
ABSTRACT
Introduction
Patients with myelofibrosis (MF) have no effective treatment option after the failure of approved JAK inhibitor (JAKi) therapy. Non-JAK inhibitors (non-JAKi) that target non-canonical molecular pathways are undergoing clinical evaluations to optimize efficacy and/or to reduce hematological toxicity of JAKi.
Area covered
This article reviews the efficacy data from completed and ongoing early phase clinical trials of non-JAKi agents for chronic phase MF. The article also illuminates some of the challenges of myelofibrosis drug development.
Expert opinion
Most non-JAKi agents tested so far have shown modest benefit in improving the efficacy of ruxolitinib. Several novel agents such as BET inhibitor- CPI-0610, activin receptor ligand trap- luspatercept, recombinant pentraxin-PRM-151, telomerase inhibitor- imetelstat and bcl-2 inhibitor- navitoclax, have shown promising activity; however, they require vigorous evaluation in randomized controlled trials to understand the clinical benefit. Drugs that target new molecular pathways (MDM2, p-selectin, TIM-3, TGF-β, aurora kinase) and immune-based strategies (CALR vaccine, anti-PD-1, allogeneic cord blood regulatory T cells) are in early phase trials. Further translational studies to target leukemic stem cells, improvement in trial designs by incorporating control arm and survival endpoints, and patient-focused collaborations among all stakeholders could pave a way for future success in MF drug development.
1. Introduction
1.1. The unmet clinical needs in myelofibrosis
Myelofibrosis (MF) is the most aggressive disease among the three classical Philadelphia negative myeloproliferative neoplasms (MPN). The clinical course of MF is extremely variable; ranging from rapid leukemic transformation (LT) to an asymptomatic state lasting many years [Citation1]. Allogeneic stem cell transplantation (allo-SCT) is the only curative treatment for patients at higher risk of LT or shorter survival [Citation2–Citation5], but has significant procedure related complications [Citation6]. Advanced age at the time of MF diagnosis, significant burden of co-morbid conditions, and lack of optimal donors limit the applicability of transplant to a small proportion of MF patients [Citation7]. For majority of MF patients, the focus of the treatment is improvement in symptom burden and quality of life with ruxolitinib, the first approved therapy for MF [Citation8,Citation9]. More recently, fedratinib has been approved by FDA in 2019, but experience outside the clinical trials is limited with fedratinib. Both ruxolitinib and fedratinib reduce symptoms of splenomegaly and cytokine related symptom burden of the disease, but have limited anti-clonal activity, and they do not prevent progression to acute myeloid leukemia (AML). Moreover, there are significant rates of discontinuation of ruxolitinib therapy. The most common reasons for discontinuation include toxicities, disease progression, or loss of response [Citation10]. Such patients with ‘ruxolitinib failure’ have poor outcome with a median survival of 14–18 months [Citation11,Citation12]. Moreover, 10-15% patients with MF are ‘ruxolitinib ineligible’ because of severe thrombocytopenia (platelet count <50 x 109/L) [Citation1]. Therefore, novel treatments are needed that could (i) provide deeper and durable responses than JAKi alone, (ii) be used for patients who are intolerant or ineligible for currently available JAKi due to anemia or thrombocytopenia, and (iii) modify disease biology by eliminating leukemic stem cells or reversing bone marrow fibrosis.
1.2. The challenges in drug development in MF
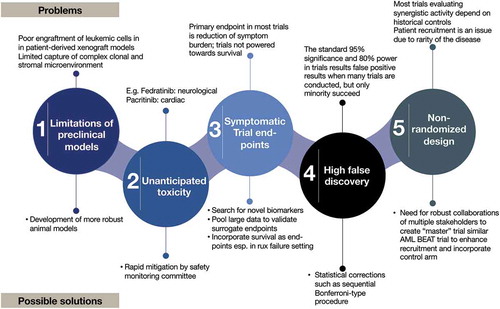
Out of many clinical trials conducted over the past decade, only two JAKi have received FDA approval and two more (pacritinib [Citation14] and momelotinib [Citation15]) are undergoing additional phase III evaluations. Numerous combination therapy clinical trials have failed in MF. Although these trials were developed based on reasonable scientific rationale; their failure highlights several challenges in drug development in MF as illustrated in . Some of these challenges include: 1) Difficulties in capturing the complex clonal and stromal microenvironment in pre-clinical patient-derived xenografts (PDX) mice models [Citation16]; 2) Emergence of unanticipated toxicities (e.g. fedratinib [Citation8] and pacritinib [Citation17]); 3) Dependence on surrogate trial endpoints such as spleen response to inform long-term efficacy in the absence of a strong biomarker [Citation18]; 4) non-incorporation of control arm when studying combination therapies; and 5) limited number of patients due to disease rarity [Citation19].
