
ABSTRACT
Introduction: Expanded access is the use of an investigational product by patients with serious medical conditions without participation in a clinical trial. It is a complicated process involving the collaboration of many parties and pharmaceutical companies. Ongoing efforts focus on accelerating expanded access procedures in the best interest of patients with cancer.
Areas covered: We review the regulatory and ethical challenges encountered in efforts to optimize expanded access.
Expert opinion: In the era of personalized medicine, patients may benefit from novel therapeutic agents that demonstrate encouraging results in early studies. However, drug approval is a lengthy and cumbersome procedure that might exceed the time frame of a life-threatening disease. Expanded access provides options to patients with unmet needs. It may provide informative safety and efficacy data to the manufacturers and the scientific and regulatory organizations.
Ongoing efforts are being made by global governmental and scientific committees, regulatory agencies, and patient organizations to address the ethical and regulatory issues and to optimize the expanded access process. Their goal is to expand access to promising novel drugs for individual patients and to accelerate the necessary procedures while preserving patient safety.
1. Introduction
Expanded access, also known as compassionate use, is the use of an investigational product by a patient with a serious and life-threatening condition without participating in a clinical trial [Citation1]. Expanded access is different than the off-label use of a drug [Citation2]. Off-label use involves the prescription of an approved drug for a condition other than that indicated in the formal approval, whereas expanded access allows for the use of a drug that has not yet been granted formal approval. Before a patient is treated under the expanded access mechanism, all alternative therapies should be exhausted, the drug should have demonstrated benefits that outweigh its potential risks, and it must not compromise its development by the sponsor [Citation3]. Thus, expanded access is a complicated process that involves the pharmaceutical companies, treating physicians, agencies responsible for approving the drug, and Institutional Review Boards (IRBs).
In 1938, the Federal Food, Drug, and Cosmetics Act required that all drugs be tested for safety before being approved by the US Food and Drug Administration (FDA) [Citation4]. In 1962, the Kefauver–Harris amendments added that potential drugs should be proven effective in addition to safe in order to be approved and added strict controls on how to use investigational drugs [Citation5,Citation6]. Although these regulations are necessary, the process for investigational drug approval by the FDA is complicated, expensive, cumbersome, and lengthy [Citation7,Citation8]. Between 1995 and 2008, the median time from an Investigational New Drug application to FDA approval was approximately 7 years [Citation7,Citation9]. The average time for approval by the regulatory authorities in Europe (European Medicines Agency, EMA) and Canada (Health Canada) was even longer [Citation10,Citation11]. During the AIDS epidemic in the 1980s, there was a need to accelerate approval of HIV drugs, which led in 1987 to the institutionalization of expanded access programs [Citation12,Citation13].
The FDA in the United States, the EMA, the Pharmaceutical Benefits Advisory Committee in Australia and Health Canada are some of the agencies formed to regulate access to drugs based on their safety and patient outcomes. Their goal is to promote and protect human health; however, there are differences between these agencies in procedures, timelines, and drug approvals [Citation14]. Criticism from the public refers to delays in their decisions, stringency on their approval criteria, firmness on the industry, and frequent denial of access to novel drugs. Some agencies work faster than others and are more lenient on their decisions [Citation10]. Every year, the FDA authorizes access to 99% of the 1000 applications received yearly through expanded access [Citation15]. The major limiting factor is the time required for the approval. It is vital that the regulatory agencies evaluate possible drug adverse events, before granting approval to that drug, in order to ensure patient safety. However, most of the decisions have to be based on limited safety data available for the investigational drugs.
Only a small proportion of patients gain access to novel experimental agents through participation in clinical studies. It is reported that of the novel agents entering clinical investigation with an Investigational New Drug application, only 3–25% are granted regulatory approval [Citation16,Citation17]. The current drug approval rate appears to be significantly lower. Expanded access is expected to make investigational drugs available to selected patients, thereby increasing the treatment options of the sickest patients.
This review focuses on the regulatory and ethical challenges encountered in offering expanded access treatments to patients and efforts to optimize the process.
