BioShield, BARDA, and Pandemic and All Hazards Preparedness Act [Citation98]. The continued success of these programs will depend largely upon whether they are funded appropriately. The DHHS and DoD are dedicated to developing and implementing medical countermeasures for both civilian and military personnel. It is encouraging to note that nine countermeasures have received US FDA approval for CB threats; five of these countermeasures have been approved for anthrax and Ciprofloxacin has approval for both anthrax and plague.
The mission of the Medical Countermeasure Initiative is to promote the development of countermeasures for CBRN threats and emerging infectious diseases. They do so by enhancing the FDA’s regulatory processes, fostering the establishment of clear regulatory pathways for medical countermeasures, and establishing effective regulatory policies and mechanisms to facilitate timely access to medical countermeasures. The current regulatory framework and the lack of regulatory science create both real and perceived barriers for developers who are keen to develop medical countermeasures. Among the most important challenges faced during medical countermeasure development assessment and approval, are the regulatory uncertainties, which are driven by gaps in scientific knowledge. Developers perceive a higher-than-typical risk for engaging in novel medical countermeasure development if they do not identify a clear development pathway. Reducing regulatory uncertainty is one of the most important challenges that the US government must successfully address.
The threat of an international anthrax outbreak, and the search for improvements to the anthrax prophylaxis tools currently available, will likely remain a subject of great interest within the realm of biodefense for the foreseeable future. Efforts to gain FDA approval for a second-generation recombinant, protective antigen-based anthrax vaccine are ongoing and significant progress has been made. Additional vaccine approaches utilizing multiple components of the B. anthracis as immune response targets are under active investigation. In brief, a multi-pronged approach targeting distinct aspects of the B. anthracis seems like a logical objective for future anthrax vaccine.
There are several encouraging medical countermeasures under advanced stages of development for CB threat agents; IMVAMUNE, the Vaccinia Ankara virus vaccine for small pox, is a live, attenuated, and modified virus that may get FDA approval in near future. It has already been approved by the European Medical Agency for human use. There are additional antivirals under development for smallpox [Citation36,Citation94].
After 11 September 2001, and subsequent anthrax attack, the DHHS initiated large-scale development of products with FDA approval for other indications and to some extent unapproved products, to be used in the event of a mass causality scenario. Prior to this action, the only option for marketing unapproved products was an IND protocol requiring Institutional Review Board approval, documented informed consent from the patients, substantial record keeping, and follow-up requirements, even in the event of an emergency. Although the IND mechanism was appropriate for a clinical study and an emergency situation for an individual, this was unsuitable for mass causality scenario, particularly when essential components of the biodefense armamentarium were either unapproved or approved for different indications at that time [Citation36,Citation98].
Project BioShield of 2004 established the Emergency Use Authorization (EUA) program which allows the FDA to approve the emergency use of drugs without previous approval or the off-label use of approved products in a well-defined emergency situation. The EUA provides physicians and public health officials with new tools for care under emergency conditions [Citation99]. It is imperative to note that countermeasures showing promising results do not need FDA approval or licensure to be produced in large quantities and then stockpiled in SNS in preparation of a mass causality scenario. Several medical countermeasures were procured for the SNS under Pandemic and All Hazards Preparedness Act, when the agents had only IND status, long before receiving FDA approval [Citation98].
Article highlights
Nine countermeasures have been approved by the FDA against chemical and biological threats following the Animal Rule.
Pyridostigmine bromide and Cyanokit (hydroxocobalamin) have received FDA approval to combat effects of exposure to nerve gas (Soman) and cyanide poisoning, respectively.The FDA has approved Heptavalent botulinum antitoxin (HBAT; equine immunoglobulin G fragments) to treat botulism resulting from Clostridium botulinum toxin.
Five countermeasures, Anthrasil (human immunoglobulin G), Raxibacumab (monoclonal antibody), Levaquin (Levofloxacin), BioThrax (vaccine) and Cipro (ciprofloxacin hydrochloride) have FDA approval for use against Bacillus anthracis.
Avelox (moxifloxacin, an antibacterial agent), Levaquin (a member of the fluoroquinolone family), and Cipro have received FDA approval for the treatment of Yersinia pestis, including pneumonic and septicemic plague.
