
ABSTRACT
Introduction: MDM2 and MDMX proteins provide the inhibition of p53 tumor suppressor, thus allowing for accelerated mutation-driven cancer microevolution. A pharmacological blockade of MDM2/X-p53 interaction results in p53 reactivation in p53wt cells, leading to cancer growth inhibition. Throughout the past 20 years, multiple chemical entities have been proposed to reactivate p53 by antagonizing MDM2/X proteins.
Areas covered: This manuscript reviews 2014–2018 therapeutic patents in the field of MDM2/X antagonists and is a continuation of previous reviews on similar matter. The patents covering the use of MDM2/X antagonists in drug combinations are also presented in this review, as they constitute an important trend in the field of cancer treatment with MDM2/X antagonists.
Expert opinion: In the years 2014–2018, several previously-known chemical scaffolds have been further developed and disclosed. Importantly, in the same time period, many lead compounds have entered clinical trials for the treatment of cancer patients. Meanwhile, several important reports have pointed to serious limitations of anticancer properties of MDM2 antagonists. As a result, many efforts have been made to seek for positive, synergistic therapeutic effects of combined anti-cancer treatment strategies. One recent example is a dual targeting of MDM2 and additional protein targets by utilizing the PROTAC technology.
Trial registration: ClinicalTrials.gov identifier: NCT02264613.
Trial registration: ClinicalTrials.gov identifier: NCT02624986.
Trial registration: ClinicalTrials.gov identifier: NCT02670044.
Trial registration: ClinicalTrials.gov identifier: NCT02545283.
1. Introduction
Cancer incidence and mortality are continuously growing worldwide. This alarming trend is observed especially in emerging economies. According to the World Health Organization, cancer has become a primary cause of premature mortality in developed countries, including the United States, Japan, and most of the European countries [Citation1]. Within the past 50 years, much effort has been done to develop and improve therapeutic methods to clinically eradicate cancer, yet with limited success. Among the four classical pillars of anticancer therapy, targeted therapy has been favored by pharmaceutical companies.
The tumor suppressor protein p53 became one of such cancer-related targets, as with time it became clear that in almost 100% of cancers the functioning of p53 is impaired [Citation2]. The p53 protein is a crucial player engaged in the response to mutations occurring within genomic DNA, providing genome stability and thus protecting from early cancerogenic events. Upon the detection of DNA damage, the activity of p53 is released. p53 acts as a transcription factor that regulates the expression of a multitude of target genes responsible for the induction of cell cycle arrest and DNA repair program, giving the cell the opportunity to self-heal the DNA-occurring lesions [Citation3]. Should the repair machinery fail, p53 induces the ultimate solution, which is the induction of apoptosis in extensively damaged cells [Citation4]. This versatile strategy enabled and integrated by the p53 protects the organism from the expansion of DNA-mutated, rebellious cells that could possibly lead to the cancer development.
Considering the anti-cancer nature of p53, it is not surprising that the inactivation of p53 is a critical step for the cancer microevolution. It has been estimated that 50% of cancers disable the p53 protein by gaining loss-of-function mutations or deletions in the TP53 gene [Citation5,Citation6]. Importantly, the remaining 50% of cancers still express the wild-type p53 protein (p53wt), but its activity is blocked by innate regulatory mechanisms, among which overexpression of MDM2 protein is the most common way to keep p53wt inactive [Citation2]. The MDM2 protein is a natural binding partner of p53 and a powerful inhibitor of p53 functioning. MDM2 binds to p53 masking its transactivation domain, targets p53 to nuclear export, and directly ubiquitinates p53 sending the protein to the proteasomal degradation [Citation7]. For this reason, blocking the MDM2-p53 interaction with MDM2 antagonists has become a promising strategy for the restoration of p53 functioning that could possibly be used for the treatment of p53wt cancers [Citation2].
The MDMX (MDM4) protein, the homolog of MDM2, is another p53 binding partner and an additional, important p53 regulator. The p53-inhibitory function of both MDMX and MDM2 proteins is similar, though the mechanism of inhibition is distinct, as MDMX does not possess the E3 ligase activity [Citation8]. Structurally, MDMX and MDM2 have remarkable similarities, especially in the N-terminal (p53 binding region) and C-terminal (RING domain) regions [Citation9]. The two MDM2/X proteins can form heterodimers, which are believed to be more stable and active in respect to p53 regulation, than their monomers [Citation7]. Therefore, simultaneous targeting of both MDM2 and MDMX may be required for effective anticancer treatment strategies [Citation7,Citation10,Citation11].
During the last years, numerous chemical scaffolds have been proposed for the design of the MDM2 antagonists. These include: cis-imidazolines, pyrrolidines, piperidines, and purines, as well as more complex multi-ring structures, such as spiroindolinones and non-small molecule moieties, such as peptides (for the review of 2006–2013 therapeutic patents on MDM2-antagonizing compounds see [Citation12,Citation13], and [Citation14]). These core structures are frequently decorated with the substituents that resemble the three critical amino acid residues of p53 that are directly engaged in its interaction with the MDM2 protein, i.e. Phe19, Trp23 and Leu26 [Citation15,Citation16]. The Phe19 side chain is commonly mimicked by 6-membered aromatic rings; benzene/indole substituents are used to mimic the Trp23 side chain and branched aliphatic residues or additional 6-membered rings are used to occupy the Leu26-binding pocket on the MDM2 protein [Citation17]. Such a structural organization of the compounds provides a classical three-point binding model, that has been successfully used for the design of the MDM2 antagonists [Citation18].
Although the mode of binding of p53 to MDMX is very similar to that of p53 binding to MDM2, with even the same p53 residues involved, the Leu26 pocket of MDMX is smaller and a unique hydrophobic area is nearby. Therefore, most of the MDM2 antagonists discovered so far are not able to efficiently disrupt the MDMX-p53 interaction, and only few several examples of dual MDM2/MDMX inhibitors are known, such as small molecule RO-5963 [Citation19], or stapled peptide ATSP-7041 [Citation20].
The chemical scaffolds of the MDM2/MDMX antagonists are continuously developed both by the scholars and big pharmaceutical companies, resulting in the annual emergence of new therapeutic patents. Previously, we have provided a review of the therapeutic patents 2011–2013 [Citation14]. This review covers the therapeutic patents from the years 2014–2018.
2. Discussion of patents on new MDM2 antagonists (2014–2018)
2.1. Mono-cyclic ring core structures
2.1.1. 5-member rings: imidazoline derivatives
Among MDM2 antagonists, the nutlins, developed and disclosed by Hoffmann-La Roche (Basel, Switzerland), were the first group of compounds that presented a real bioactivity both in in vitro and in vivo studies [Citation21]. The nutlins are cis-imidazolines (), where the core central imidazoline ring is decorated with phenyl-derived substituents, that resemble a classical three-point interaction of p53 with MDM2, provided by the three crucial p53 amino acids, Phe19, Trp23 and Leu26 [Citation15,Citation16]. The most potent representative of first-generation cis-imidazolines, Nutlin-3 (1), became a proof-of-concept molecule in demonstrating the anticancer potential of pharmacological reactivation of p53 by antagonizing MDM2 protein. Further optimization of Nutlin-3 structure led to the discovery of RG7112 (2), which presented improved activity and was the first MDM2 antagonist that entered clinical evaluation [Citation22]. While RG7112 presented sufficient clinical activity to significantly induce p53 and p21 levels in the treated liposarcoma patients, serious adverse hematological effects were observed in most of the treated individuals making the long-term treatments schemes challenging [Citation23].
Figure 1. Structure and in vitro activity of exemplary MDM2 inhibitors based on a 5 membered ring: cis-imidazoline scaffold. The IC50 value for nutlin 3 was is from [Citation21] and for RG-7112 from [Citation24].
![Figure 1. Structure and in vitro activity of exemplary MDM2 inhibitors based on a 5 membered ring: cis-imidazoline scaffold. The IC50 value for nutlin 3 was is from [Citation21] and for RG-7112 from [Citation24].](/cms/asset/64574e19-6708-42ac-98a3-a9c3b2617b8c/ietp_a_1582645_f0001_oc.jpg)
In the year 2014 Hoffmann-La Roche has published one additional patent, covering further optimization trials of the cis-imidazoline structures () [Citation25]. As in RG7112, in the compounds covered by the patent, the two 4-chlorophenyl substituents at the cis configuration on the imidazoline ring are preserved, together with the two methyl groups at the same positions, likely representing the optimal frontiers of the structure optimization at these positions (3). The main optimization efforts reported in the patent were concentrated on the changes in the N-1 and C-2 positions (). The N-1 4-(3-methyanesulfonylpropyl)piperazine-1-carbonyl substituent, which was the most striking improvement made from nutlin-3 to RG7112, has been explored with aliphatic linkers of different lengths, solubility tags (carboxyl, amid, sulfonyl groups), or a heterocycle (piperidine) [Citation25]. Another structure variation was the introduction of a second, substituted sulphonyl group in the phenyl ring at the C-2 position of imidazoline. The resulting compounds were subjected to HTRF assay resulting in IC50 constants of around 5 nM against MDM2 protein and 0.2–6 µM against MDMX protein [Citation25].
Table 1. MDM2/X antagonists with monocyclic core structures.
The last patent covering the optimization trial of cis-imidazoline compounds was disclosed in 2015 by St. Jude Children’s Research Hospital () [Citation26]. The disclosed compounds differ from the nutlins at all substituted positions within the imidazole ring (4), resulting in the compounds of improved in vitro properties compared to Nutlin-3. The most striking difference lies in the halogen positioning and the diversity of cis-3,4-phenyl substituents of imidazoline, including the substitution of one of the phenyl rings to the bioisoster of Trp23: 6-chloroindole (). Interestingly, all of the 9 tested compounds present similar, two-three digit nanomolar binding affinities towards both MDM2 and MDMX proteins, unlike Nutlin-3, representing a promising class of dual inhibitors of these two proteins [Citation26].
2.1.2. 5-member rings: pyrrolidine and pyrrole derivatives
Serious adverse effects observed in clinical trials for the imidazoline-based compound RG7112, and relatively high doses required to obtain a clinical effect have prompted the discovery of RG7388 (5) (idasanutlin, RO5503781) () [Citation27]. The compound invented and disclosed by Hoffmann-La Roche, presented much-improved activity in pre-clinical and clinical studies, allowing for lower dosing schedules and resulting in milder side effects. RG7388 is currently undergoing several clinical examinations, including phase III clinical trial in combination with cytarabine for the treatment of relapsed and refractory acute myeloid leukemia (AML) patients [Citation22].