2. Expanded access: types and regulation
Currently, expanded access approval is granted for three types of use: (1) individual use, the most common category, in which access is granted for a single person with a serious disease and no viable alternative option; (2) limited use, in which access is granted for an intermediate-size patient population not exceeding 1000; and (3) widespread use, in which access is granted to a large patient population on the basis of a successful clinical trial result, since the drug has not yet been approved for public access.
The regulations governing expanded access are based on various criteria and are intended to (1) ensure the safety of patients who are vulnerable because they have a serious illness that lacks valid treatment options, (2) provide faster access to potentially beneficial treatments, and (3) facilitate the clinical development and acquisition of scientific data related to the drug.
In the initial expanded access regulations (1987), the mandatory requirements for all categories of use included that the FDA should determine that the disease is serious and life-threatening to justify the risk of taking an investigational drug. Additionally, there should be no available reasonable (similar or satisfactory) alternative therapeutic option. The FDA should also determine that expanded access to the drug will not affect the initiation, conduction, or completion of clinical trials of the drug. Additional specific requirements are included for each type of expanded access use [Citation13]. For approval of expanded access for individual use, the physician must provide evidence demonstrating that the benefit of the investigational drug is greater than the associated risk, and the FDA must ensure that the patient cannot access the drug in limited or widespread use of expanded access programs or the clinical trial setting. The duration of treatment should be specified. For limited use, the manufacturer should explain why a clinical trial is not an option for these patients. For widespread use, the FDA should determine whether there are adequate safety and effectiveness data (in controlled clinical trials) and that the manufacturer is working on marketing final approval. The investigators and the manufacturer have the obligation to provide accurate safety and efficacy data to the responsible authorities in a timely matter.
From 2005 to 2014, 10,939 requests for expanded access to investigational new drugs were submitted to the Center of Drug Evaluation and Research. The number of these requests increased over time [Citation18]. For instance, the requests received in 2014 increased by twofold compared to those received in 2005. The vast majority of the expanded access requests were for single patient use and included emergency (n = 5511) and non-emergency (n = 5284) applications. Cancer represented approximately a quarter of the diagnoses. Overall, 99.7% of the submitted requests for expanded access were approved to proceed. Of 1033 investigational drugs used for expanded access, serious adverse events, eventually leading to death, were reported in 2 (0.2%) patients who were treated with 2 investigational drugs. Two clinical trials associated with these drugs (one each) were placed on clinical hold. In one trial, hold actions were removed 2 months later, after specific protocol modifications to mitigate risks. In the other trial, patients with similar characteristics were excluded from future enrollment. The development of both investigational drugs continued [Citation18].
3. Benefits for patients and industry
The main benefit of expanded access for patients is the opportunity to receive potentially beneficial investigational treatments. For example, emerging data support the use of targeted treatments for patients whose tumors harbor specific alteration(s) [Citation19,Citation20]. We have demonstrated that matched therapy against specific tumor molecular aberrations is associated with higher rates of response, progression-free survival, and overall survival compared to non-matched therapy [Citation21–Citation23]. However, access to these investigational treatments has been limited. Thus, expanded access programs are critical for patients with rare tumors or alterations for whom the evidence for the benefit of a drug is anecdotal, as well as for patients who have exhausted approved available treatments. Additionally, expanded access may bridge the time gap between the developmental phase and the final approval of a drug.
Data from expanded access programs demonstrating treatment benefit in such patients may lead to increased expanded use of the drug. Efficacy and safety data from patients treated under expanded access programs may benefit the manufacturer by accelerating approval of the drug or treatment.
4. Challenges of expanded access
Patients who have a life-threatening disease are particularly vulnerable because they have few treatment options and limited data to make an informed decision. Most investigational drugs fail to receive FDA approval. According to FDA data, at least one-third of phase I drugs are withdrawn for safety concerns, and only about one-fifth are approved [Citation24]. The cost–benefit risk ratio associated with expanded access of a drug is usually unknown. Additionally, patients are sometimes responsible for paying for their expanded access treatment out of pocket, because the manufacturer is not obligated to provide the drugs at no cost, and Medicare and most of the private insurers only cover ‘reasonable and necessary’ treatments [Citation25].