Several additional countermeasures against CB threats have been patented and some are under advance development using Animal Rule.This box summarizes key points contained in the article.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Acknowledgments
The opinions or assertions contained herein are the professional views of the authors and do not necessarily represent the Armed Forces Radiobiology Research Institute, the Uniformed Services University of the Health Sciences, or the Department of Defense, USA. Authors are thankful to Victoria L Newman for editorial help and to Patricia L.P. Romaine for patent searches. Mention of specific therapeutic agents does not constitute endorsement by the U.S. Department of Defense, and trade names are used only for the purpose of clarification. We apologize to those having contributed substantially to the topics discussed herein that we were unable to cite due to space constraints.
Additional information
Funding
References
- Vogel H. Weapons of mass destruction, WMD. Eur J Radiol. 2007;63:205–213.
- Prockop LD. Weapons of mass destruction: overview of the CBRNEs (Chemical, Biological, Radiological, Nuclear, and Explosives). J Neurol Sci. 2006;249:50–54.
- Salem H. Issues in chemical and biological terrorism. Int J Toxicol. 2003;22:465–471.
- Maher C, Hu-Primmer J, MacGill T, et al. Meeting the challenges of medical countermeasure development. Microb Biotechnol. 2012;5:588–593.
- U.S. Food and Drug Administration. FDA’s Medical Countermeasures Initiative Year‐1 Status Report — September 2011. 2011. [cited 2016 Jun 17]. Available from: http://www.fda.gov/downloads/EmergencyPreparedness/MedicalCountermeasures/UCM270750.pdf.
- Thiermann H, Worek F, Kehe K. Limitations and challenges in treatment of acute chemical warfare agent poisoning. Chem Biol Interact. 2013;206:435–443.
- Timbie JW, Ringel JS, Fox DS, et al. Systematic review of strategies to manage and allocate scarce resources during mass casualty events. Ann Emerg Med. 2013;61:677–689 e101.
- Gronvall GK, Trent D, Borio L, et al. The FDA Animal Efficacy Rule and biodefense. Nat Biotechnol. 2007;25:1084–1087.
- Singh VK, Newman VL, Berg AN, et al. Animal models for acute radiation syndrome drug discovery. Expert Opin Drug Discov. 2015;10:497–517.
- Aebersold P. FDA experience with medical countermeasures under the Animal Rule. Adv Prev Med. 2012;2012:507571.
- Singh VK, Newman VL, Romaine PL, et al. Use of biomarkers for assessing radiation injury and efficacy of countermeasures. Expert Rev Mol Diagn. 2016;16:65–81.
- Russell PK. Project BioShield: what it is, why it is needed, and its accomplishments so far. Clin Infect Dis. 2007;45(Suppl 1):S68–S72.
- Need JT, Mothershead JL. Strategic National Stockpile program: implications for military medicine. Mil Med. 2006;171:698–702.
- Esbitt D. The Strategic National Stockpile: roles and responsibilities of health care professionals for receiving the stockpile assets. Disaster Manag Response. 2003;1:68–70.
- Gronvall G. Biodefense countermeasures: the impact of Title IV of the US Pandemic and All-Hazards Preparedness Act. Emerg Health Threats J. 2008;1:e3.
- Wright LK, Lee RB, Vincelli NM, et al. Comparison of the lethal effects of chemical warfare nerve agents across multiple ages. Toxicol Lett. 2016;241:167–174.
- Newmark J. Nerve agents. Neurologist. 2007;13:20–32.
- Bailey AM, Baker SN, Baum RA, et al. Being prepared: emergency treatment following a nerve agent release. Adv Emerg Nurs J. 2014;36:22–33. quiz 34-5
- Brett J, Murnion B. Management of benzodiazepine misuse and dependence. Aust Prescr. 2015;38:152–155.
- Nachon F, Brazzolotto X, Trovaslet M, et al. Progress in the development of enzyme-based nerve agent bioscavengers. Chem Biol Interact. 2013;206:536–544.
- Leadbeater L, Inns RH, Rylands JM. Treatment of poisoning by soman. Fundam Appl Toxicol. 1985;5:S225–S231.
- Von Bredow JD, Adams NL, Groff WA, et al. Effectiveness of oral pyridostigmine pretreatment and cholinolytic-oxime therapy against soman intoxication in nonhuman primates. Fundam Appl Toxicol. 1991;17:761–770.
- U.S. Food and Drug Administration. FDA approves pyridostigmine bromide as pretreatment against nerve gas. 2003. [cited 2016 Jan 12]. Available from: http://www.fda.gov/Drugs/EmergencyPreparedness/BioterrorismandDrugPreparedness/ucm130342.htm.