Figure 1. Challenges and possible solutions in drug development in MF.
1.3. The current understanding of pathobiology guiding the novel therapies
We provide a synopsis of pathophysiology of MF to understand the targets of investigational drugs in MF [Citation20,Citation21]. MF is a clonal disorder of hematopoiesis that arises in the hematopoietic stem cells (HSC) from driver mutations (50% JAK2 [Citation22–Citation24], 30% CALR [Citation25] or 8% MPL [Citation26]). These activate JAK/STAT signaling, which cross talks with non-canonical pathways such as PI3K or Ras-Raf-MAPK [Citation27]. Together, they promote downstream transcription of genes regulating cellular proliferation, apoptosis, migration, and differentiation. Additional mutations in epigenetic regulators (TET2, DNMT3A, ASXL1, IDH1/2, EZH2) and in spliceosome components (SF3B1, U2AF1, SRSF2) favor clonal dominance, change disease phenotype, and promote progression of myelofibrosis and leukemic transformation [Citation28]. The microenvironment maintains the HSC in its niche and increases bone marrow fibrosis (BMF) as a reaction to the clonal hematopoiesis by releasing pro-fibrotic (transforming growth factor β1 [TGF-β1] etc.), angiogenic and pro-inflammatory cytokines [Citation29]. Based on these notions, the current investigational agents either target 1) the molecular pathways in HSC to suppress the malignant clone, or 2) the microenvironment to reduce BMF. This distinction is not consistent because the drugs could have multiple mechanisms of action. The goals of treatment are to modify the disease biology (reduce fibrosis/eliminate leukemic stem cells), prevent leukemic transformation and improve overall survival.
In this article, we review the current early phase (I and II), non-JAKi investigational agents for chronic phase MF and discuss how these could impact treatment landscape in near future.
2. Methods
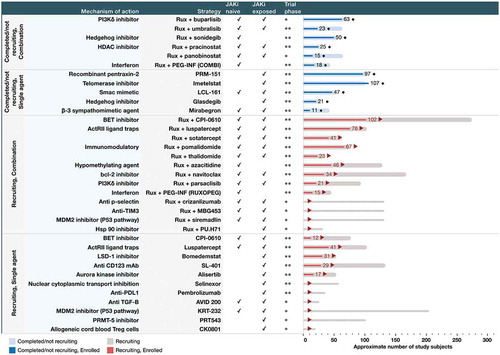
We searched the clinical trials databases (International clinical trials registry platform (ICTRP), ClinicalTrials.gov, Health Canada’s clinical trials database, and European union clinical trials register) using the terms ‘myelofibrosis’ and ‘phase I’ and ‘phase II’ trials between 1 January 2010 till 12 December 2019. We searched Medline and EMBASE databases using the terms ‘myelofibrosis’, ‘phase I’, ‘phase II’ and abstracts of American Society of Clinical Oncology, European Hematology Association, and American Society of Hematology. We classified trials of interest into two main strategies: a) Clinical trials evaluating non-JAKi agents in combination with ruxolitinib (add-on strategy) and b) Clinical trials evaluating non-JAKi agents as monotherapy in ruxolitinib ineligible or relapsed/refractory patients. We have shown an overview of completed and ongoing trials of novel non-JAKi agents, their mechanism of action, the strategy used for evaluation (add-on vs single agent), and enrollment status in .
Figure 2. Snapshot of early phase clinical trials of non-JAK inhibitor drugs in MF: Strategy, mechanism of action, phase of development, and enrollment status.