Another challenge of expanded access use is the time and effort that the treating physician must dedicate to this process. Physicians must be willing to be actively involved in the expanded access process and must be familiar with the investigational drug [Citation26]. In general, physicians infrequently request expanded access use of a treatment because of the complexity of the process. The physician faces the pressure of helping the patient make the decision to receive an investigational treatment, frequently without having adequate data. This is a time-consuming process, and the physician is uncompensated for this work. Legal issues are also of concern, since there are no clear boundaries regarding whether the physician will be held responsible in the case of a fatal adverse event and whether the patient’s informed consent will be considered adequate. Finally, the physician has to obtain approval from an IRB. The IRB review can be time consuming, since these submissions are often evaluated by people who are not familiar with expanded access programs. Notably, physicians who work at institutions without an IRB must address their request to an independent IRB, thus increasing the costs.
The treating physician is also responsible for obtaining the manufacturer’s agreement to provide the experimental drug. Drug manufacturers play a key role in expanded access programs and do not always agree to provide the drug. The major concern is that access to an investigational treatment might interfere with its FDA approval. Patients requesting access to a drug under development are usually frailer, given the advanced stage of their disease and the multiple lines of prior therapy. Therefore, they are at high risk of toxicity. Once reported to the FDA, these events might ultimately negatively affect the approval process [Citation27]. Additionally, participation in clinical trials might be jeopardized if a drug is easily accessible through an expanded access program. Such interference with phase II/III clinical trials might lead to a significant decrease in the company’s profits. Even if the manufacturer is willing to provide an experimental drug for expanded access, the drug might be too expensive and difficult to produce to allow for administration without participation in a clinical trial. The expanded access process involves the manufacturer’s regulatory, business, and production sectors, and this consumption of human and financial resources may become a significant burden, especially to a smaller manufacturer [Citation13,Citation28]. Finally, because of the high cost of drugs, manufacturers have the authority to charge patients or their third party payers for the direct cost of manufacturing and shipping, in addition to the costs of monitoring and reporting [Citation29], which is usually lower than the price of the drug after it is approved by the FDA and available on the market. According to an FDA report, if the insurer declines to cover the cost, most manufacturers will not charge the patient [Citation30].
5. Legal and ethical issues
The difficulty in accessing investigational drugs has led to legal actions. Some patients have claimed that the FDA violated their constitutional rights by not allowing them access to an investigational agent [Citation31,Citation32]. One such case was that of the Abigail Alliance for Better Access to Developmental Drugs v. von Eschenbach [Citation32], in which the FDA was sued to allow Abigail Burroughs, a college student with advanced head and neck cancer, to have access to cetuximab treatment. At that time, cetuximab was an investigational agent in clinical trials for colon cancer. In May 2006, the US Court of Appeals for the District of Columbia ruled in favor of the Abigail Alliance. The FDA requested that the Court of Appeals rehear the case. On 1 March 2007, the US Court of Appeals for the District of Columbia reheard and on 7 August 2007, the Court issued an 8-2 decision against the Abigail Alliance, reversing the previous panel decision. Frank Burroughs, Abigail’s father, vowed to pursue an appeal, but in 2008, the Supreme Court declined to review the case.
Patients enrolled on clinical trials may also request access to an investigational drug after the clinical trial ends and before FDA approval of the drug [Citation33]. In a legal case, in 2007, the court determined that despite a provision in the consent form, stating that subjects could elect to continue taking the study drug for up to 2 years after the trial ended, the sponsor had no obligation to provide the drug [Citation34].
The ethical debate about using investigational drugs in vulnerable patients with serious diseases includes individual autonomy and informed consent. Those in favor of expanded access programs argue that patients should have the right to minimize their suffering and improve their well-being. The major stakeholders are the patients themselves (who are presumed to be capable of making well-informed treatment decisions) and their treating physicians. Therefore, patients should be allowed to assess their own risk–benefit thresholds. On the other hand, the advocates for limited access to unapproved medications argue that the probability of significant clinical benefit from these investigational compounds in their early development stages is minimal (<10%) [Citation35,Citation36]. In addition, access to information about investigational drugs, including both clinical and pharmacological characteristics, is limited (owing to proprietary claims by the manufacturer). Therefore, the risk perception is usually falsely low, especially in sick patients [Citation37,Citation38]. In addition, granting expanded access in an equitable manner is a major ethical challenge, as these programs generally favor the rich and/or well-connected over the poor [Citation39].