- Lawson-Smith P, Jansen EC, Hyldegaard O. Cyanide intoxication as part of smoke inhalation -A review on diagnosis and treatment from the emergency perspective. Scand J Trauma Resusc Emerg Med. 2011;19:14.
- U.S. Food and Drug Administration. FDA approves drug to treat cyanide poisoning. 2006. [cited 2016 Jan 13]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2006/ucm108807.htm.
- Borron SW, Stonerook M, Reid F. Efficacy of hydroxocobalamin for the treatment of acute cyanide poisoning in adult beagle dogs. Clin Toxicol (Phila). 2006;44(Suppl 1):5–15.
- Jenner J, Graham SJ. Treatment of sulphur mustard skin injury. Chem Biol Interact. 2013;206:491–495.
- Gosden C, Gardener D. Weapons of mass destruction–threats and responses. BMJ. 2005;331:397–400.
- Pohanka M. Antioxidants countermeasures against sulfur mustard. Mini Rev Med Chem. 2012;12:742–748.
- Zapanta PE, Ghorab S. Age of bioterrorism: are you prepared? Review of bioweapons and their clinical presentation for otolaryngologists. Otolaryngol Head Neck Surg. 2014;151:208–214.
- Gluodenis T, Harrison S. Homeland security and bioterrorism applications: detection of bioweapon pathogens by microfluidic-based electrophoretic DNA analysis. MLO Med Lab Obs. 2004;36:34–38.
- Pettineo C, Aitchison R, Leikin SM, et al. Biological and chemical weapons of mass destruction: updated clinical therapeutic countermeasures since 2003. Am J Ther. 2009;16:35–43.
- Espelund M, Klaveness D. Botulism outbreaks in natural environments – An update. Front Microbiol. 2014;5:287.
- Horowitz BZ. Type E botulism. Clin Toxicol (Phila). 2010;48:880–895.
- Chudzicka A. Intoxication of botulinum toxin. Pol Merkur Lekarski. 2015;39:153–156.
- Centers for Disease Control and Prevention. Investigational heptavalent botulinum antitoxin (HBAT) to replace licensed botulinum antitoxin AB and investigational botulinum antitoxin E. MMWR Morb Mortal Wkly Rep. 2010;59:299.
- U.S. Food and Drug Administration. FDA approves first Botulism antitoxin for use in neutralizing all seven known botulinum nerve toxin serotypes. 2013. [cited 2016 Jan 14]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm345128.htm.
- World Health Organization. Anthrax in humans and animals. 4th ed. Geneva: World Health Organization; 2008.
- Duncan EJ, Kournikakis B, Ho J, et al. Pulmonary deposition of aerosolized Bacillus atrophaeus in a Swine model due to exposure from a simulated anthrax letter incident. Inhal Toxicol. 2009;21:141–152.
- Schneemann A, Manchester M. Anti-toxin antibodies in prophylaxis and treatment of inhalation anthrax. Future Microbiol. 2009;4:35–43.
- Zakowska D, Bartoszcze M, Niemcewicz M, et al. New aspects of the infection mechanisms of Bacillus anthracis. Ann Agric Environ Med. 2012;19:613–618.
- Twenhafel NA. Pathology of inhalational anthrax animal models. Vet Pathol. 2010;47:819–830.
- Williamson ED, Dyson EH. Anthrax prophylaxis: recent advances and future directions. Front Microbiol. 2015;6:1009.
- Cangene Corporation/Emergent BioSolution. Highlights of prescribing information - Anthrasil. 2015. [cited 2016 Jan 17]. Available from: http://emergentbiosolutions.com/sites/default/files/AIG_PI_2015.pdf.
- U.S. Food and Drug Administration. FDA approves treatment for inhalation anthrax. 2015. [cited 2016 Jan 17]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm439752.htm.
- Mazumdar S. Raxibacumab. mAbs. 2009;1:531–538.
- U.S. Food and Drug Administration. FDA approves Raxibacumab to treat inhalational anthrax. 2012. [cited 2016 Jan 17]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm332341.htm.
- Tsai CW, Morris S. Approval of Raxibacumab for the treatment of inhalation anthrax under the US Food and Drug Administration “Animal Rule”. Front Microbiol. 2015;6:1320.
- Corey A, Migone TS, Bolmer S, et al. Bacillus anthracis protective antigen kinetics in inhalation spore-challenged untreated or levofloxacin/Raxibacumab-treated New Zealand white rabbits. Toxins (Basel). 2013;5:120–138.