3. Results and discussion
3.1. Ruxolitinib based combinations or ‘add-on’ strategies
3.1.1. Bromodomain and extra-terminal (BET) inhibitor (CPI-0610) ± ruxolitinib
NF-κB is an important mediator of chronic inflammation in MPN, as shown by RNA-seq and CHIP-seq data in two different mouse models [Citation30]. It is constitutively active in both mutant and non-mutant hematopoietic cells and co-regulates transcription of key target genes along with activated STAT3. The BET proteins that ‘read’ the acetylated lysine residues in histone tails recruit the transcriptional machinery for gene expression, including NF-κB [Citation31]. Inhibition of the BET protein in mice engrafted with JAK2 V617F mutant MPN cells reduces NF-κB transactivation, inflammatory cytokine production, and improves survival compared to vehicle-control [Citation30]. Further, combined BET/JAK inhibition synergistically reduces leucocytes and spleen weights, extramedullary hematopoiesis, mutant allele burden, bone marrow fibrosis, leukemic stem cells and delays persistence associated with JAKi [Citation30]. A phase II clinical trial (MANIFEST, NCT02158858) is testing BET inhibitor (CPI-0610) in MF as an add-on to ruxolitinib for suboptimal response or progression; and as monotherapy in patients who are ruxolitinib resistant, refractory, intolerant or ineligible. They stratified patients based on whether they are transfusion dependent (TD) or independent (TI). In the preliminary analysis, most treated patients had high-risk mutations and substantial symptom burden () [Citation32]. In the combination arm, encouraging responses in reduction of splenomegaly and improvement in patient-reported outcomes were seen. In addition, improvement in anemia and transfusion requirements, bone marrow fibrosis and reduction in inflammatory cytokine levels were observed (). Patients tolerated CPI-0610 well; thrombocytopenia was the only overlapping toxicity with ruxolitinib but was non-cumulative and reversible. An additional cohort is recruiting JAKi naïve patients to receive CPI-0610/ruxolitinib combination, and showed encouraging preliminary activity [Citation33]. Confirmation of the preliminary results in a larger number of patients would potentially pave the way for a randomized phase III trial to understand the clinical benefit of CPI-0610 in combination with ruxolitinib.
Table 1. Completed early phase clinical trials of non JAK inhibitor drugs in myelofibrosis.
Table 2. Preliminary results of early phase clinical trials of non JAK inhibitor drugs in myelofibrosis.
3.1.2. Azacitidine + ruxolitinib
Azacitidine is a DNA hypomethylating agent that has shown limited efficacy as a single agent in MF [Citation34]. Its synergistic activity with ruxolitinib has been evaluated in a phase II trial in JAKi naïve patients with MF. A sequential dosing schedule was used to mitigate the myelosuppression related to azacitidine [Citation35]. In interim results, the spleen volume reduction (SVR) and total symptom score (TSS) improvement were comparable to historical responses in COMFORT trials of ruxolitinib but 21% patients achieved responses after the addition of azacitidine (). A higher incidence of cytopenia was noted. The reduction in bone marrow fibrosis seen in about 57% patients at 24 months was encouraging. The implications of this finding remain unclear till long-term data is available. This combination could have a role in aggressive MF where more robust cytoreduction is warranted but will need a randomized trial to understand the additional benefit of azacitidine.
3.1.3. Histone deacetylases inhibitors (HDACi) + ruxolitinib
Histone deacetylation by histone deacetylases (HDAC) is associated with repression of gene expression. In MPN, HDAC expression is increased and down-regulates expression of SOCS1/3, a negative regulator of JAK/STAT signaling, enhancing the activity of this pathway [Citation36]. JAK2 V617F cell lines are sensitive to treatment with HDAC inhibitor as shown by reduced colony formation at threefold lower doses compared to JAK2 wild type [Citation37]. HDACi downregulates the hematopoietic transcription factors NF-E2, C-MYB and STAT5, both in JAK2 V617F cell lines and CD34+ cells from MPN patients. In addition, it induces pro-apoptotic genes and downregulates JAK2 activity via inhibition of hsp90, a molecular chaperone for JAK2 [Citation38]. Despite this strong preclinical activity, HDACi have shown limited clinical efficacy in MF. (). Pracinostat, a pan-HDACi, evaluated as an add-on strategy after a run-in phase of ruxolitinib, did not improve its responses [Citation39]. Patients experienced frequent dose interruptions after starting pracinostat because of anemia (≥ grade 2 in 75% patients) and thrombocytopenia (≥ grade 2 in 25% patients). Panobinostat, another HDACi, when combined with ruxolitinib, showed modest efficacy (). This drug was well tolerated. Forty seven percent patients had worsening anemia but no treatment emergent neutropenia, or thrombocytopenia were observed. Gastrointestinal toxicity occurred in over two-third of patients [Citation40]. To the best of our knowledge, there are no planned phase III trials with HDACi in MF.