Recently, drug approval delays have become a legislative concern. To date, 37 states have decided to take action to expedite drug approvals. The US Congress passed the ‘Right to Try’ bill, which allows terminally ill patients to access investigational drugs, bypassing the time-consuming FDA review procedures [Citation40,Citation41]. Eligibility criteria include the presence of a terminal illness with no other approved treatment options, a written informed consent form signed by the patient, and documentation from the treating physician that this patient is eligible for the treatment under consideration. This law protects treating physicians and manufacturers from professional discipline, claims of negligence, or legal responsibility in the case of any harms caused by the investigational drugs, assuming the treating physicians are making ‘good faith’ recommendations [Citation42–Citation44]. The ‘right-to-try’ laws do not oblige the insurance companies or the manufacturers to cover the cost of the investigational drugs. In addition, the ‘right-to-try’ laws cannot prevent the Drug Enforcement Administration from revoking the registration of physicians prescribing experimental drugs [Citation45,Citation46]. However, by abolishing the review of safety and ethical considerations, this law exposes a vulnerable population to potential risks. The balance of accelerating the drug approval processes to benefit the patients versus compromising their safety remains to be determined.
6. Future perspectives
Governments have acknowledged the challenges faced by all parties throughout the process of offering expanded access to investigational drugs. Various strategies are being assessed to simplify these processes and reduce the time to approval. Online petitions and social media campaigns to allow and expand access to experimental treatments are dramatically increasing [Citation47–Citation51].
Recently, US Senators introduced the ‘Enhanced Clinical Trial Design Act of 2017’ to render investigational drugs more widely available to higher risk and more diverse patient populations through clinical trials [Citation52]. They plan to update current processes to include more patients by expanding the eligibility criteria for clinical trials. Their specific aims are to (a) organize a collaboration between the FDA and the National Institutes of Health to expand inclusion study criteria, (b) carry out studies addressing the challenges of drug approval processes, (c) allow manufacturers to offer drugs on the basis of phase I data, and (d) facilitate IRB procedures. It is encouraging that the FDA has recently approved drugs/strategies for rare tumors and immunotherapeutic agents across tumor types.
Recent published data reevaluated eligibility criteria for clinical trials [Citation53,Citation54]. Despite the known advantages of participating in therapeutic clinical trials, the National Cancer Institute Clinical Trials Program Review Group reported that the percentage of eligible patients with cancer entering clinical research studies was estimated to be less than 5% [Citation55]. Age, history of prior malignancy, comorbidities, elevated laboratory test values are some of the criteria which can render a patient ineligible for participating in a clinical trial. Thus, the American Society of Clinical Oncology (ASCO), Friends of Cancer Research, and the FDA formed working groups to modify and broaden the eligibility criteria while protecting the safety of patients. The consensus included recommendations for patients and the scientific community [Citation53,Citation54].
Many expanded access requests are based on evidence demonstrating that treatment matched to a patient’s tumor molecular/biologic aberrations is associated with superior clinical outcomes compared to treatment not matched to patients’ alterations [Citation21,Citation23,Citation56]. We are conducting a randomized study evaluating molecular profiling and targeted therapy in metastatic cancer, Initiative for Molecular Profiling and Advanced Cancer Therapy 2 [Citation57,Citation58]. The end point of the study is to compare the progression-free survival of patients with advanced cancer who receive treatment based on their molecular profile to that of patients treated without taking into consideration their molecular profile [Citation59]. Subsequently, to learn from the real-world practice, the Targeted Agent and Profiling Utilization Registry (TAPUR) Study [Citation60] and the Drug Rediscovery Protocol (DRUP) were initiated [Citation61]. TAPUR is a non-randomized clinical trial that aims to test FDA-approved targeted treatments in different types of advanced cancers harboring a genomic variant known to be a drug target. It is being conducted by the ASCO in collaboration with pharmaceutical companies and multiple institutions and practices. Approved treatments are provided at no cost to the patients or their insurance companies. This study will provide important data on the efficacy of currently used drugs in various tumor types. Ultimately, it will benefit patients who will have access to novel drugs based on their molecular profile, as well as manufacturers, and regulatory agencies, who will receive data on drug use, safety, and outcomes. Following the TAPUR design, DRUP was designed in Europe to describe efficacy and toxicity of commercially available, targeted anticancer drugs when prescribed across tumor types based on tumor profile.