- Migone TS, Bolmer S, Zhong J, et al. Added benefit of Raxibacumab to antibiotic treatment of inhalational anthrax. Antimicrob Agents Chemother. 2015;59:1145–1151.
- GSK group of companies. Highlights of prescribing information - Raxibacumab. 2015. [cited 2016 Jan 16]. Available from: https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Raxibacumab/pdf/RAXIBACUMAB-PI-PIL.PDF
- Subramanian GM, Cronin PW, Poley G, et al. A phase 1 study of PAmAb, a fully human monoclonal antibody against Bacillus anthracis protective antigen, in healthy volunteers. Clin Infect Dis. 2005;41:12–20.
- Meaney-Delman D, Rasmussen SA, Beigi RH, et al. Prophylaxis and treatment of anthrax in pregnant women. Obstet Gynecol. 2013;122:885–900.
- Tanaka M, Onodera Y, Uchida Y, et al. Inhibitory activities of quinolones against DNA gyrase and topoisomerase IV purified from Staphylococcus aureus. Antimicrob Agents Chemother. 1997;41:2362–2366.
- Janssen Pharmaceuticals Inc. Highlights of prescribing information - Levaquin for plague and anthrax. 2013. [cited 2016 Jan 17]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/020634s065,020635s071,021721s032lbl.pdf.
- U.S. Food and Drug Administration. Levaquin (levofloxacin) information for inhalational anthrax. 2016. [cited 2016 Jan 18]. Available from: http://www.fda.gov/Drugs/EmergencyPreparedness/BioterrorismandDrugPreparedness/ucm133678.htm.
- U.S. Food and Drug Administration. FDA approves vaccine for use after known or suspected anthrax exposure. 2015. [cited 2016 Jan 19]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm474027.htm.
- Emergent BioSolutions. BioThrax (anthrax vaccine adsorbed). 2015. [cited 2016 Jan 20]. Available from: http://www.fda.gov/downloads/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/LicensedProductsBLAs/UCM074923.pdf.
- U.S. Food and Drug Administration. Cipro (ciprofloxacin hydrochloride) for inhalation anthrax. 2015. [cited 2016 Jan 29]. Available from: http://www.fda.gov/Drugs/EmergencyPreparedness/BioterrorismandDrugPreparedness/ucm130709.htm.
- Kelly DJ, Chulay JD, Mikesell P, et al. Serum concentrations of penicillin, doxycycline, and ciprofloxacin during prolonged therapy in rhesus monkeys. J Infect Dis. 1992;166:1184–1187.
- Friedlander AM, Welkos SL, Pitt ML, et al. Postexposure prophylaxis against experimental inhalation anthrax. J Infect Dis. 1993;167:1239–1243.
- Bayer Pharmaceuticals Corporation. Medication guide - Ciprofloxacin hydrochloride. 2015. [cited 2016 Jan 29]. Available from: http://www.fda.gov/downloads/Drugs/DrugSafety/ucm246794.pdf.
- Bayer Pharmaceuticals Corporation. CIPRO. 2005. [cited 2016 Jun 19]. Available from: http://www.fda.gov/ohrms/dockets/ac/05/briefing/2005-4152b1_03_05_02_Cipro%20label%20injection%20FDA%201-7-05.pdf.
- Ligon BL. Plague: a review of its history and potential as a biological weapon. Semin Pediatr Infect Dis. 2006;17:161–170.
- Steward J, Lever MS, Russell P, et al. Efficacy of the latest fluoroquinolones against experimental Yersinia pestis. Int J Antimicrob Agents. 2004;24:609–612.
- U.S. Food and Drug Administration. FDA approves additional antibacterial treatment for plague. 2015. [cited 2016 Jan 15]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm446283.htm.
- U.S. Food and Drug Administration. Ciprofloxacin - Supplemental NDA approved to add indication for treatment and prophylaxis of plague due to Yersinia pestis in adults and pediatric patients. 2015. [cited 2016 Jan 29]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2015/019537Orig1s083,019847Orig1s055,019857Orig1s063,020780Orig1s041ltr.pdf.
- Carlsbad Technology Inc. Ciprofloxacin. 2016. [cited 2016 Jun 19]. Available from: https://www.drugs.com/pro/ciprofloxacin.html.
- Madsen JM. Toxins as weapons of mass destruction. A comparison and contrast with biological-warfare and chemical-warfare agents. Clin Lab Med. 2001;21:593–605.
- Ulu-Kilic A, Doganay M. An overview: tularemia and travel medicine. Travel Med Infect Dis. 2014;12:609–616.