Figure 2. Structure and in vitro activity of exemplary MDM2 inhibitors based on 5 membered ring: pyrrolidine and pyrrole scaffolds. The IC50 value for RG7388 was is from [Citation28].
![Figure 2. Structure and in vitro activity of exemplary MDM2 inhibitors based on 5 membered ring: pyrrolidine and pyrrole scaffolds. The IC50 value for RG7388 was is from [Citation28].](/cms/asset/4c0e087d-5f09-4f10-a2e8-c4886d79d3dd/ietp_a_1582645_f0002_oc.jpg)
The structure of RG7388 was modified by the addition of a polyethylene glycol solubilizing linker by the esterification of the carboxylic group of the 3-methoxy benzoic acid, substituted at the C-2 position of the pyrrolidine ring (6), as disclosed by Hoffmann-La Roche in 2015 [Citation29]. The compound was successfully used in combination with cytarabine in disseminated MOLM-13 AML model in immunocompromised SCID-beige mice, presenting more than additive therapeutic effects [Citation29]. Similar polyethylene glycol linker was added to the structure of RG7388 by the company Arivinas Inc. [Citation30,Citation31]. The modification, however, went one step further resulting in the generation of bi-functional drug candidates, that utilize a Proteolysis Targeting Chimera (PROTAC) technology to target MDM2 protein for proteasomal degradation (see section 2.5 for more details).
Bi-specific MDM2/MDMX inhibitors designed around the 5-membered ring core structure of 3-pyrrolin-2-on were patented in 2015 by Adamed Sp. z o.o. () [Citation32]. The core structure of the compounds is decorated with halogenated aryl substituents (), including 4-chlorophenyl groups (7), characteristic also for bioactive representatives of other classes of MDM2 antagonists, such as Nutlin-3 or AMG-232. The activity of the compounds was monitored by the fluorescence polarization (FP) assay, resulting in millimolar Ki constants of MDM2/p53 disruption and millimolar Ki constants of the disruption of MDMX/p53 complex.
2.1.3. 6-membered ring core structures – piperidinone and morpholinone derivatives
In 2014 Amgen Inc. released a series of patents covering structure optimization of previously reported 2-piperidinones, and core-modified derivatives, morpholinones () [Citation33–Citation35]. Importantly, one of the representatives of 2-piperidinones, AMG-232 (8) (), is currently undergoing clinical trials for the treatment of multiple myeloma, metastatic cutaneous melanoma and acute myeloid leukemia patients [Citation22].
Figure 3. Structure and in vitro activity of exemplary MDM2 inhibitors based on 6 membered ring: piperidinone and morpholinone scaffolds. The IC50 value for AMG-232 was is from [Citation94].
![Figure 3. Structure and in vitro activity of exemplary MDM2 inhibitors based on 6 membered ring: piperidinone and morpholinone scaffolds. The IC50 value for AMG-232 was is from [Citation94].](/cms/asset/15676538-72f1-4561-9eca-3239d99d26dc/ietp_a_1582645_f0003_oc.jpg)
The US 2014/0243372 patent claims the structure of only one compound (9) which is highly similar to AMG-232 [Citation33]. The main difference is the introducing of the amide (from 4-amino-2-metoxybenzoic acid) substituent instead of the acetic acid moiety placed in the C-3 position of piperidinone ring (). Other changes are at the N-substituent (replacement of isopropyl group with cyclopropyl and tert-butyl) and C-6 phenyl ring (additional fluorine atom at the 3-position of phenyl substituent). The remaining two patents by Amgen Inc. are focused on morpholinone instead of piperidinone core [Citation34,Citation35]. The examples presented in the patents cover a broad scope of compounds designed around the morpholinone-cored AMG-232-like general structure, with the optimization of substituents leading to the best-performing representative compounds (10) from WO 2014/130470 and the (11) from WO 2014/151863 (), as shown by the HTRF in vitro assay. The most important characteristic feature of the compounds disclosed in the patent WO 2014/130470 [Citation34] is the 5,6-cis configuration of the halogenated phenyl substituents, which is in the contrast to the 5,6-trans configuration present in AMG-232 and other two patents mentioned above [Citation33,Citation35]. Additional differences are located all over the core structure, including C-5 and C-6-phenyl substituents (e.g. various halogens, ethers, trifluoromethyl, indole.), N-1 substituent (shortened compered to AMG-232, aliphatic or benzyl, including amide and others functional groups), and exchange of C-2 acetic acid to other substituents (e.g. benzyl, alcohol, amine or aldehyde). Similar substituent modifications, but with much lower diversity and much fewer examples given, was covered by the patent WO 2014/151863 [Citation35].
Additional Amgen Inc. patent from 2014 claims the process of the production of AMG-232 compound and its intermediates, securing the field of compound synthesis for any further clinical use. This potential use includes various cancer treatments and combinations with other drugs, such as inhibitors of PI3K, mTOR or Akt [Citation36]. In fact, several drug combinations involving AMG-232 compound and its derivatives were patented by Amgen Inc. in a 2015 world patent [Citation37] (see section 3 for more details).
2.2. Bicyclic core structures
Besides well exploited MDM2 antagonists designed around a central, single ring core structures, additional scaffolds were proposed, containing bicyclic cores. Among these structures are: (i) 6 + 5-membered rings cores: purines, pyrrolopyrimidines, and isoindolinones from Merck Sharp & Dohme Corp. and Astex Therapeutics Limited, (ii) 5 + 5-membered rings cores: pyrazopyrrolidinones and imidazopyrrolidinon from The Second Military Medical University and Novartis Ag, respectively, and (iii) 6 + 6-membered rings cores – tetrahydro-isoquinolin-3-ones from Novartis Ag (). From all these classes, only two compounds owned by Novartis Ag, namely HDM201 (5 + 5-membered rings core) and CGM097 (6 + 6-membered rings core), have entered clinical trials, so far. Recently, Novartis has disclosed the invention related to the dosing and regimen for the use of these molecules for the treatment of hematological tumors [Citation38,Citation39].
Table 2. MDM2/X antagonists with bicyclic and multicyclic core structures.
2.2.1. Purine, pyrrolopyrimidine and isoindolinone derivatives (6 + 5)
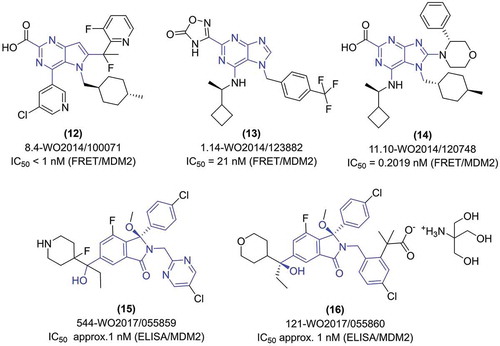
In 2014 Merck Sharp & Dohme Corp. have released three world patents, covering a long list of MDM2 antagonists built around the pyrrolo[3,2-d]pyrimidine [Citation41] and purine [Citation42,Citation43] cores (). In all the three patents multiple examples of compound modifications were presented, with some clear trends in the substitution patterns [Citation41]. Most commonly the pyrrolo[3,2-d]pyrimidine core was substituted at carbons atoms: C-2, C-4, C-6/C-7, and the nitrogen atom N-5, and purine core at the corresponding carbon atoms C-2, C-6, C-8, and the nitrogen atom N-7 (). Similar trends are observed in all patents, with the highest diversity of the tested compounds presented in the case of 2,6,7,8-tetrasubstituted purines [Citation42]. First, from all of the introduced substituents, the one placed at the C-2 position of the pyrrolo[3,2-d]pyrimidine core (accordingly C-2 in purine) seems to be introduced in order to increase solubility in a water medium. The substituents at the C-2 position are, among others: 4,5-dihydro-1,2,4,-oxadiazol-5-one, carboxylic acid, tetrazole, sulphonamide, or sulphoxide. The rest of the positions of the core rings are decorated with the substituents characteristic also for many other MDM2 inhibitors. Position C-4 (accordingly C-6 in purine) is usually substituted with aliphatic amines (i.e. 1-cyclobuthylethanamine, cyclohexylamine, benzylamine), aliphatic ethers (i.e. 1-cyclobutylethoxyl), halogenated (chlorinated or fluorinated) phenyl, pyridine and others moieties (i.e. morpholine, 2-pyridone). At the position N-5 (accordingly N-7 in purine) aromatic: phenyl or methylpyridine (substituted with i.e. halogen atoms, trifluoromethyl, short aliphatic chain, and ethers), or aliphatic rings (i.e. 4-methylcyclohexyl, spiro[2.5]octanyl) are introduced. In all cases, the ring is separated from the molecule core by the methylene group. Substitution at C-6 position (accordingly C-8 in purine) was extensively studied, especially with the purine core [Citation42]. The substituents presented there include aromatic rings directly attached to the C-6/C-8 carbon atom (i.e. substituted phenyl, pyrimidine, imidazole), or attached via a one-atom bridge (i.e. methylene, cyclopropyl). Aromatic rings used were usually substituted with fluorine or short aliphatic chains and ethers. Moreover, in this position aliphatic rings (3-phenylmorpholine, cyclopropyl, octahydro-4H-1,4-benzoxazine) were tested. Also, in this case, a one-bond bridge was introduced and found in several examples. Besides various ring structures, aliphatic chain substituents with different functional groups (amines, alcohols, ethers) can be found in the patents. Taking all this data together, in all cases the substitutions are composed of at least one aromatic ring and one aliphatic, and when possible, a third ring, being either aliphatic or aromatic. The activity of compounds present in all the mentioned Merck Sharp & Dohme Corp. patents was assessed by the fluorescence resonance energy transfer assay (FRET). In the cases of pyrrolo[3,2-d]pyrimidine, IC50 values start from several micromoles, and go down to low nanomolar values, as for the exemplary compound (12), with the most potent having IC50 below 1 nM. Generally, slightly worse IC50 values were achieved by 2,6,7-trisubstituted purines [Citation43], where the best binding example (13) had IC50 values equal 21.58 nM. The best Mdm2 inhibitors from these patents were examples of 2,6,7,8-tetrasubstituted purines, where for multiple examples the IC50 value below 1 nM was reported , i.e compound (14) with IC50 of 0.2019 nM (). Altogether, Merck Sharp & Dohme Corp present extensive structure-activity relationship studies on the presented scaffolds, with exceptionally good results and tremendous diversity of substitution pattern.
Figure 4. Structure and in vitro activity of exemplary MDM2 inhibitors based on bicyclic core (5 + 6): purine, pyrrolopyrimdine, isoindolinone scaffolds.