3.1.4. PI3Kδ/mTOR/AKT inhibitors + ruxolitinib
The phosphatidylinositol 3-kinase integrates cues from cytokines and growth factors and transmits them through AKT and mTOR to effector molecules that control protein synthesis, growth, survival, and proliferation (PI3K/mTOR/AKT pathway) [Citation41]. JAK2 V617F mutated cells from MPN patient samples and mice models show hyper phosphorylation of AKT and sensitivity to inhibition of PI3K/mTOR/AKT pathway at doses lower than the controls [Citation42]. Combined PI3K inhibitior/ruxolitinib reduces erythropoietin-independent colony growth and spleen weight in mice engrafted with Ba/F3 cells expressing JAK2 V617F [Citation43]. Umbralisib (TGR-1202) is a selective PI3Kδ inhibitor that showed robust activity in preclinical testing in myeloid malignancies [Citation44]. In a phase II trial in patients with MF, umbralisib was added after ≥8 weeks of run-in phase of ruxolitinib. Most patients showed clinical improvement and two patients achieved complete remission [Citation45]. The drug was well tolerated with asymptomatic elevation of lipase and amylase, and diarrhea being the commonest grade 3 adverse events (). Final results from this trial are pending. Parsaclisib, another PI3Kδ selective inhibitor was combined with ruxolitinib in MF patients for suboptimal response. Although well tolerated, clinical benefits were modest [Citation46]. Buparlisib, an oral pan-PI3K inhibitor in combination with ruxolitinib (HARMONY) showed a modest risk-benefit profile in a phase 1b trial in both JAK naïve and JAKi treated patients and is unlikely to be developed further [Citation47].
Proviral-integration site for Moloney-murine leukemia virus (PIM) kinases regulate different cancerous pathways and interact with the PI3K/mTOR/AKT pathway. In MPN, overexpression of PIM kinases causes activation of JAK/STAT and PI3K/AKT pathways [Citation48]. In MPN cell lines and patient samples, PIM inhibitor INCB053914 synergized with JAKi to reduce cell proliferation and erythropoietin- independent colony formation, and sensitized JAKi resistant cell lines to JAK inhibition [Citation49]. A phase I/II study of PIM inhibitor is enrolling advanced MF patients who failed JAKi as an add-on treatment.
3.1.5. Hedgehog pathway inhibitor (Hhi) ± ruxolitinib
Hedgehog signaling is a conserved pathway involved in HSC self-renewal and differentiation. The expression of PTCH1 (a transmembrane receptor that binds hedgehog molecules) and GLI1 (a transcription factor that controls downstream target genes in hedgehog pathway) is increased up to 100-fold in granulocytes in MPN patients and mice models [Citation50]. Combined ruxolitinib/HHi shows a greater reduction of the mutant allele burden and bone marrow fibrosis than ruxolitinib alone in the MPN mice model. Hh inhibitors- vismodegib [Citation51] and sonidegib [Citation52] in combination with ruxolitinib were evaluated in patients with MF. Although well tolerated, they did not have impressive activity (). Glasdegib, another potent and selective HHi was tested in a Phase Ib/II trial as a single agent in patients with suboptimal response to JAKi [Citation53]. Efficacy was modest (). All patients had one or more AEs (serious in 23%), most common were dysgeusia, muscle spasms, and alopecia. They terminated the trial early due to a lack of efficacy.
3.1.6. Bcl-2 inhibitor, navitoclax + ruxolitinib
B cell leukemia-2 (Bcl-2) family of proteins has a central role in apoptosis. In patients with MF, the JAK2 V617F mutation is associated with dysregulation of Bcl-2 proteins [Citation54]. The mRNA levels pro-apoptotic proteins (BAX, BAD, and BIK) are lower and anti-apoptotic (A1, MCL1, BCL-W, and BCL-XL) higher in JAK2 V617F mutated cases than controls. The ‘BH3-only’ proteins are a subset of Bcl-2 proteins that neutralize pro-survival proteins to trigger apoptosis [Citation55]. In preclinical mice models, BH3 mimetic drug navitoclax synergistically induced apoptosis with ruxolitinib, prevented fibrosis, and overcame resistance to JAKi [Citation56]. In a phase II trial, navitoclax was combined with ruxolitinib as an add-on to improve the responses to JAKi. Patients were heavily pre-treated. The median duration of prior ruxolitinib was 21 months () [Citation57]. Thirty percent patients achieved >35% SVR (SVR35), including those with high-risk molecular mutations, and 35% achieved >50% TSS reduction (TSS≥50). The bone marrow fibrosis reduced by ≥1 grade in 25% patients, possibly signifying disease modification. Leukoreduction was robust and 60% patient achieved transfusion-independence. This combination was well tolerated. Thrombocytopenia was common AE but plateaued into a safe range (nadir mean platelet count 95 x 109/L) in 6–8 weeks with no bleeding events. Given the encouraging results, this study is being expanded further.