- El-Mahallawy HS, Lu G, Kelly P, et al. Q fever in China: a systematic review, 1989–2013. Epidemiol Infect. 2015;143:673–681.
- Weiss RA, Esparza J. The prevention and eradication of smallpox: a commentary on Sloane (1755) ‘An account of inoculation’. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140378.
- Jones T. IMVAMUNE, an attenuated modified vaccinia Ankara virus vaccine for smallpox infection. Curr Opin Mol Ther. 2008;10:407–417.
- Pita R, Romero A. Toxins as weapons: a historical review. Forensic Sci Rev. 2014;26:85–96.
- Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13:16–34.
- Wesołowski A, Płusa T. [Saxitoxins and tetrodotokxins as a new biological weapon]. Pol Merkur Lekarski. 2015;39:173–175.
- Albuquerque EX, Pereira EF, Aracava Y, et al. Effective countermeasure against poisoning by organophosphorus insecticides and nerve agents. Proc Natl Acad Sci USA. 2006;103:13220–13225.
- Hilmas CJ, Poole MJ, Finneran K, et al. Galantamine is a novel post-exposure therapeutic against lethal VX challenge. Toxicol Appl Pharmacol. 2009;240:166–173.
- Lilienfeld S. Galantamine–a novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer’s disease. CNS Drug Rev. 2002;8:159–176.
- Albuquerque EX, Adler M, Pereira EFR. Method of treating organophosphorous poisoning. US9132135B2. 2015.
- Grunwald J, Raveh L, Doctor BP, et al. Huperzine A as a pretreatment candidate drug against nerve agent toxicity. Life Sci. 1994;54:991–997.
- Shaikh S, Verma A, Siddiqui S, et al. Current acetylcholinesterase-inhibitors: a neuroinformatics perspective. CNS Neurol Disord Drug Targets. 2014;13:391–401.
- Marks JD, Amersdorfer P, Geren I, et al. Therapeutic monoclonal antibodies that neutralize botulinum neurotoxins. US7999079B2. 2011.
- Parikh SM. Methods and compositions for the treatment and diagnosis of systemic anthrax infection. US8685393 B2. 2014.
- Chen Z, Purcell RH, Schneerson R, et al. Monoclonal antibodies that react with the capsule of Bacillus anthracis. US8501182 B2. 2013.
- Casey LS. Methods of preventing or treating anthrax using anti-anthrax antibodies. US8617548 B2. 2013.
- Lien E, Goguen JD. Modified pathogens for use as vaccines. US9138468 B2. 2015.
- Lowell GH, Burt DS, Jones DH, et al. Compositions and methods for activating innate and allergic immunity. US8709447 B2. 2014.
- Ann J, Samant M, Rheaume C, et al. Adjuvanted inactivated influenza A(H3N2) vaccines induce stronger immunogenicity in mice and confer higher protection in ferrets than unadjuvanted inactivated vaccines. Vaccine. 2014;32:5730–5739.
- Yusibov V, Mett V, Musiychuk K. Yersinia pestis antigens, vaccine compositions, and related methods. US8945580 B2. 2015.
- Mizel SB, Honko AN. Use of flagellin in the immunotherapy of Yersinia pestis. CA2589553 C. 2014.
- Baker JR, Makidon PE, Mank NJ, et al. Multivalent nanoemulsion vaccines. US8877208 B2. 2014.
- Matheny J, Mair M, Smith B. Cost/success projections for US biodefense countermeasure development. Nat Biotechnol. 2008;26:981–983.
- Cohen J. Biodefense: 10 years after reinventing Project BioShield. Science. 2011;333:1216–1218.
- Singh V, Gupta D, Arora R. NF-κB as a key player in regulation of cellular radiation responses and identification of radiation countermeasures. Discoveries. 2015;3:e35.
- Singh VK, Romaine PL, Seed TM. Medical countermeasures for radiation exposure and related injuries: characterization of medicines, FDA-approval status and inclusion into the Strategic National Stockpile. Health Phys. 2015;108:607–630.
- Singh VK, Newman VL, Romaine PL, et al. Radiation countermeasure agents: an update (2011–2014). Expert Opin Ther Pat. 2014;24:1229–1255.
- Kels CG. Dispensing medical countermeasures: emergency use authorities and liability protections. Health Secur. 2015;13:139–151.
- Nightingale SL, Prasher JM, Simonson S. Emergency use authorization (EUA) to enable use of needed products in civilian and military emergencies, United States. Emerging Infect Dis. 2007;13:1046–1055.