Additional efforts are being made by the French National Cancer Institute. In 2013, they developed the AcSé (Accès sécurisé à des thérapies ciblées innovantes) Program to evaluate the efficacy of targeted drugs used outside of their approved indications [Citation62]. Through this program, patients with cancers with different histologies have access to many centers, where their tumors are assessed for possible molecular alterations. On the basis of the detected tumor alterations, the patients receive the appropriate targeted treatment. In 2016, more than 7000 patients had already participated in AcSé-led trials carried out at 183 French clinical sites. Examples of phase II clinical trials currently recruiting patients with advanced malignancies include those involving BRAF genomic alterations and treatment with vemurafenib and molecular alterations on ALK, MET, or ROS1 genes and treatment with crizotinib [Citation63].
In 2014, the EMA, with members of the Committee for Medicinal Products for Human Use (CHMP), introduced a strategy to facilitate accelerated assessment of drugs that seem promising for patients with unmet medical needs (PRIority Medicines [PRIME]) [Citation64]. PRIME involves only unapproved products in the earliest stages of development. The majority of the drugs reviewed have been tested in phase I/II studies and have only preliminary data available. PRIME also focuses on ‘advanced treatments’ such as gene therapies, Chimeric Antigen Receptor (CAR) T-cell treatments, and oncolytic viruses. In order to further facilitate access to investigational treatments, PRIME offers advice and expertise on clinical trial design to less experienced manufacturers and academic institutions [Citation65]. Finally, this program works closely with regulatory agencies to improve drug access and pricing of novel drugs.
Global regulatory agencies share the same goals and oversee the same processes. Therefore, a collaboration would facilitate the exchange of expertise and accelerate the production of more robust data. The FDA and the EMA have formed ‘clusters,’ i.e. collaborative groups, to advance and strengthen international collaborations [Citation66]. Global regulators, including Health Canada, the Australian Therapeutic Goods Administration, and the Japanese Pharmaceuticals and Medical Devices Agency, joined the ‘Oncology-hematology medicinal product’ cluster. In addition, the FDA and EMA expanded their cooperation and formed another cluster in 2014, which focuses on pharmacovigilance [Citation67]. The benefits, challenges, and ongoing efforts to improve expanded access programs are summarized in .
Table 1. Benefits, challenges, and ongoing efforts to improve expanded access programs.
7. Conclusions
Expanded access programs for investigational drugs are vital for selected patients in need. However, these programs are complicated, and time to access of novel drugs often exceeds the time limits defined by the severity of the disease. Efforts of all involved parties have to be harmonized to create a patient-centered drug approval process. Legislative and regulatory agencies must evaluate a delicate balance between drug safety and efficacy, while accelerating the lengthy procedures. Equally important is an increase in patient awareness and involvement in the therapeutic management of their disease. Worldwide collaborations, trial guideline modifications, and new legislation are expected to expedite expanded access to novel treatments with promising antitumor activity.
8. Expert opinion
Expanded access programs involve time-consuming, complicated procedures that involve various parties. It is imperative that these processes remain patient centered, without the influence of potential personal or corporate benefit.
In general, phase I clinical trials are designed to assess the safety profile and pharmacokinetics/pharmacodynamics, and to determine the maximum tolerated toxicity or phase II recommended dose of novel agents. These trials provide treatment options for patients with metastatic disease who have failed standard-of-care therapy, for patients whose disease is not curable with established therapy, or for patients who decline to receive standard-of-care therapy. The antitumor activity noted with the use of novel agents in phase I clinical trials is encouraging, particularly with the use of the ‘precision medicine’ approach.
Recent advances in the pharmaceutical industry have led to the production of a large number of novel anticancer agents with potential antitumor activity. Multiple studies have shown a clear survival benefit and increased response with the use of innovative treatments. However, even though patients have the right to access novel drugs, they often face numerous obstacles, including suffering from their disease, while waiting for the completion of several lengthy procedures.