In 2017, Astex Therapeutics Ltd. has released two world patents covering a large panel of the compounds, designed around the central isoindolin-1-one core structure () [Citation44,Citation45]. The compounds possess a large diversity of substituents at several positions of the isoindolin-1-one bicycle, including N-substitutions, and a featured double substitution at the C-3 position (). This special double substitution provides a stereo-geometry of the compound similar to the geometry of spriro-oxindole compounds, that have been extremely exploited by several leading companies in the recent years (see section 2.4 for more details on spiro-oxindole-based compounds). The C-3 bi-substitutions in all the presented compounds are composed of one aromatic substituent (4-chlorophenyl), and a short aliphatic chains or rings, substituted with various functional groups such as hydroxyl, amide, sulphonamide, or carboxyl, in some cases even deuterated. The additional aromatic ring (phenyl, pyridine or piperidine substituted usually with halogen, sulphonyl, nitrile or short aliphatic ethers) is linked by a methylene bridge at the N-2 position of the central ring. At the C-6 position, usually, aliphatic rings with a hydroxyl group are introduced (i.e. 2-hydroxypropan-2-yl, 1-(4-fluorooxazn-4-yl)-1-hydroxyethyl), although also several heterocycles were tested (i.e. 1-methyl-1H-pyrazole-4-carbonyl). A small diversity is presented in the case of the C-4 atom, which is almost universally a fluorine atom. The resulting compounds covered by the Astex Therapeutics Ltd. patents have been evaluated by an ELISA assay for testing of the blockade of MDM2-p53 interaction. Most of the compounds, for which ELISA results were reported in the patents WO 2017/055859 A1 [Citation44] and WO 2017/055860 A1 [Citation45] (almost 600 and 150 different compounds, respectively) presented IC50 values in low nM range, with multiple individuals, exemplified by the compounds (15, 16), having IC50 values around or even below 1 nM. The activity of most of these hundreds of compounds was further evaluated with the use of two MDM2 amplified isogenic matched osteosarcoma cell lines, SJSA-1 (with wild-type p53 status) and SN40R2 (p53-mutated). The compounds presented excellent selectivity in the two orthogonal cell viability assays performed (total protein staining with SRB dye and CellTiter-Glo® assay), and high anti-cancer activities with IC50 values of SJSA-1 growth inhibition going down to two-digit nM scale for the best-performing representatives. Altogether, the data provided in the patents confirm the successful development of isoindolin-1-one MDM2 antagonists and justifies any further transition of this class of compounds to further pre-clinical trials.
2.2.2. Pyrrolidonopyrazoles and pyrrolidonoimidazoles (5 + 5)
In the years 2014–2018 there were two patents covering MDM2 antagonists that contain a 5 + 5 bicyclic core structure (). The first patent, assigned by The Second Military Medical University in China presents an attempt to designing MDM2-targeting compounds based on dihydropyrrolo[3,4-c]pyrazol-6-one core structure () [Citation46]. The patent includes around 25 compounds such as (17) tested in the biological assays, presenting Ki values of the inhibition of p53-MDM2 interaction ranging from 0.06 to around 100 µM. Further biological data is somehow confusing, as the given IC50 values of the inhibition of the growth of p53wt and p53null cells suggest no selectivity towards p53 compromised cancer cells. Notably, since 2014 no additional structure optimization has been patented by The Second Military Medical University, suggesting that the project might have been closed.
Figure 5. Structure and in vitro activity of exemplary MDM2 inhibitors based on bicyclic core (5 + 5 and 6 + 6): pyrrolidonopyrazole, pyrrolidonoimidazole, dihydro-isoquinolin-3-one scaffolds. The IC50 value for CGM097 was is from [Citation48].
![Figure 5. Structure and in vitro activity of exemplary MDM2 inhibitors based on bicyclic core (5 + 5 and 6 + 6): pyrrolidonopyrazole, pyrrolidonoimidazole, dihydro-isoquinolin-3-one scaffolds. The IC50 value for CGM097 was is from [Citation48].](/cms/asset/f03e275e-cb69-4790-bd23-c242c6ffd825/ietp_a_1582645_f0005_oc.jpg)
Contrarily, a previous successful development of another 5 + 5 bicyclic-cored compound, HDM201 (18), based on the dihydropyrrolo[3,4-d]imidazol-4-one core (), encouraged its owner, Novartis Ag, to disclose its clinical use in combination with B-Raf inhibitors [Citation47] (see section 3 for more details on drug combinations). Importantly, the HDM201 compound is one of the few MDM2 antagonists, that have been progressed to the clinical trials in recent years [Citation22].
2.2.3. Dihydro-isoquinolin-3-one (6 + 6)
Dihydro-isoquinolin-3-ones (), the compounds with the core structure composed of two 6-membered rings, were exploited by the Novartis Ag company. The structure optimization process led to the discovery of CGM097 compound (19) () [Citation48], which presented excellent activity and in vivo profile, and thus was directed to the evaluation in the clinical trials [Citation22]. Since then, the already optimal structure of CGM097 was not further developed. Instead, Novartis Ag has focused on patenting the use of CGM097 in combinations with the inhibitors of several additional therapeutic targets. For a more detailed description of these therapeutic strategies see section 3 of this review article.
2.3. MDMX inhibitors
In the recent years, the importance of the design of dual inhibitors of MDM2 and MDMX proteins got more and more attention. Therefore, the compounds designed for MDM2 are routinely checked also for their activity towards MDMX. Such an approach is presented in several patents released in the years 2014–2018 [Citation25,Citation26,Citation32,Citation49–Citation51]. From these, half patents concern peptidomimetics, though for macrocyclic peptides patented in 2016 by Aileron Therapeutics Inc. no data confirming the claim about the anti-MDMX activity is presented [Citation50,Citation51].
The other class of peptidomimetic molecules with MDMX-antagonizing activity were published by the Agency for Science, Technology and Research from Singapore as a continuation of SAH-p53 peptides optimization [Citation49]. The presented molecules have very good activity at low micromolar to nanomolar range, both for MDM2 and MDMX. Most often the activity towards MDMX is similar or only slightly weaker than it is for MDM2, as exemplified for the peptide (20: sMTide-05), for which the KD value measured by the T22 cell p53 β-galactosidase assay is similar for MDMX (26.3 nM), and for MDM2 (29.9 nM).
The non-peptidomimetic dual MDM2/X antagonists patented over the years 2014–2018 are based on either 3-pyrrolin-2-one [Citation32], or cis-imidazolinone [Citation25,Citation26] scaffolds. The activity of the 3-pyrrolin-2-one compounds, patented by Adamed in 2015, is rather low compared to the other potent MDM2/X antagonists. The described compounds were evaluated by TR-FRET method, and showed micro- to millimolar Ki values, with around 10–100 times stronger inhibition observed for MDM2, than for MDMX. The most optimal antagonist (20) had Ki of 3.42 mM for MDMX and 0.38 mM for MDM2 () [Citation32].
Figure 6. Structure and in vitro activity of exemplary MDMX inhibitors.
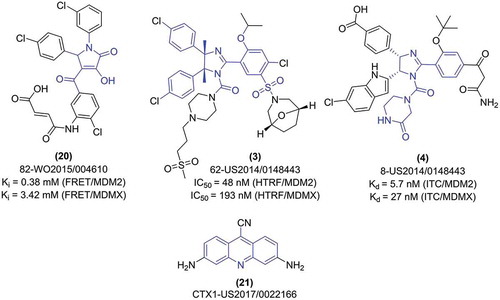
Markedly better results were presented for cis-imidazoline compounds. The patent presented in 2014 by Hoffman-La Roche describes multiple examples of compounds with the modified Nutlin-3a structure of 4,5-dimethyl-2-(3-sulfonylphenyl)-4,5-diphenyl-imidazoline-1-carboxamide derivative with the activity towards MDMX in low micromolar range [Citation25]. Likewise, for other dual antagonists, the activity towards MDM2 was significantly better than for MDMX, with the best compounds active at low nanomolar range. This is represented by one of the most active compounds (3), for which IC50 value of 4.8 nM for MDM2, and 193 nM for MDMX, as assessed by the HTRF technique, was depicted.
The second patent disclosing cis-imidazoline compounds, more specifically the substituted derivatives of 4-(2,4,5-triphenylimidazoline-1-carbonyl)piperazin-2-one, was presented by St. Jude Children’s Research Hospital in 2015 [Citation26]. The activity of these compounds was evaluated by either FP or ITC, resulting in a low micromolar range of KD for MDMX, and the values around 10 times better for MDM2 protein. As a prominent example, the compounds (4) has the KD (ITC) 5.7 nM for MDM2 and 27 nM for MDMX.
Notably, in the reviewed years there was only one patent application claiming the compounds developed specifically for MDMX [Citation52] The scaffold, disclosed by Mirx Pharmaceuticals, is based on the substituted acridine (). Changeable substituents were mostly placed at C-3, C-6 and C-9 atoms of the central heterocyclic core, with the C-3 and C-6 usually being a nitro, amino or N-acetylamino group. The most variations were placed on the C-9 position, with the versatile substituents like halogen, nitrile, carboxylic acid, sulfonyl, azide or N-aryl (e.g. phenyl, indole, thioindole, benzothiazole) amines. Most of the examples were tested on the OCI cell line, resulting in IC50 values ranging from over 15 μM to below 1 μM. From all the compounds, the compound CTX1 (21) was the most studied, including additional in vitro assays performed on several cell lines, and in vivo examination using NSG mice injected with primary human acute myeloid leukemia cells. The improved survival of tumor-bearing animals following the treatment with CTX1 may encourage the grant assignee to provide further structure optimization and progress to further pre-clinical studies.
2.4. Spiro-oxindoles
Since the initial design of spiro-oxindole compounds [Citation53], they became one of the most exploited and potent scaffolds for MDM2 antagonists, with three prominent exemplary compounds tested currently in several clinical trials. During the last years, this scaffold was extensively tested and heavily patented. Notably, all of the presented compounds possess the 6-chloro substituent in the 2-oxindole (isoindolin-1-one) core, which is consistent also with other MDM2 antagonist classes and is justified by the anchoring character of 6-chloroindole ( and ) (with the only one exception in the 2017 patent by King Saud University [Citation54]).
Figure 7. Structure and in vitro activity of exemplary MDM2 inhibitors based on spiro-oxindoles connected with 5-membered ring: pyrrolidine and pyrrolinone. The IC50 value for MI-77301 was is from [Citation96] and for MI-1061 from [Citation59].
![Figure 7. Structure and in vitro activity of exemplary MDM2 inhibitors based on spiro-oxindoles connected with 5-membered ring: pyrrolidine and pyrrolinone. The IC50 value for MI-77301 was is from [Citation96] and for MI-1061 from [Citation59].](/cms/asset/5c6173ec-eb9e-4ea1-88b3-305cd4044cc4/ietp_a_1582645_f0007_oc.jpg)
Figure 8. Structure and in vitro activity of exemplary MDM2 inhibitors based on spiro-oxindoles connected with multiple rings.