3.1.7. Immunomodulatory drugs (IMiD) + ruxolitinib
IMiD are thalidomide analogues with anti-proliferative, anti-angiogenic and anti-inflammatory effects in MPN through inhibition of bFGF and VEGF [Citation58]. Based on their single agent activity in achieving transfusion independence and improvements in platelet count, thalidomide (NCT03069326) and pomalidomide (NCT01644110) are undergoing evaluation in combination with ruxolitinib in patients with MF [Citation59,Citation60]. In both relapsed and JAKi naïve patients, ruxolitinib + thalidomide showed a significant increase in platelet count and clinically meaningful SVR in half the treated cohort (). Grade ≥3 AE were limb edema, diverticulitis, hypertension, and syncope [Citation61]. A phase III trial (RESUME) comparing pomalidomide vs placebo in patients with red blood cell (RBC)-transfusion dependent MF showed similar rates of conversion to transfusion-independence [Citation62]. Pomalidomide is being evaluated in a phase II trial in combination with ruxolitinib (MPNSG-0212) (). The interim results showed a clinical benefit in improving hemoglobin. The combination was well tolerated [Citation63]. Even though pomalidomide and thalidomide show improvement in anemia and thrombocytopenia respectively, they have modest synergistic activity for SVR or TSS reduction. The development of lenalidomide in MF is no longer pursued because of myelosuppression. We did not come across any active phase III trial evaluating IMiDs as monotherapy or in combination with JAKi therapy in patients with MF.
3.1.8. Activin receptor ligand traps (sotatercept and luspatercept) ± ruxolitinib
TGF-β signaling plays a major role in development of fibrosis in MF. It is highly expressed in the bone marrow of patients with MF and in animal models and released in large amounts by the megakaryocytes. Its inhibition reactivates normal hematopoiesis, particularly erythropoiesis [Citation64]. Luspatercept and sotatercept are recombinant fusion proteins comprising of the extracellular domain of activin receptor type IIB (ACVR2B) and type IIA and linked to the human immunoglobulin G1 (IgG1) Fc domain. They act as ‘traps’ for TGF-β superfamily ligands and enhance late-stage erythropoiesis [Citation65]. In a phase 2 study, patients with MF received luspatercept as monotherapy or as combination with ruxolitinib. They were further stratified according to transfusion dependency [Citation66] (). A higher number of patients in the combination arm converted from transfusion-dependence (TD) to independence (TI) than monotherapy arm. The most common AEs were hypertension, bone pain and diarrhea. The combination arm continuing to recruit patients. Sotatercept has shown comparable efficacy and safety profile [Citation67]. Compared to sotatercept, luspatercept has a more selective activity on GDF-11, a key inhibitor of late-stage erythroid differentiation [Citation68]. Also, it does not bind to other members of the TGF-β superfamily, such as activin A which has important roles in physiological processes such as islet β-cell proliferation and stem cell self-renewal and differentiation [Citation69]. Luspatercept is presently preferred over sotatercept for further phase III trials.
3.1.9. Interferon-α-2a (IFN- α-2a) + ruxolitinib
Several small, including retrospective studies suggested a role of interferon-α-2a (IFNα −2a) in an early phase MF for targeting malignant HSC clone [Citation70,Citation71]. But immunological and hematological toxicities led to its rapid discontinuation in over 50% patients. Combining ruxolitinib (for anti-inflammatory activity) with IFN (to target HSC) could provide a synergistic effect. In the phase II COMBI study, MF patients with dynamic international prognostic scoring (DIPSS) low, intermediate-1 or −2 risk were treated using a combination of ruxolitinib and low-dose PEG-IFNα −2a [Citation72] (). All 18 patients were DIPSS low- or intermediate-1-risk. Complete remission and partial remission were achieved in 20% patients each. The median % JAK2 V617F allele burden decreased from 45% to 18% at 12 months (p <.0001). Hematologic toxicities were the most common AE, and arthralgia and/or myalgia were most common non-hematological AE. The combination was well tolerated with most patients completing ≥12 months of treatment. Another Bayesian phase 1/2 adaptive trial of ruxolitinib and pegylated IFNα −2a combination (RUXOPEG) is ongoing [Citation73] (). Interim results in 15 patients showed a decrease in spleen size at 6 months, improvement in blood counts, partial remission in three patients and, hematological improvement in seven. JAK2 V617F allele burden decreased from 75% at baseline to 46% at 6 months. These results confirm the feasibility of combining low-dose IFN with JAKi; further studies are required to understand the efficacy of this combination.