To date, there are several ongoing efforts led by global governmental and scientific committees to simplify and shorten the expanded access process, which include collaborations of regulatory agencies to expedite data assessment, modifications of eligibility criteria to increase patient access to clinical trials testing novel therapeutic agents, and legislative adjustments to expand access to new treatments. It is important that the scientific community, along with the patients, continue to work with all involved parties to optimize the process. We propose a simplified algorithm for expanded access to investigational drugs ().
Figure 1. Proposed algorithm for expanded access to investigational drugs.
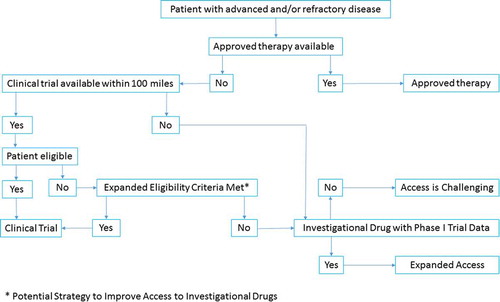
On the other head, enthusiasm for emerging treatments must not obscure the need to assess the possible adverse events of these drugs, and therefore the patient’s cost–benefit risk ratio of the drug under consideration should be assessed.
Article highlighted box
Expanded access is the use of an investigational product by a patient with a serious and life-threatening condition who cannot participate in a clinical trial.
Expanded access to an investigational drug offers a treatment option to a patient with unmet needs, and requires approval by the drug manufacturer and regulatory agencies.
Pharmaceutical companies, treating physicians, patient advocates, regulatory agencies, and institutional review boards are involved in the complicated process of expanded access.
Ongoing efforts focus on overcoming the safety, regulatory, legal, and ethical challenges associated with expanded access.
Global scientific communities, governmental and regulatory agencies, and patient advocates are committed to improve expanded access procedures.
Optimization of laws and policies, development of specialized regulatory agencies, harmonization of policies among regulatory agencies, and broadening of inclusion criteria in clinical studies are expected to accelerate expanded access to investigational drugs.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Holbein ME, Berglund JP, Weatherwax K, et al. Access to investigational drugs: FDA expanded access programs or “Right-to-Try” legislation? Clin Transl Sci. 2015;8(5):526–532.
- Saiyed MM, Ong PS, Chew L. Off-label drug use in oncology: a systematic review of literature. J Clin Pharm Ther. 2017;42(3):251–258.
- Code of federal regulations title 21. SERVICES DOHAH, editor; 2017. Available from: https://www.ecfr.gov/cgi-bin/text-idx?SID=3ee286332416f26a91d9e6d786a604ab&mc=true&tpl=/ecfrbrowse/Title21/21tab_02.tpl
- The history of Food and Drug Administration Agency. Available from: https://www.fda.gov/AboutFDA/WhatWeDo/History/default.htm
- 21 U.S.C. § 355. PLN-, 76 Stat. 780. [Safety and efficacy amendments to the Federal Food, Drug and Cosmetic Act]; 1962.
- 21 U.S.C. ch. 9 § 301 et seq. [Kefauver-Harris Amendment].
- Richey EA, Lyons EA, Nebeker JR, et al. Accelerated approval of cancer drugs: improved access to therapeutic breakthroughs or early release of unsafe and ineffective drugs? J Clin Oncol. 2009;27(26):4398–4405.
- Kaitin KI, DiMasi JA. Pharmaceutical innovation in the 21st century: new drug approvals in the first decade, 2000–2009. Clin Pharmacol Ther. 2011;89(2):183–188.
- Lanthier ML, Sridhara R, Johnson JR, et al. Accelerated approval and oncology drug development timelines. J Clin Oncol. 2010;28(14):e226–e227. author reply e8.
- Downing NS, Aminawung JA, Shah ND, et al. Regulatory review of novel therapeutics–comparison of three regulatory agencies. N Engl J Med. 2012;366(24):2284–2293.
- Ns D, Ad Z, Ross JS. Regulatory review of new therapeutic agents - FDA versus EMA, 2011–2015. N Engl J Med. 2017;376(14):1386–1387.
- FDA. Investigational new drug, antibiotic, and biologic drug product regulations: treatment use and sale. Fed Regist. 1987;52:19466–19467.
- Food and Drug Administration, HHS. Expanded access to investigational drugs for treatment use: final rule. Fed Regist. 2009;74(155):40900–40945.