2.4.1. Spiro-oxindoles with a 5-membered ring
The closest to the original design are the compounds disclosed in the three patents from 2015. The disclosed compounds are based on the core structure of 6-chloro-spiro[indoline-3,3ʹ-pyrrolidine]-2-one (, ). The first patent, from Hudson Biopharma Inc., presents structure-activity studies focused on the position C-2ʹ and C-5ʹ of pyrrolidine ring [Citation55]. The C-5ʹ was substituted with carboxamide (similarly to the previous potent compound, MI-77301 (22) (SAR405838), derived from various anilines containing halogen and/or carboxylic or amide group. The C-2ʹ was tested with various short fluorinated aliphatic chains. In all cases at the C-4ʹ position of the pyrrolidine ring, a 2-fluoro-3-chlorophenyl group was introduced, which is characteristic for most of the examples of spiro-oxindole MDM2 antagonists. The activity of the presented compounds was assessed by TR-FRET method, with the IC50 values at low nM range, exemplified by the compound (23) with IC50 ≤ 3.6 nM.
Table 3. MDM2/X antagonists based on spiro-oxindole core structures.
The second group of compounds based on the structure of 6-chloro-spiro[indoline-3,3ʹ-pyrrolidine]-2-one (, ), was presented by the University of Michigan (The Regents of the University of Michigan) in the two 2015 patents [Citation57,Citation58]. Nine compounds showed in the patents derive from a continued optimization of the MI-1061 structure (24), which is the second generation of the spiro-oxindole derivatives with improved chiral stability of the inhibitors, acquired by replacing the short aliphatic chain in the C-2ʹ position to another spiro-structure (with cyclohexane) [Citation59]. The compounds present not only various new C-2ʹ spiro-structures (substituted cyclohexanes) but mainly are focused on replacing the C-5ʹ carboxamide, derived from 4-aminobenozic acid, to other benzoic acid bicyclic bioisosters (i.e. bicycle [Citation2,Citation2]octane-1-carboxylic acid). The tested compounds were tested for MDM2 inhibition (by the FP assay and various cell-line analysis), and chiral stability of the compound, with the final N-1ʹ-alkylated compound demonstrating complete and persistent tumor regression in mice bearing the SJSA-1 xenograft tumors (26; IC50(FP) = 3.8 nM; IC50(SJSA-1) = 70 nM).
The last patent in this category of spiro-oxindole conjunct with the 5-membraned ring was published also in 2015 by Adamed Sp. z o.o., based on the tetrahydrospiro[indole-3,2ʹpyrrole]-2,5ʹ-dione (, ) [Citation60]. The compounds present here seem to be the continuation of a previous patent disclosed by this company, presenting structures built around the 3-pyrrolin-2-one core (see secion 2.1.2). It is easily noticeable that the introduction of 6-chloro-2-oxindole in the spiro configuration to the scaffold makes the designed compounds significantly more active than the original structures. The examples shown in the patent could be subdivided into the two separate classes: one with only 3-pyrrolin-2-one (1ʹH-pyrrol-5ʹ-one), and the second with the bicyclic formation of pyrrolo[3,4-c]pyrazole-6-one or pyrrolo[3,4-c]pyrrol-3-one. Both classes have aromatic ring (usually halogenated phenyl or pyridine) substituents at the N-1ʹ position of the 3-pyrrolin-2-one ring. In the 1ʹH-pyrrol-5ʹ-one ring (numbering according to the one used in the whole tetrahydrospiro[indole-3,2ʹpyrrole]-2,5ʹ-dione structure), the C-4ʹ position is usually substituted with hydroxyl. The most prominent changes are present in the C-3ʹ position which is tested with various short aliphatic ketones, substituted phenyl attached directly or via a sulfide bridge. In the second group of compounds, the C-3ʹ and C-4ʹ substitutions were exchanged to a substituted ring (pyrazole or pyrrole). The huge diversity of substitutions placed on this additional ring was tested, most commonly with two substituents, one of which was a short aliphatic chain (i.e. isopropyl), and the second an aromatic ring. The activity of the compounds was tested in the FP assay, and in SJSA-1 cell survival study. Clearly the most potent compounds were those with the pyrrolo[3,4-c]pyrazole-6-one or pyrrolo[3,4-c]pyrrol-3-one ring, which is illustrated with the compounds (26) (one ring) and (27) (two rings), exhibiting IC50(SJSA-1) = 21.4 μM/Ki = 2.4 nM, and IC50(SJSA-1) = 0.53 μM/Ki = 4.7 nM, respectively.
2.4.2. Spiro-oxindole with a bicyclic fragment
In the years 2014–2018, four patents were published presenting bicyclic molecules attached to the 6-chloro-2-oxindole structure. One of them, describing the pyrrolo[3,4-c]pyrazole-6-one and pyrrolo[3,4-c]pyrrol-3-one rings [Citation60], was a part of the research on the tetrahydrospiro[indole-3,2ʹpyrrole]-2,5ʹ-dione scaffold and was described in section 2.4.1.
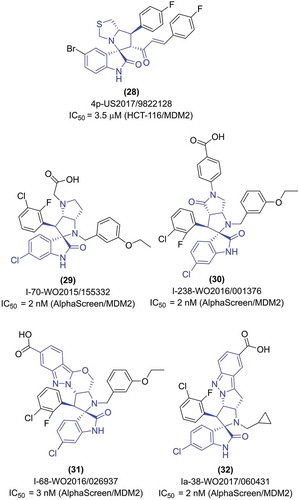
The second, relatively short patent, was disclosed in 2017 by the King Saud University [Citation54] and describes structures based on the tetrahydro-3H’-spiro[indolin-3,5ʹ-pyrrolo[1,2-c]thiazol]-2-one (). Probably, the requirements of the synthesis led to compounds symmetrically substituted with aryl component. One of the mentioned aryls was placed directly at the C-7ʹ position of the pyrrolo[1,2-c]thiazol, and the second at its C-6ʹ position via the acryloyl bridge. The suitable aryls were chosen from a short list (total number of around 20 compounds were tested), including substituted phenyl (with various halogens, trifluoromethyl, methoxyl) or heteroaryl (thiophene, furane). Interestingly, unlike in the other patents describing spiro-oxindole scaffold, in the King Saud University patent, the C-6 position of 2-oxindole was not substituted. Moreover, in the case of relatively active compound (28), the bromine atom was introduced in an unusual C-5 position of the 2-oxindole core (). All compounds were tested on numerous human cell lines, including colon cancer cells (HCT-116) and hepatocellular carcinoma cells (HepG2), with overall activity in the low micromolar range.
The biggest effort in the case of the spiro-oxindoles research was done by Boehringer Ingelheim International Gmbh, with a total of four bulk patents on this subject disclosed in the years 2014–2018. Three of them present bicyclic cores, linked with 2-oxindole core via a spiro formation (, ) [Citation61–Citation63]. The first two describe mostly the 6-chloro-hexahydro-1ʹH-spiro[indolin-3,2ʹ-pyrrolo[3,2ʹ-b]pyrrol]-2one core, though also the 6-membered analogues were disclosed (with hexahydro-pyrrolo[3,2-b]pyridine) [Citation61,Citation62]. The latter patent describes quite similar compounds [Citation63]. The main scaffold is changed by introducing another carbonyl group leaving the core of 6-chloro-hyexahydro-4ʹH-spiro[indolin-3,2ʹ-pyrrolo[3,4-b]pyrrol]2,4ʹ-dione. Significantly more examples of the 6-membered analogues (with octahydro-pyrrolo[3,2-c]pyridine-4-one) can be found throughout this patent. The substitution pattern presented in structure-relationship studies of both patents is very similar. The substitutions at the N-1ʹ and C-3ʹ position of the disclosed compounds were similar to the corresponding positions in the MI-77301 compound (N-1ʹ: usually a short aliphatic, less often a benzyl group; C-3ʹ: a halogenated phenyl, usually the 2-fluoro-3-chlorophenyl). The largest diversity was placed at C-5ʹ atom and N-4ʹ atom of a bicyclic fragment. Most commonly, different versions of carboxylic acid or its derivative (amide, ethyl ester) could be found, either of aromatic (i.e. 4-carboxyphenyl, benzyl, pyridine) or aliphatic character. Other substitutions with known bioisosters of carboxylic acid could be found (tetrazole, sulfonyl). In all Boehringer Ingelheim patents describing the MDM2 spiro-oxinodle inhibitors the AlphaScreen (Amplified Luminescent Proximity Homogeneous Assay) was used to validate compounds activity. The resulting IC50 values were starting with low micromolar for the weak compounds, going down to 2 nM for the most potent ones, as for the compounds (29) and (30).
2.4.3. Spiro-oxindole with multicyclic fragments
The multicycle-containing spiro-oxindole derivatives were disclosed solely by the Boehringer Ingelheim International Gmbh as the continuation of the work on the scaffold with bicyclic fragment (see section 2.4.2). In the two patents, the multicycle fragment attached to the 6-chloro-2-oxindole is composed of four different rings (, ) [Citation64,Citation65]. Like in the previous Boehringer Ingelheim patents, the substitutions around the first pyrrolidine ring (attached directly through a sprio formation to the 6-chloro-2-oxindole) are similar in both patents, and also similar to the general trends observed for MI-compounds (like in the case of MI-77301, see section 2.4.2). The most prominent changes here were done by the introduction of a long pattern of cumulated rings, which ends with the carboxylic group or other similar groups (amide, ester, methylsulfonyl, halogen atoms). The multicyclic formation is composed of pyrrolidine, aliphatic 5–7 membered ring (in the WO 2016/026937 A1 patent it is the tetrahydropyran or oxepane, while in the WO 2017/060431 A1 patent it is the cyclopentane and cyclohexane), a 5-membered heteroaromatic (WO 2016/026937 A1 – pyrazole; WO 2017/060431 A1 – pyrazole or imidazole), and, at the end, the substituted benzene (for the sake of clarity the aliphatic rings were named without nitrogen, which is shared between them and heteroaromatic ring). Disclosed patents described altogether a long list of potential, highly active MDM2 inhibitors, with the activity of the most potent compounds, represented by the IC50 values, reaching 2 nM in the AlphaScreen assay, as for the exemplary compounds (31) with IC50 = 3 nM, and (32) with IC50 = 2 nM.