3.1.10. Siremadlin or crizanlizumab or MBG453 + ruxolitinib
A randomized, open-label, phase I/II platform study (NCT04097821) is evaluating safety and efficacy of combining ruxolitinib with 3 novel compounds-siremadlin, crizanlizumab, and MBG453 in MF subjects who have been treated with ruxolitinib for at least 24 weeks and have splenomegaly. This trial includes progression-free survival as a secondary end point and has a control arm. It provides an opportunity for disease modification through three different experimental agents: 1) Siremadlin: is a p53-MDM2 inhibitor. In MPN, JAK2 V617F increases translation of MDM2 because of accumulation of E3 ubiquitin ligase. This increased MDM2 affects the p53 response to DNA damage, as shown in Ba/F3-EPOR and CD34+ cells from MPN patients [Citation74]. Inhibition of p53-MDM2 interaction reduces JAK2 V617F hematopoietic progenitor cells in long-term cultures and JAK2 V617F allele burden in mice indicating depletion of MPN HSC. 2) Crizanlizumab is a recombinant anti-p-selectin monoclonal antibody. The spleen of MF patients contains increased numbers of HSC and megakaryocytes. These megakaryocytes express high levels of p-selectin that triggers TGF-β release causing disease progression. When p-selectin is deleted, survival of MF mice increases by 3 months with reduction in marrow fibrosis and splenomegaly [Citation75]. 3) MBG453: This is a high-affinity, ligand blocking, humanized anti-T-cell immunoglobulin domain and mucin domain-3 (TIM-3) antibody. It works as a checkpoint inhibitor that blocks the binding of an immune checkpoint receptor TIM-3 to phosphatidylserine. Most leukemic stem cells and progenitors in acute myeloid leukemia (AML) express TIM-3 that with its ligand, galectin-9, constitutes an autocrine loop critical for self-renewal of leukemic stem cells [Citation76]. Up-regulation of TIM-3 is associated with leukemic transformation in MPN [Citation77], besides its complex role in immune system regulation [Citation78].
3.1.11. CK0801 (cord blood regulatory T cells) + ruxolitinib
In MF patients, regulatory T cells (Treg) produce soluble IL2Rα which induces CD8 + T cell proliferation causing bone marrow dysfunction [Citation79]. In fact, IFN is known to result in expansion of Treg cells resulting in clearance of a malignant HSC clone [Citation80]. Based on this rationale and function of Treg cells, a phase I clinical trial examining the role of the single infusion of CK0801, an allogeneic, fresh cord blood Treg product is ongoing (NCT03773393). In results reported on two patients with MF, one achieved cytogenetic remission [Citation81].
3.2. Monotherapies that target alternative pathways in MPN HSC
3.2.1. Lysine-specific demethylase 1 (LSD1) inhibitor (bomedemstat)
LSD1 is an enzyme that modifies chromatin by removing methyl groups from histones and is critical for myeloid maturation, self-renewal of leukemia initiating cells. LSD1 bound to GFIb, a key transcription factor for hematopoiesis, has a specific role in the maturation of megakaryocytes [Citation82]. In MPN mice models, inhibition of LSD1 using IMG-7289 improved blood counts; reduced spleen volumes, bone marrow fibrosis, and mutant allele burden, increased expression of p53 and pro-apoptotic factor PUMA; and enhanced survival [Citation83]. In a phase II trial of bomedemstat (NCT03136185) in patients with refractory MF, a novel method of targeting a platelet count (between 50 and 100 x 109/L) as a biomarker of effective thrombopoiesis [Citation84]. Bomedemstat was well tolerated with 85% patients completing the first 12 weeks of treatment (). SVR and TSS responses and reduction in BMF were modest.
3.2.2. Telomerase inhibitor (imetelstat)
Telomerase is a ribonuclear protein complex made up of a reverse transcriptase catalytic protein subunit, an RNA template, and specialized proteins that extends the length of telomeres, a process necessary for maintaining the replicative potential of cells. In MPN, HSCs have shorter telomere lengths along with increased telomerase activity compared to that of healthy controls which allows them to maintain increased proliferative capacity [Citation85]. Imetelstat is an oligonucleotide that directly and competitively inhibits telomerase activity leading to cell cycle arrest and apoptosis. In MF, it targets leukemia stem cells and has a long residence time in bone marrow, spleen, and liver and promotes apoptosis in splenic CD34+ cells and megakaryocyte colony-forming units but spares normal CD34+ cells [Citation86]. In a phase II trial of imetelstat (NCT02426086) in patients with refractory MF, responses in SVR, TSS, and BMF were encouraging [Citation87,Citation88] (). The median survival was 30 months, twofold compared to historical controls treated with JAKi. SRSF2 mutation predicted responses to imetelstat. Overall, imetelstat was well tolerated. The commonest AE was reversible and dose-related myelosuppression. Grade ≥3 liver function test (LFT) elevations were observed in 7 patients on study. An independent review committee reviewed all LFT and concluded that they were unrelated to imetelstat.