- Trotta F, Leufkens HG, Schellens JH, et al. Evaluation of oncology drugs at the European Medicines Agency and US Food and Drug Administration: when differences have an impact on clinical practice. J Clin Oncol. 2011;29(16):2266–2272.
- FDA. Available from: https://blogs.fda.gov/fdavoice/index.php/2017/10/expanded-access-fda-describes-efforts-to-ease-application-process/
- DiMasi JA. Risks in new drug development: approval success rates for investigational drugs. Clin Pharmacol Ther. 2001;69:297–307.
- Martell RE, Sermer D, Getz K, et al. Oncology drug development and approval of systemic anticancer therapy by the U.S. Food and Drug Administration. Oncologist. 2013;18(1):104–111.
- Jarow JP, Lemery S, Bugin K, et al. Access of investigational drugs: the experience of the center of drug evaluation and research over a 10-year period. Ther Innov Regul Sci. 2016;50(6):705–709.
- Planchard D, Smit EF, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–1316.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–697.
- Tsimberidou AM. Initiative for molecular profiling and advanced cancer therapy and challenges in the implementation of precision medicine. Curr Probl Cancer. 2017;41(3):176–181.
- Tsimberidou AM, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res. 2012;18(22):6373–6383.
- Tsimberidou AM, Wen S, Hong DS, et al. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: validation and landmark analyses. Clin Cancer Res. 2014;20(18):4827–4836.
- FDA expanded access. Available from: https://www.fda.gov/downloads/ForIndustry/DevelopingProductsforRareDiseasesConditions/OOPDNewsArchive/UCM294794.pdf.
- Black L. Experimental breast cancer treatments and health insurance coverage. The Virtual Mentor. 2007;9(1):34–37.
- Falit BP, Gross CP. Access to experimental drugs for terminally ill patients. Jama. 2008;300(23):2793–2795.
- Mack GS. Expanded access rules pose quandary for drug developers. Nat Biotechnol. 2009;27(10):871–872.
- Kroll D 2014 March 12. Available from: https://www.forbes.com/sites/davidkroll/2014/03/12/why-everyone-has-a-stake-in-the-chimerix-drug-offered-to-josh-hardy-cmx001-brincidofovir/#6d2a1c7a40da
- Electronic Code of Federal Regulations, Part 314.510. US Food and Drug administration silver spring. MD US Food and Drug Administration; 2008. Available from: https://www.ecfr.gov/cgi-bin/text-idx?SID=3ee286332416f26a91d9e6d786a604ab&mc=true&tpl=/ecfrbrowse/Title21/21tab_02.tpl
- Jc D. Connecting patients with experimental drugs. Wall Street J. 2012, October22. Available from: https://www.wsj.com/articles/should-the-fda-relax-rules-on-compassionate-access-to-new-drugs-1491962521
- United States v. Rutherford, 442 U.S. 544; 1979.
- Abigail Alliance for Better Access to Developmental Drugs v. von Eschenbach, 495 F.3d 695 (D.C. Cir. 2007).
- Dahl v. HEM Pharms, 7 F.3d 1399 (9th Cir. 1993).
- Mello MM, Joffe S. Compact versus contract–industry sponsors’ obligations to their research subjects. N Engl J Med. 2007;356(26):2737–2743.
- Horstmann E, McCabe MS, Grochow L, et al. Risks and benefits of phase 1 oncology trials, 1991 through 2002. N Engl J Med. 2005;352(9):895–904.
- Freireich EJ, Kurzrock R. The role of investigational therapy in management of patients with advanced metastatic malignancy. J Clin Oncol. 2009;27(2):304–306.
- Woloshin S, Schwartz LM, Welch HG. Patients and medical statistics. Interest, confidence, ability. J General Internal Med. 2005;20(11):996–1000.
- Kolva E, Rosenfeld B, Brescia R, et al. Assessing decision-making capacity at end of life. Gen Hosp Psychiatry. 2014;36(4):392–397.
- Leonard EW. Right to experimental treatment: FDA new drug approval, constitutional rights, and the public’s health. J Law, Med Ethics. 2009;37(2):269–279.