2.5. The PROTAC technology utilizing MDM2 antagonists
Potent compounds targeting MDM2 are now utilized in experimental cancer treatment strategies not only in a classical way, using MDM2 antagonists as simple disruptors of MDM2/p53 complexes, but also in the creation of so-called PROTAC (Proteolysis Targeting Chimeric) compounds. PROTAC technology was firstly discovered in early XXI century starting a new chapter in small-molecule-driven inhibition of protein targets for therapeutic use [Citation66–Citation68]. In general, PROTAC compounds are composed of three distinguished parts: the fragment with E3 ubiquitination ligase recognition ability, a small molecule designed to selectively bind to the targeted protein, and the aliphatic linker, which couples both compounds to create a single, bi-functional compound [Citation69,Citation70]. Thanks to this bi-functionality, PROTAC compounds have the ability to recruit ubiquitinating enzyme, an E3 ligase, to the target protein by simultaneous binding of the two proteins. The recruited E3 ligase marks the targeted protein with a poly-ubiquitin chain, directing the protein to proteasomal degradation, thus eliminating the molecular target.
PROTAC technology is highly patented, licensed by Arvinas Inc. – a life-science company that originates from Yale University. In the years 2015–2018 several patents by Arvinas Inc. have been released, that utilize well-characterized compounds with a strong MDM2 binding capability in a search for therapeutic PROTAC probes ().
Figure 9. Structure and activity of exemplary compounds utilizing PROTAC technology and MDM2 E3 ligase activity.
In the initial patent, dated 2015 [Citation30], the PROTAC technology was used to attract a cereblon E3 ubiquitin ligase to a set of therapeutic targets: androgen receptor (AR), estrogen receptor (ER) α, bromodomain-containing protein 4 (BRD4), TANK-binding kinase 1 (TBK1) and c-Myc. The proteins are targeted by designing two-headed compounds composed of target-interacting molecules, a linker, and a cereblon E3 ubiquitin ligase-binding ligand. This attraction of cereblon ligase would lead to ubiquitination of protein targets and their proteasomal degradation. Besides the tested interactions, the patent covers the use of other molecular targets, including MDM2 protein, for which nutlins have been proposed as target-specific probes. However, because no bioactivity data for the nutlin-directed cereblon-mediated degradation of MDM2 is provided, the use of nutlins is somehow theoretical in this patent.
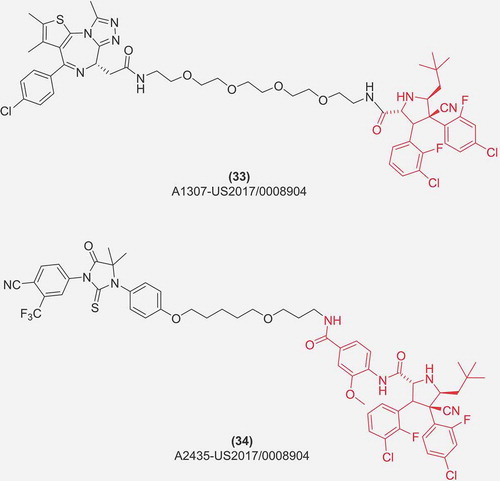
In the following five patents by Arvinas Inc., MDM2 protein is not only used as a therapeutic molecular target but also its ubiquitinating properties are employed to target additional proteins of therapeutic potential: BRD4 (33) [Citation31], Androgen Receptor (AR) (34) [Citation31], c-Jun N-terminal kinases (JNK) [Citation31], Enhancer of Zeste Homolog 2 (EZH2) [Citation31,Citation71], ER [Citation72,Citation73], or Rapidly Accelerated Fibrosarcoma (RAF) proteins [Citation74]. Noteworthy, though the MDM2 ligase compounds are mentioned and claimed in all of the listed patents, the examples of such compounds are rare. The coverage of MDM2 antagonist-based PROTAC compounds is basically only presented in the US 2017/0008904 A1 patent [Citation31]. The examples presented there are derivatives of Hoffmann-La Roche cis-imidazolines (Nutlin-3a; RG7112) and pyrrolidine (RG7388, idasanutlin), linked to the target-specific compounds. Importantly, the designed compounds have the ability of both: (i) the induction of p53-response characteristic for MDM2 antagonists, and (ii) the degradation of PROTAC-targeted proteins (BRD4, AR, JNK or EZH2), as exemplified by cell line data for the BRD4-targeted compound [Citation31]. The activity of the disclosed compounds was assessed by a suitable cell-line analysis and the results were categorized in three classes of activity, giving the best result in the category of the degradation of the targeted protein larger the 50% at 1 μM concertation. This simultaneous targeting of the two important cancer-related proteins, namely p53 and BRD4, was recently shown by the Arvinas R&D employees to give much improved results in in vitro cell line-based assays, as compared with a corresponding VHL-utilizing PROTAC with similar potency and efficacy to degrade BRD4 [Citation75].
It is worth mentioning, that the five above-mentioned patents cover the use of almost every known potent MDM2 antagonist in the PROTAC technology, living very limited space for any competitors in the field of medicinal chemistry. Recently, the spectrum of uses of PROTAC technology for addressing of the target proteins for proteasomal degradation was broaden even further with the new WO patent by Arvinas, Inc. [Citation76]. Given the promising results of the initial in vitro experiments based on the PROTAC technology, and the high attention given to disclosing any possible drug conjugates, we can expect more therapeutic uses and Arvinas patents covering MDM2 antagonists coming out in the nearest future.
2.6. Peptide derivatives
Peptide derivatives are another class of molecules, engineered to provide drug candidates for the treatment of cancers by blocking of protein-protein interactions. In a case of a MDM2-p53 system, initially p53-derived peptides were utilized in attempts to block the interaction between these two molecules. Soon it became clear, that stabilization of peptides by molecule cyclization provides improved activities due to several effects, including increased cell penetration, stability and target affinity [Citation77].
In 2014 and 2015, the Agency for Science, Technology and Research from Singapore patented a group of staple peptide-inhibitors of MDM2-p53 interaction [Citation49,Citation78]. The peptides sequences were selected based on a phage display technology. Both, the WO [Citation49] and the US [Citation78] patents cover the same bunch of compounds and present the same results to support their in vitro activity. In fact, the presented stapled peptides interact with MDM2, as exemplified by the crystal structure of MTide-1 compound bound to MDM2 protein. At higher doses, the peptides also induce the expression of p53 and its target, MDM2 protein, but this effect can be observed only in the absence of Fetal Bovine Serum. Additionally, the peptides inhibit the growth of cancer cell lines by the induction of cell cycle arrest, but the selectivity towards p53wt HCT116 cells in comparison to p53null HCT116 cells is very limited. No further optimization of the stapled peptides by the Agency for Science, Technology and Research has been patented in the next years.
In 2016, the Aileron Therapeutics Inc. company has secured the field of MDM2-antagonizing peptidomimetic macrocycles in the two world patents [Citation50,Citation51]. By combining both natural and artificial amino acids and different chemical linkers the patents covered a huge spectrum of modifications of the selected active peptide sequences. The in vitro results, exemplified for SP-154 and SP-763 compounds, show an excellent selectivity of the peptides towards p53wt cells in cell viability assay [Citation51]. Importantly, one representative of the peptidomimetic macrocycles family disclosed by Aileron Therapeutics Inc., the compound named ALRN-6924, is currently undergoing Phase 1/Phase 2 clinical evaluation for the treatment of solid tumors, lymphoma and peripheral T-cell lymphoma (ClinicalTrials.gov Identifier: NCT02264613). Besides the list of potential therapeutics, the patents of Aileron Therapeutics Inc. present dosing schemes and toxicology assessments, resulting from the Phase 1 clinical trials [Citation50,Citation51].
One last patent, presenting peptide-based probes specific for the MDM2, was released in 2018 by the Sorrento Therapeutics Inc [Citation79]. The MDM2-targeting peptide is linked via a dibenzylocyclooctyl (DBCO) click chemistry reagent and an ethyleneglycolphosphoramidite linker with a short nucleotide sequence (35) (). Such a construction of the probe is said to provide cell permeability, preserving the p53-inducing potential, as presented by the NanoBRET technology in the transfected HeLa cells [Citation79]. The patent represents an innovative approach to targeting MDM2-p53 interaction in living cells, but more in vitro and in vivo validation is required to fully judge on its therapeutic potential.
Figure 10. Exemplary peptide inhibitors of MDM2/X protein.
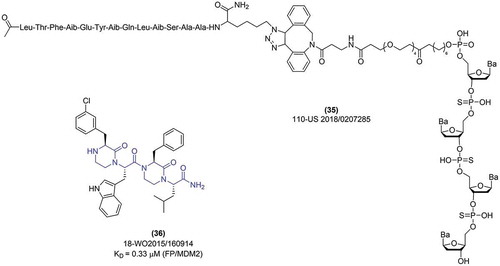
Another approach to peptidomimetic compounds was shown by the researchers from New York University [Citation80]. The patent from 2015 describes the development and characterization of oligo-oxopiperazine compounds. The structure was designed, modeled with MDM2 protein and optimized with Rosetta algorithm in order to mimic the three p53 amino acids crucial for MDM2 binding (Phe19, Trp23, Leu27). The initial screening led to the synthesis of 20 dimeric oxopiperazines – with four amino acid side chains included, and 2 trimeric oxopiperazine – with six amino acid side chains. The compounds were tested against MDM2 with the Fluorescence Polarization assay, revealing the best binding properties of (O-Me)Tyr-Trp-Phe-Leu (example 17 in [Citation80]), and 3-ClPhe-Trp-Phe-Leu (36) (example 18 in [Citation80]), with KD equal to 0.32 and 0.33 μM, respectively. The binding of the latter was additionally tested by HSQC-NMR (heteronuclear single quantum coherence) titration methodology.
2.7. Fused multicyclic compounds
Several patents described cumulated multicyclic structures with MDM2/X-binding activity. Most of them are based on the spiro-oxindole scaffolds (see Section 2.4). Another example of such compounds are acridine derivatives with moderate MDMX activity (see Section 2.3). The last one describes the beta-caboline-based compounds claimed in 2016 by Texas Tech University System [Citation81] ().
3. MDM2 antagonists in drug combinations
Following the successful development of the optimized, active and safe MDM2 antagonists, several pharmaceutical companies, such as Hoffmann-La Roche, Amgen Inc., and Novartis, have stopped further structure optimization trials of their lead compounds. Instead, the companies have progressed for the patents covering positive effects of combinations of these drugs with additional therapeutic treatments.