3.2.3. Second mitochondrial-derived activator of caspases (Smac) mimetic (LCL161)
In MPN, TNF-α has been shown to promote clonal dominance of JAK2 V617F-expressing cells over normal hematopoietic cells [Citation89]. This resistance to apoptosis by TNF-α is mediated by inhibition of caspases by the inhibitor of apoptosis (IAP) proteins [Citation90]. Smac is an endogenous antagonist of IAP and thus promotes apoptosis [Citation91]. Smac-mimetics are compounds with N-terminal four amino acid stretch of the endogenous IAP antagonist Smac. LCL161, an orally bioavailable Smac mimetic, in high-risk MF patients refractory to or ineligible for ruxolitinib showed long-term responders (), but limited single agent activity in controlling symptom burden [Citation92].
3.2.4. Monotherapies targeting MPN HSC in early recruitment stage
PRT543, a novel and selective protein arginine methyl transferases (PRMT) inhibitor is being tested in a phase I trial in MF (NCT03886831). PRMT plays an important role in histone methylation. It is strongly phosphorylated by JAK2, reducing its methylation activity and PRMT5 inhibition activated p53 in JAK2-mutant progenitors [Citation93]. KRT-232 is an MDM2 inhibitor being evaluated in patients with relapsed or refractory MF (NCT03662126) in subjects not achieving at least a partial remission (PR) after 3 cycles. Selinexor, an oral, first-in-class selective inhibitor of nuclear export (SINE) compound is undergoing evaluation in refractory MF in a phase II trial (ESSENTIAL, NCT03627403). It inhibits nuclear–cytoplasmic transport (NCT), essential for the survival of JAK2 V617F mutant cells [Citation94]. PU-H71 is another molecule in phase Ib testing (NCT03935555) that inhibits heat shock protein 90 (Hsp90), the chaperones for JAK2 protein [Citation95]. In JAK inhibitor persistent cells, inhibition of Hsp90 using PU-H71 led to degradation of JAK2 and abrogation of downstream JAK2 signaling [Citation96].
3.3. Monotherapies that target HSC niche/microenvironment/fibrosis
3.3.1. Anti-CD-123 fusion protein, SL-401 (tagraxofusp)
CD123 is the α subunit of IL-3 receptor (IL-3R) to which IL-3 binds and results in cell survival and proliferation [Citation97]. CD123 is not expressed on normal HSC but highly expressed in leukemic stem cell compartment in AML [Citation98]. In MF, 1- 2% of circulating cells are CD123+ and 30 to 50% of CD123+ cells co-express CD13, CD16 or CD11b, representing monocytes and immature myeloid cells [Citation99]. Tagraxofusp, a targeted therapy directed to CD123, comprises recombinant IL-3 fused to a truncated diphtheria toxin payload [Citation100]. In the interim results of a phase I/II trial in relapsed MF (), tagraxofusp showed modest single agent activity, particularly in patients with an associated monocytosis and manageable safety profile [Citation101] (NCT02268253).
3.3.2. Aurora kinase inhibitor (alisertib)
The Aurora kinases take part in chromosomal segregation during endomitosis, the unique cell cycle process that leads to the polyploidization of megakaryocytes [Citation102]. An aurora kinase inhibitor- alisertib in the MPN models preferentially induced apoptosis and differentiation of mutant megakaryocytes, reduced TGF-β secretion, and improved BMF [Citation103]. Clinically, alisertib showed an impressive reduction in BMF () and increased GATA-1 staining suggestive of megakaryocyte differentiation [Citation104].
3.3.3. Recombinant pentraxin-2 (PRM-151)
PRM-151 is a recombinant form of pentraxin-2, an endogenous human protein that induces macrophage differentiation to prevent and reverse fibrosis [Citation105]. PRM-151 as monotherapy in relapsed refractory MF reduced BMF, reduced transfusion requirements, with only modest benefits in symptom control (). With its ability to target BMF and non-overlapping toxicity, PRM-151 could be combined with drugs targeting HSC clone for its disease modification effect [Citation106].
3.3.4. Monotherapies targeting microenvironment in early recruitment stage
Mirabegron, a β-3 sympathomimetic agonist that restored nestin-positive cells within the stem cell niche and improved BMF in a mouse model of JAK2 V617F positive MPN [Citation107]. In a phase II trial, the primary endpoint of reduction of JAK2 V617F allele burden was not reached in any of the patients with MF [Citation108]. TGF-β pathway plays a dual role in promoting myelofibrosis and myeloproliferation in MF [Citation109]. AVID200 is a drug that targets both TGF-β1 and TGF-β3. A first in human, phase I/Ib trial of AVID200, in relapsed refractory MF is underway (NCT03895112). The only immune check point inhibitor in clinical development in MF is pembrolizumab (NCT03065400), based on preclinical studies showing higher PD-L1 expression on primary patient cells compared to controls, and prolongation survival in MPN xenograft models with PD-L1 inhibition [Citation110]. The only vaccine strategy in MF, using the CALR peptide sequence, is based on the induction of specific T-memory immune responses against the epitopes in the mutant CALR peptide [Citation111]. This trial has completed enrollment and results are expected (NCT03566446).