- Right to try model legislation. Available from: https://goldwater-media.s3.amazonaws.com/cms_page_media/2016/1/5/GoldwaterInstituteRighttoTryModel.pdf
- Dennis B, Cha AE “Right to try” laws spur debate over dying patients’ access to experimental drugs. Washington Post. May 16, 2014
- H.R. 891, Gen. Assemb., Reg. Sess. (La. 2014) [“right-to-try” laws, manufactures can provide experimental medicines to terminally ill patients without FDA authorization].
- H.R. 1685, Gen. Assemb., Reg. Sess. (Mo. 2014) [“right-to-try” laws, manufactures can provide experimental medicines to terminally ill patients without FDA authorization].
- H.R. 1281, Gen. Assemb., Reg. Sess. (Colo. 2014) [“right-to-try” laws, manufactures can provide experimental medicines to terminally ill patients without FDA authorization].
- 21 U.S.C. § 823 [United States Code (USC) Controlled Substances Act].
- 21 C.F.R. § 1301.36 [Code of Federal Regulations, suspension or revocation of registration].
- Walker MJ, Rogers WA, Entwistle V. Ethical justifications for access to unapproved medical interventions: an argument for (limited) patient obligations. Am J Bioethics. 2014;14(11):3–15.
- Chapman A. Proposal for patient obligations for access to unapproved medical interventions: both too much and not enough. Am J Bioethics. 2014;14(11):25–26.
- Darrow JJ, Sarpatwari A, Avorn J, et al. Practical, legal, and ethical issues in expanded access to investigational drugs. N Engl J Med. 2015;372(3):279–286.
- Magnus D. Compassion and research in compassionate use. Am J Bioethics. 2014;14(11):1–2.
- Mackey TK, Schoenfeld VJ. Going “social” to access experimental and potentially life-saving treatment: an assessment of the policy and online patient advocacy environment for expanded access. BMC Med. 2016;14:17.
- Enhanced Clinical Trial Design Act of 2017. S. 1048, H.R. 2430.
- Kim ES, Bruinooge SS, Roberts S, et al. Broadening eligibility criteria to make clinical trials more representative: American Society of Clinical Oncology and friends of cancer research joint research statement. J Clin Oncol. 2017;35(33):3737–3744.
- Jin S, Pazdur R, Sridhara R. Re-evaluating eligibility criteria for oncology clinical trials: analysis of investigational new drug applications in 2015. J Clin Oncol. 2017;35(33):3745–3752.
- Scoggins JF, Ramsey SD. A national cancer clinical trials system for the 21st century: reinvigorating the NCI Cooperative Group Program. J Natl Cancer Inst. 2010;102(17):1371.
- Korphaisarn K, Kopetz S. BRAF-directed therapy in metastatic colorectal cancer. Cancer J. 2016;22(3):175–178.
- Tsimberidou AM, Eggermont AM, Schilsky RL. Precision cancer medicine: the future is now, only better. Am Soc Clin Oncol Educ Book. 2014;61–69.
- Tsimberidou AM, Hong DS, Ye Y, et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): an MD Anderson Precision Medicine Study. JCO Precis Oncol. 2017;2017. doi: 10.1200/PO.17.00002.
- IMPACT 2. Available from: https://www.clinicaltrials.gov/ct2/show/NCT02152254
- TAPUR Study. Available from: https://clinicaltrials.gov/ct2/show/NCT02693535
- The Drug Rediscovery Protocol (DRUP Trial) (DRUP). Available from: https://clinicaltrials.gov/show/NCT02925234
- Buzyn A, Blay JY, Hoog-Labouret N, et al. Equal access to innovative therapies and precision cancer care. Nat Rev Clin Oncol. 2016;13(6):385–393.
- https://clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT02034981?term=crizotinib&draw=1&rank=1
- Mullard A. PRIME time at the EMA. Nat Rev Drug Discov. 2017;16(4):226–228.
- PRIME. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000660.jsp
- Em A. Cluster activities. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/partners_and_networks/general/general_content_000655.jsp&mid=WC0b01ac0580953d98.
- Dal Pan GJ, Arlett PR. The US Food and Drug Administration-European Medicines Agency collaboration in pharmacovigilance: common objectives and common challenges. Drug Saf. 2015;38(1):13–15.