In 2014, Hoffmann-La Roche patented the use of nutlin-3 in a combination with CD20 antibodies for the treatment of CD20-expressing cancers [Citation82]. The use of nutlin-3 in combination with obinutuzumab (GA101, afucosylated type II anti-CD20 antibody B-HH6-B-KV1 GE) or rituximab (RTX) provided much improved in vivo antitumor activity than monotherapy [Citation82]. Each of the treatments alone resulted in only a partial control over the growth of human Z138 mantle cell lymphoma xenografts in SCID beige mice, while anti-CD20 treatments combined with 150 mg/kg nutlin-3 orally administered 3 times weekly completely blocked the growth of xenografts. A similar combination of GA101 with a new generation MDM2 antagonist RG7388 was patented by the Hoffmann-La Roche in 2016 [Citation83]. Again, the treatment of mice, bearing DOHH-2 human diffuse large B-cell lymphoma xenografts, with the combination of 30 mg/kg RG7388 (oral administration) with obinutuzumab provided an improved control over the tumor growth and much improved survival rate [Citation83,Citation84]. The pre-clinical success of the MDM2 antagonists/anti-CD20 combination strategy has encouraged Hoffmann-La Roche to proceed to the clinical trials with the treatment of Relapsed/Refractory Follicular Lymphoma patients with RG7388 and obinutuzumab, or Relapsed/Refractory Diffuse Large B-Cell Lymphoma patients with RG7388 and rituximab (ClinicalTrials.gov Identifier: NCT02624986). The study has finished the recruitment, but the results have not been posted yet.
In addition to anti-CD20 treatment, the 2016 patent Hoffmann-La Roche proposes one additional combination, targeting anti-apoptotic Bcl-2 protein with the use of venetoclax (GDC-0199) [Citation83]. Not only the combination of RG7388 with venetoclax provided improved control over the tumor growth in a mice xenograft model, but was also able to decrease the initial tumor volume suggesting a successful tumor eradication by the drug combination. Moreover, the triple combination of RG7388, venetoclax and obinutuzumab resulted in tumor eradication and 100% of animal survival over the period of 130 days of the study [Citation83]. Again, the corresponding clinical trial for the treatment of Relapsed/Refractory Acute Myeloid Leukemia patients with the combination of RG7388 and venetoclax has been launched in 2016 and is during the recruitment process (ClinicalTrials.gov Identifier: NCT02670044).
Another beneficial combination dedicated to RG7388, that was patented in 2015, was the combination with cytarabine (cytosine arabinoside, ara-C) [Citation29]. Cytarabine is an antimetabolite used in a chemotherapy of leukemia patients. The treatment of SCID-beige mice bearing MOLM-13 AML xenografts with cytarabine alone did not bring any therapeutic effects, while the treatment with an esterified derivative of RG7388 inhibited the growth of tumors. The combination of both compounds potentiated the therapeutic effect of the treatment with MDM2 antagonists, suggesting positive modulatory effects of cytarabine on the outcome of the treatment with the MDM2 antagonist [Citation29]. With time, the use of cytarabine became a favored strategy for the combined cancer treatments with MDM2 in the clinical trials. The most advanced program is the treatment of Relapsed or Refractory Acute Myeloid Leukemia with RG7388 and cytarabine, that is sponsored by RG7388 owner, Hoffmann-La Roche (ClinicalTrials.gov Identifier: NCT02545283). This treatment regime has reached phase 3 of clinical trials and is now recruiting patients for the 3.5 years overall survival study. Besides RG7388, multiple additional MDM2 antagonists are involved in completed or recruiting clinical trials in combination with cytarabine, including RG7112 (NCT01635296), MK-8242 (NCT01451437), DS-3032b (milademetan, NCT03634228) and ALRN-6924 (NCT03654716).
Another two big companies that put much effort into testing and patenting positive drug interactions of their lead MDM2 antagonists were Amgen Inc. and Novartis. First, in a single patent from 2015, Amgen Inc. has reported a broad screening of drug combinations of the lead MDM2 antagonist, AMG-232, with a panel of anticancer drugs [Citation37]. The combinations were first tested in vitro with the use of multiple different cell lines, and synergistic effects were calculated based on cell viability CellTiter-Glo (Promega) and ATPLite (Perkin Elmer) assays. Then, the selected treatments were used in in vivo tumor xenograft studies, utilizing nude mice bearing the xenografts of appropriate cancer cells. Altogether, the reported in vivo studies comprised the combinations of AMG-232 with: (a) MEK inhibitor AMG 1009089 in RKO, A375sq2 and MOLM-13 xenografts, (b) B-Raf inhibitor AMG 2112819 in RKO and A375sq2 xenografts, (c) cisplatin in H460 and HCT-116 xenografts, (d) doxorubicin in SJSA-1 and MOLM-13 xenografts, (e) topoisomerase-1 inhibitor irinotecan (CPT-11) in HCT-116 xenografts, (f) cytarabine in MOLM-13 xenografts, (g) tyrosine protein kinases inhibitor sorafenib in MOLM-13 xenografts, (h) 5-aza-2ʹ-deoxycytidine (decitabine) in MOLM-13 xenografts, (i) PI3K inhibitor AMG 2520765 in U87 xenografts, and finally (j) double and triple combinations with B-Raf inhibitor AMG 2112819, and PI3K inhibitor AMG 2539965 in RKO xenografts [Citation37]. Almost every reported drug combination provided improved anticancer effects as compared to single treatments, justifying the amenability of these drug interactions for testing in clinical trials. In fact, three clinical trials involving combined treatment with AMG-232 and MEK 1/2 inhibitor trametinib, with B-Raf inhibitor dabrafenib, or with dexamethasone are currently in the patient recruitment stage (NCT02016729, NCT02110355, and NCT03031730). In addition, dexamethasone is evaluated also in combination with RG7388 from Roche (NCT02633059), trametinib with HDM201 from Novartis (NCT03714958), and pimasertib, another MEK 1/2 inhibitor has been tested in combination with SAR405838 (NCT01985191).
In the years 2015–2018 Novartis was successively patenting a series of drug combinations of CGM097 compounds with therapeutics, targeting B-Raf protein [Citation47], tyrosine kinase (nilotinib) [Citation85], PI3K [Citation86], MEK [Citation87], Bcl-2 [Citation86,Citation87], PKC [Citation88], and Bcl-xL [Citation89]. From this list, synergistic effects of CGM097 with B-Raf inhibitor, MEK inhibitor, Bcl-2 inhibitor, and PKC inhibitor have been verified in in vivo mice models, where combined treatments presented much improved anticancer effects than monotherapies [Citation47,Citation87,Citation88]. For the other combinations, only cell-based in vitro assays were performed and the in vivo outcomes remain to be evaluated.
In a single patent by the University of the Science of Philadelphia, a unique protein entity is presented, that provides simultaneous modulation of the activity of several cancer-related targets. The protein is composed of the MDM2-binding peptide, linked via a caspase cleavage sequence with either a whole human serum albumin or its fragments, that retain the fatty acid (FA)-binding capability [Citation90]. The patent assignee claims that such a protein provides good transport of MDM2-antagonizing peptide to the cell, and can be used for co-transportation of additional, FA-linked drugs, such as MTX, for combined anticancer treatments [Citation90]. Additionally, the patent reports unexpected interaction and blockade of two additional targets, Bcl-xL and MCL, by the pure protein entity, without loading of FA-linked drugs. This observation, although very interesting, requires additional experimental verification, as for now the mechanism explaining this surprising action remains elusive.
The recent patent by Daiichi Sankyo Ltd. covers the use of their DS-3032 (milademetan) in combination with DNA methyltransferase inhibitor (azacitidine) [Citation91]. The drug combination has been tested in in vivo model using NOD-SCID mice implanted with MOLM-13 cells. The combination of 50 mg/kg of DS-3032 and 2.5 or 4 mg/kg of azacitidine provided 90% tumor growth inhibition, compared to untreated control, while single therapies allowed for 28% and 72% of growth inhibition for azacytidine and DS-3032, respectively [Citation91]. Along with the Daiichi Sankyo Inc. patent the combination of DS-3032 with azacitidine is currently undergoing clinical investigation in acute myelogenous leukemia and high-risk myelodysplastic syndrome patients, sponsored by Daiichi Sankyo Inc. (NCT02319369).
4. Expert opinion
Since the discovery of the nutlins, first-in-class bioactive MDM2 antagonists, the structure optimization of imidazolines and the discovery of additional chemical scaffolds led to the development of extremely strong MDM2-binding and selective second and third generation antagonists. These optimized compounds are able to reactivate the wild-type p53 by antagonizing MDM2 and/or MDMX proteins, to selectively inhibit the growth of the p53 wild-type cells and provide good control over the growth of p53wt tumors in animal models. Importantly, in recent years, several representative compounds have successfully passed through pre-clinical studies and entered the clinical trials (reviewed in [Citation22], see also –). These include the nutlin-3a derivative RG7112 [Citation92] and its pyrrolidine relative, RG7388 (idasanutlin) from Roche [Citation27], the piperidinone compound AMG-232 from Amgen [Citation93,Citation94], the bicyclic-core compounds CGM097 and HDM201 from Novartis [Citation48,Citation95], three spiroindolinone derivative compounds, SAR405838 from Sanofi [Citation96], APG-115 from Ascentage Pharma Group Inc [Citation97]., DS-3032b from Daiichi Sankyo [Citation98], a multicyclic-core compound BI 907828 from Boehringer Ingelheim [Citation99], and a stapled peptide ALRN-6924 from Aileron Therapeutics [Citation100]. Among these, RG7388 (idasanutlin, RO5503781) is now most advanced, as it has recently progressed to the phase III clinical trials in combination with cytarabine in patients with relapsed or refractory acute myeloid leukemia [Citation22].
Additionally, new classes of MDM2-antagonizing molecules appear sporadically, employing both new chemistry and new biochemical mechanisms of action. These new classes include peptidomimetic compounds, such as stapled peptides, and PROTAC compounds. The latter strategy, utilizing bi-specific probes composed of MDM2 antagonists and the compounds specific to additional, cancer-related molecular targets, provides very promising pre-clinical results and may contribute much to the development of future anticancer therapies.
It has been initially believed, that the reactivation of p53 by antagonizing MDM2 protein will alone be sufficient to eradicate the p53wt cancers. However, in the past decade, the growing evidence has undermined this hypothesis. While antagonists of MDM2 provide good control over the proliferation of multiple p53wt cancer cell lines both in vitro and in vivo, it is now clear that even more than 50% of p53wt cancer cells fail to comply with this rule [Citation101]. Additionally, only in a subset of p53wt cells that respond to MDM2 antagonists, the apoptosis program is induced, allowing for the successful cancer cell elimination [Citation6,Citation102,Citation103]. Moreover, the induction of cell quiescence with limited cell elimination by apoptosis leads to the selection of drug-resistant clones, as first shown for nutlin-3 [Citation104–Citation106], and then for the more recent, clinically-evaluated MDM2 antagonists, such as SAR405838 [Citation107,Citation108], HDM201 [Citation109] and RG7388 [Citation110]. For this reasons, there is now a clear trend for the design of drug combination studies that would involve MDM2 antagonists. Such an approach gives a chance to take an advantage of the growth-inhibitory potential of the MDM2 antagonists against p53wt cells and provide additional anti-cancer effects by targeting additional molecular targets, such as CD20, MEK 1/2, B-Raf, TK, PI3K, Bcl-2 or Bcl-xL, or by adding standard chemotherapy agents, such as doxorubicin cytarabine or 5-aza-2ʹ-deoxycytidine. Around 30% of the therapeutic patents on MDM2 antagonists that were disclosed in the years 2014–2018 cover at least one drug combination strategy for improved treatment of human cancers. These combinations are provided by combining two chemical entities in the dosing scheme, or by the chemical conjugation of the two entities, which results in bi-functional combinatorial drugs. It seems clear, that now, when several excellent MDM2-antagonizing, orally available and relatively safe compounds have been developed, the discovery of positive drug interactions will proceed and will result in new patents on the treatment regimens for anticancer therapy.
Article highlights
The liberation of p53 activity with MDM2 antagonists provides a non-genotoxic strategy for the treatment of p53-wild type tumors.
The progress in the development of MDM2 antagonists patented between 2014 and 2018 is reviewed.
The optimization of multiple chemical scaffolds has led to the discovery of low picomolar MDM2 antagonists.
Several MDM2 antagonists are currently being evaluated in clinical trials.
The limited activity of MDM2 antagonists justifies the need for the development of synergistic combined therapies.
New PROTAC technology has been harnessed to target MDM2 protein for degradation, or to utilize its ubiquitinating properties for the elimination of other protein targets.
This box summarizes key points contained in the article.
Declaration of interest
L. Skalniak has received Sonata Grant No. UMO-2016/21/D/NZ7/00596 from the National Science Centre, Poland. T.A. Holak has received Symphony Grant No. UMO-2014/12/W/NZ1/00457 from the National Science Centre, Poland. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Additional information
Funding
References
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
- Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217–236.
- Brady CA, Attardi LD. p53 at a glance. J Cell Sci. 2010;123:2527–2532.
- Zhang X-P, Liu F, Wang W. Two-phase dynamics of p53 in the DNA damage response. Proc Natl Acad Sci. 2011;108:8990–8995.
- Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12:303–312.
- París R, Henry RE, Stephens SJ, et al. Multiple p53-independent gene silencing mechanisms define the cellular response to p53 activation. Cell Cycle. 2008;7:2427–2433.
- Wade M, Li Y-C, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96.
- Toledo F, Wahl GM. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int J Biochem Cell Biol. 2007;39:1476–1482.
- Khoury K, Popowicz GM, Holak TA, et al. The p53-MDM2/MDMX axis - A chemotype perspective. Medchemcomm. 2011;2:246–260.
- Marine J-C, Francoz S, Maetens M, et al. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006;13:927–934.
- Karni-Schmidt O, Lokshin M, Prives C. The roles of MDM2 and MDMX in cancer. Annu Rev Pathol Mech Dis. 2016;11:617–644.
- Weber L. Patented inhibitors of p53-Mdm2 interaction (2006–2008). Expert Opin Ther Pat. 2010;20:179–191.
- Kamal A, Mohammed AA, Shaik TB. p53-Mdm2 inhibitors: patent review (2009–2010). Expert Opin Ther Pat. 2012;22:95–105.
- Zak K, Pecak A, Rys B, et al. Mdm2 and MdmX inhibitors for the treatment of cancer: a patent review (2011-present). Expert Opin Ther Pat. 2013;23:425–448.
- Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993;13:4107–4114.
- Böttger A, Böttger V, Garcia-Echeverria C, et al. Molecular characterization of the hdm2-p53 interaction. J Mol Biol. 1997;269:744–756.
- Popowicz GM, Dömling A, Holak TA. The structure-based design of Mdm2/Mdmx-p53 inhibitors gets serious. Angew Chem Int Ed Engl. 2011;50:2680–2688.
- Estrada-Ortiz N, Neochoritis CG, Dömling A. How to design a successful p53-MDM2/X interaction inhibitor: a thorough overview based on crystal structures. ChemMedChem. 2016;11:757–772.
- Graves B, Thompson T, Xia M, et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc Natl Acad Sci U S A. 2012;109:11788–11793.
- Chang YS, Graves B, Guerlavais V, et al. Stapled α-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A. 2013;110:E3445–E3454.
- Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848.
- Tisato V, Voltan R, Gonelli A, et al. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol. 2017;10:133.
- Ray-Coquard I, Blay J-Y, Italiano A, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13:1133–1140.
- Vu B, Wovkulich P, Pizzolato G, et al. Discovery of RG7112: A small-molecule MDM2 Inhibitor in clinical development. ACS Med Chem Lett. 2013;4:466–469.
- Chu X-J, Lovey AJ, Thanh VB, et al., inventor; Hoffmann-La Roche Inc., assignee. Novel Imidazolines as dual inhibitors of MDM2 and MDMX. patent US 2014/0148443 A1.
- Guy RK, inventor; St. Jude Children’s Research Hospital, assignee. Aryl-substituted imidazoles and methods of making and using same. patent WO 2015/184383 A1.
- Ding Q, Zhang Z, Liu -J-J, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013;56:5979–5983.
- Zhang Z, Chu X-J, Liu -J-J, et al. Discovery of potent and orally active p53-MDM2 inhibitors RO5353 and RO2468 for potential clinical development. ACS Med Chem Lett. 2014;5:124–127.
- Higgins B, Nichols G, Packman K, inventor; Hoffmann-La Roche Inc., assignee. Novel combination treatment for acute myeloid leukemia (AML). patent US 2015/0157603 A1.
- Crew AP, Crews C, Dong H, et al., inventor; Arvinas Inc., assignee. Imide-based modulators of proteolysis and associated methods of use. patent US 2015/0291562 A1.
- Crew AP, Crews CM, Dong H, et al., inventor; Arvinas, Inc., assignee. MDM2-based modulators of proteolysis and associated methods of use. patent US 2017/0008904 A1.
- Feder M, Dubin G, Bulkowska U, et al., inventor; Adamed Sp. z o.o., assignee. 1,5-dihydropyrrol-2-one derivatives as inhibitors of p53-MDM2/MDM4 protein-protein interaction. patent WO 2015/004610 A1.
- Rew Y, inventor; Amgen Inc., assignee. Benzoic acid derivative MDM2 inhibitor for the treatment of cancer. patent US 2014/0243372 A1.
- Bartberger MD, Beck HP, Degraffenreid MR, et al., inventor; Amgen Inc., assignee. Cis-morpholinone and other compounds as MDM2 inhibitors for the treatment of cancer. patent WO 2014/130470 A1.
- Gonzalez Buenrostro A, Li Y, Medina JC, et al., inventor; Amgen Inc., assignee. Heteroaryl acid morpholinone compounds as MDM2 inhibitors for the treatment of cancer. patent WO 2014/151863 A1.
- Bio M, Caille S, Cochran B, et al., inventor; Amgen Inc., assignee. Processes of making and crystalline forms of a MDM2 inhibitor. patent US 2014/0364455 A1.
- Caenepeel S, Canon J, Hughes P, et al., inventor; Amgen Inc., assignee. Combination therapy including an MDM2 inhibitor and one or more additional pharmaceutically active agents for the treatment of cancers. patent WO 2015/070224 A2.
- Ferretti S, Guerreiro N, Jeay S, et al., inventor; Novartis Ag, assignee. Dose and regimen for HDM2-p53 interaction inhibitors. patent WO 2018/092020 A1.
- Ferretti S, Guerreiro N, Jeay S, et al., inventor; Novartis Ag, assignee. Dose and regimen for an HDM2-p53 interaction inhibitor in hematological tumors. patent WO 2018/178925 A1.
- Zhang R, Wang W, inventor; Texas Tech University System, assignee. Cancer treatment utilizing SP-141 to bind with MDM2 and act as an inhibitor of MDM2 expression. patent US 2017/0283413 A1.
- Dinsmore C, Fradera Llinas FX, Kudale AA, et al., inventor; Merck Sharp & Dohme Corp., assignee. Substituted pyrrolopyrimidines as HDM2 inhibitors. patent WO 2014/100071 A2.
- Cammarano CM, Christopher MP, Dinsmore C, et al., inventor; Merck Sharp & Dohme Corp., assignee. 2,6,7,8 substituted purines as HDM2 inhibitors. patent WO 2014/120748.
- Christopher MP, Fradera Llinas FX, Machacek M, et al., inventor; Merck Sharp & Dohme Corp., assignee. 2,6,7 substituted purines as HDM2 inhibitors. patent WO 2014/123882 A1.
- Chessari G, Howard S, Buck IM, et al., inventor; Astex Therapeutics Limited, assignee. Isoindolinone inhibitors of the MDM2-p53 interaction having anticancer activity. patent WO 2017/055859 A1.
- Chessari G, Howard S, Buck IM, et al., inventor; Astex Therapeutics Limited, assignee. Isoindolinone inhibitors of the MDM2-p53 interaction having anticancer activity. patent WO 2017/055860 A1.
- Zhang W, Miao Z, Zhuang C, et al., inventor; Second Military Medical University, assignee. Pyrrolidonopyrazole compounds and use thereof as drugs. patent WO 2014/134968 A1.
- Emerson E, Halilovic E, Wang H-Q, et al., inventor; Novartis Ag, assignee. Combination of MDM2 inhibitor and BRAF inhibitor and their use. patent WO 2015/084804 A1.
- Holzer P, Masuya K, Furet P, et al. Discovery of a dihydroisoquinolinone derivative (NVP-CGM097): A highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J Med Chem. 2015;58:6348–6358.
- Joseph TL, Verma CS, Lane DP, et al., inventor; Agency for Science, Technology and Research, assignee. Peptides and methods for treating cancer. patent WO 2014/055039 A1.
- Annis DA, Darlak K, Rhodes C, et al., inventor; Aileron Therapeutics Inc., assignee. Peptidomimetic macrocycles and formulations thereof. patent WO 2016/049355 A1.
- Chen H, Annis DA, Chang Y, et al., inventor; Aileron Therapeutics Inc., assignee. Peptidomimetic macrocycles and uses thereof. patent WO 2016/049359 A1.
- Wald D, Agarwal M, Xia Z, et al., inventor; MiRx Pharmaceuticals, LLC, assignee. HDMX inhibitors and their use for cancer treatment. patent US 2017/0022166 A1.
- Ding K, Lu Y, Nikolovska-Coleska Z, et al. Structure-based design of potent non-peptide MDM2 inhibitors. J Am Chem Soc. 2005;127:10130–10131.
- Barakat A, Islam MS, Al-Majid AM, et al., inventor; King Saud University, assignee. Substituted spirooxindoles. patent US 9822128 B1.
- Chen Y, Ding Q (Jack), Sun Y, inventor; Hudson Biopharma Inc., assignee. Spiropyrrolidines as MDM2 inhibitors. patent US 2015/0322076 A1.
- Debussche L, Nicolas J-P, Rowley S, et al., inventor; Sanofi, assignee. Compositions and methods using hdm2 antagonist and mek inhibitor. patent EP 2 752 191 A1.
- Wang S, Aguilar A, Liu L, et al., inventor; The Regents of the Universityy of Michigan, assignee. MDM2 inhibitors and therapeutic methods using the same. patent WO 2015/161032 A1.
- Wang S, Aguilar A, Liu L, et al., inventor; The Regents of the Universityy of Michigan, assignee. MDM2 inhibitors and therapeutic methods using the same. patent US 2015/0299211 A1.
- Aguilar A, Sun W, Liu L, et al. Design of chemically stable, potent, and efficacious MDM2 inhibitors that exploit the retro-mannich ring-opening-cyclization reaction mechanism in spiro-oxindoles. J Med Chem. 2014;57:10486–10496.
- Feder M, Kalinowska I, Jaszczewska JA, et al., inventor; Adamed Sp. z o.o., assignee. Compounds comprising 1,1ʹ,2,5ʹ-tetrahydrospiro[indole-3,2ʹ-pyrrole]-2,5ʹ-dione system as inhibitors p53-MDM2 protein-protein interaction. patent WO 2015/189799 A1.
- Gollner A, Kofink C, Ramharter J, et al., inventor; Boehringer Ingelheim International GmbH, assignee. Spiro[3H-indole-3,2′-pyrrolidin]-2(1H)-one compounds and derivatives as MDM2-P53 inhibitors. patent US 2015/0291611 A1.
- Gollner A, Kofink C, Ramharter J, et al., inventor; Boehringer Ingelheim International GmbH, assignee. Spiro[3H-Indole-3,2ʹ-Pyrrolidin]-2(1H)-one derivatives and their use as Mdm2-P53 inhibitors. patent WO 2015/155332 A1.
- Weinstabl H, Gollner A, Ramharter J, et al., inventor; Boehringer Ingelheim International GmbH, assignee. New spiro[3h-indole-3,2´-pyrrolidin]-2(1h)-one compounds and derivatives as MDM2-p53 inhibitors. patent WO 2016/001376 A1.
- Ramharter J, Broeker J, Gille A, et al., inventor; Boehringer Ingelheim International GmbH, assignee. New spiro[3h-indole-3,2´-pyrrolidin]-2(1h)-one compounds and derivatives as MDM2-p53 inhibitors. patent WO 2016/026937 A1.
- Gollner A, Broeker J, Kerres N, et al., inventor; Boehringer Ingelheim International GmbH, assignee. Spiro[3h-indole-3,2´-pyrrolidin]-2(1h)-one compounds and derivatives as MDM2-p53 inhibitors. patent WO 2017/060431 A1.
- Cyrus K, Wehenkel M, Choi E-Y, et al. Two-headed PROTAC: an effective new tool for targeted protein degradation. Chembiochem. 2010;11:1531–1534.
- Sakamoto KM, Kim KB, Verma R, et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003;2:1350–1358.
- Zhang D, Baek SH, Ho A, et al. Degradation of target protein in living cells by small-molecule proteolysis inducer. Bioorg Med Chem Lett. 2004;14:645–648.
- Churcher I. Protac-induced protein degradation in drug discovery: breaking the rules or just making new ones? J Med Chem. 2018;61:444–452.
- Collins I, Wang H, Caldwell JJ, et al. Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochem J. 2017;474:1127–1147.
- Crew AP, Snyder LB, Wang J, et al., inventor; Arvinas, Inc., assignee. Compounds and methods for the targeted degradation of enhancer of zeste homolog 2 polypeptide. patent US 2018/0177750 A1.
- Crew AP, Qian Y, Dong H, et al., inventor; Arvinas, Inc., assignee. Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degraders. patent US 2018/0155322 A1.
- Qian Y, Crew AP, Crews CM, et al., inventor; Arvinas, Inc., assignee. Modulators of estrogen receptor proteolysis and associated methods of use. patent WO 2018/140809 A1.
- Crew AP, Hornberger KR, Wang J, et al., inventor; Arvinas, Inc., assignee. Compounds and methods for the targeted degradation of rapidly accelerated fibrosarcoma polypeptides. patent US 2018/0179183 A1.
- Hines J, Lartigue S, Dong H, et al. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 2019;79:251–262.
- Crew AP, Dong H, Wang J, et al., inventor; Arvinas, Inc., assignee. Modulators of proteolysis and associated methods of use. patent WO 2018/226542 A1.
- Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012;503:3–33.
- Joseph TL, Verma CS, Lane DP, et al., inventor; Agency for Science, Technology and Research, assignee. Peptides and methods for treating cancer. patent US 2015/0246946 A1.
- Fu Y, Kaufmann GF, inventor; Sorrento Therapeutics, Inc., assignee. Cell penetrating peptide inhibitors of p53-MDM2 interaction. patent US 2018/0207285 A1.
- Arora PS, Lao BB, Guarracino D, et al., inventor; New York University, assignee. Oxopiperazine helix mimetics as inhibitors of the p53-MDM2 interaction. patent WO 2015/160914 A1.
- Zhang R, Wang W, inventor; Texas Tech University System, assignee. Cancer treatment utilizing SP-141 to bind with MDM2 and act as an inhibitor of MDM2 expression. patent WO 2016/049453 A1.
- Eric E, Frank H, Christian K, et al., inventor; Roche Glycart AG, assignee. Combination therapy of an afucosylated CD20 antibody with a MDM2 inhibitor. patent US 2014/0140988 A1.
- Klein C, Herting F, Dangl M, inventor; Hoffmann-La Roche Inc., assignee. Combination therapy of an anti CD20 antibody with a BCL-2 inhibitor and a MDM2 inhibitor. patent WO 2016/188935 A1.
- Herting F, Herter S, Friess T, et al. Antitumour activity of the glycoengineered type II anti-CD20 antibody obinutuzumab (GA101) in combination with the MDM2-selective antagonist idasanutlin (RG7388). Eur J Haematol. 2016;97:461–470.
- Bhatia R, inventor; Novartis Ag, assignee. Pharmaceutical combinations and their use. patent WO 2016/035023 A1.
- Caponigro G, Horn-Spirohn T, Lehar J, inventor; Novartis Ag, assignee. Combination therapy using PI3K inhbitor and MDM2 inhibitor. patent WO 2017/037586 A1.
- Halilovic E, Caponigro G, Horn-Spirohn T, et al., inventor; Novartis Ag, assignee. MDM2 inhibitors and combinations thereof. patent WO 2017/037579 A1.
- Halilovic E, Emery C, inventor; Novartis Ag, assignee. MDM2 inhibitors for treating uveal melanoma. patent WO 2017/029588 A2.
- Chapeau E, Durand E, Gembarska A, et al., inventor; Novartis Ag, assignee. Combinations of MDM2 inhibitors and BCL-XL inhibitors. patent WO 2018/092064 A1.
- Li Z, Parker M, inventor; University of the Sciences of Philadelphia, assignee. Compositions comprising serum albumin and p53 peptides fusion proteins. patent US 2016/0145314 A1.
- Seki T, inventor; Daiichi Sankyo Company, Limited, assignee. Combination therapy method using MDM2 inhibitor and DNA methyltransferase inhibitor. patent WO 2018/074387 A1.
- Tovar C, Graves B, Packman K, et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013;73:2587–2597.
- Gonzalez AZ, Eksterowicz J, Bartberger MD, et al. Selective and potent morpholinone inhibitors of the MDM2-p53 protein-protein interaction. J Med Chem. 2014;57:2472–2488.
- Sun D, Li Z, Rew Y, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem. 2014;57:1454–1472.
- Furet P, Masuya K, Kallen J, et al. Discovery of a novel class of highly potent inhibitors of the p53-MDM2 interaction by structure-based design starting from a conformational argument. Bioorg Med Chem Lett. 2016;26:4837–4841.
- Wang S, Sun W, Zhao Y, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014;74:5855–5865.
- Yi H, Yan X, Luo Q, et al. A novel small molecule inhibitor of MDM2-p53 (APG-115) enhances radiosensitivity of gastric adenocarcinoma. J Exp Clin Cancer Res. 2018;37:97.
- Arnhold V, Schmelz K, Proba J, et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget. 2018;9:2304–2319.
- Rinnenthal J, Rudolph D, Blake S, et al. Abstract 4865: BI 907828: A highly potent MDM2 inhibitor with low human dose estimation, designed for high-dose intermittent schedules in the clinic. Cancer Res. 2018;78:4865.
- Carvajal LA, Neriah DB, Senecal A, et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Transl Med. 2018;10:eaao3003.
- Jeay S, Gaulis S, Ferretti S, et al. A distinct p53 target gene set predicts for response to the selective p53-HDM2 inhibitor NVP-CGM097. Elife. 2015;4:1–23.
- Tovar C, Rosinski J, Filipovic Z, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A. 2006;103:1888–1893.
- Tsao CC, Corn PG. MDM-2 antagonists induce p53-dependent cell cycle arrest but not cell death in renal cancer cell lines. Cancer Biol Ther. 2010;10:1315–1325.
- Michaelis M, Rothweiler F, Barth S, et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011;2:e243.
- Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator nutlin-3. Oncogene. 2011;30:4678–4686.
- Wei SJ, Joseph T, Sim AYL, et al. In vitro selection of mutant HDM2 resistant to nutlin inhibition. PLoS One. 2013;8:e62564.
- Hoffman-Luca CG, Ziazadeh D, McEachern D, et al. Elucidation of acquired resistance to Bcl-2 and MDM2 inhibitors in acute leukemia in vitro and in vivo. Clin Cancer Res. 2015;21:2558–2568.
- Jung J, Lee JS, Dickson MA, et al. TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat Commun. 2016;7:12609.
- Chapeau EA, Gembarska A, Durand EY, et al. Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf-/- mouse model. Proc Natl Acad Sci U S A. 2017;114:3151–3156.
- Skalniak L, Kocik J, Polak J, et al. Prolonged idasanutlin (RG7388) treatment leads to the generation of p53-mutated cells. Cancers (Basel). 2018;10:1–17.