4. Conclusion
Our review describes the non-JAK inhibitor drugs in early phase clinical trials for chronic phase myelofibrosis. These agents broadly target MPN HSC using non-canonical molecular pathways and/or bone marrow microenvironment. Most new agents as an add-on to ruxolitinib have shown limited efficacy in prolonging or deepening responses (HDACi/hedgehog inhibitors/interferons/PI3Kδ inhibitors) or improving toxicity (IMiD). They are unlikely to undergo further development. Others such as BETi (CPI-0610) and activin receptor ligand traps (luspatercept and sotatercept) that target anemia, have shown benefit when combined with ruxolitinib and are likely to be evaluated in further phase III trials. PRM-151 and imetelstat improved or reversed BM fibrosis as single agents and are attractive as a disease modification strategy in combination with JAKi. A number of new molecular targets (MDM2, P-selectin, TIM-3, Bcl-2, TGF-β, aurora kinase, hsp90, CALR vaccine, and allogeneic cord blood regulatory T cells) are early into patient enrollment and the results are keenly awaited. In addition, preclinical strategies, such as combining targeted agents with JAKi, will bring in an enthusing drug development pipeline in myelofibrosis.
5. Expert opinion
The JAK-STAT signaling is the most commonly targeted molecular pathway in myelofibrosis. The JAKi drugs ruxolitinib and fedratinib have significantly improved the care and the quality of life for patients with MF. The rapid advances in the basic and translational research has shown contribution of many other complex molecular pathways to MF pathogenesis. Based on strong preclinical evidence, the drugs inhibiting these non JAK-STAT pathways are evaluated to deepen or prolong the responses of JAKi, reduce its side effects, provide an option for treatment in the setting of anemia and thrombocytopenia, modify the disease biology, reduce leukemic transformation and improve progression-free and overall survival.
From clinical standpoint, the results of BET inhibitor (CPI-0610) in combination with ruxolitinib in the frontline setting would be keenly followed. This appears a promising therapy which can potentially change the standard of care for upfront treatment. JAKi- and disease-induced anemia continues to be a challenge in patients with MF and results in significant impairment in quality of life and complications related to transfusional iron overload. Activin receptor ligand (luspatercept) and BET inhibitor (CPI-0610) may provide an add-on option for preventing or rescuing JAKi-induced anemia or as monotherapy when symptom control with JAKi is not required. Another common reason for JAKi to fail is suboptimal or loss of response. The BET inhibitor, Bcl-2 inhibitor, and azacitidine in combination with ruxolitinib have encouraging activity in this setting. Several drugs have shown improvement in bone marrow fibrosis including PRM-151, azacitidine, BET inhibitor, navitoclax, imetelstat and alisertib. Demonstration of impact of this finding on improving cytopenia and disease progression would be important as it could usher in a new strategy of combining drugs for its dual effects. Results of several new disease modification strategies such as MDM-2 inhibitor and anti-p-selectin and anti-TIM-3 monoclonal antibodies would be important to see if they reduce disease progression. None of the current treatments under evaluation have shown any evidence for reducing leukemic transformation.
In addition to evaluation of newer agents, a priority for MPN experts and industry alike is to revisit the many challenges that exist for developing combinational treatments in MF and formulate solutions. First, ensuring a strong scientific rationale based on robust preclinical models for the combination in terms of increased efficacy. The clinical studies should include biomarkers indicative of inhibition of targeted molecular pathways. Second, validation of surrogate endpoints that show an association with reduced transformation to AML or improved overall survival is required. Many patients with MF live longer than a decade and incorporating mortality end points could result in excessively long clinical trials or poor predictability due to fewer events. On the other hand, patients with ‘ruxolitinib failure’ have a short median OS and time-to event endpoints are feasible. Notwithstanding, ‘ruxolitinib failure’ itself needs a consensus definition so that trial results are comparable. Previous studies have shown that patients with high DIPSS score, pre-treatment transfusion dependence, and number and type of mutations (ASXL1/EZH2) are associated with shorter time to treatment failure in MF [Citation112,Citation113]. Identification of these patients who are unlikely to benefit from JAKi therapy is desirable, and these patients should preferably be enrolled in clinical trials.
Finally, the need to optimize the trial designs. If the evaluation of the synergy of the combinations will be the strategy henceforth, we cannot be relying on ‘contemporary’ control of COMOFRT trials. It needs no emphasis that patients with MF are heterogeneous and the trials therefore should have a control arm. It is even more important when the drugs are evaluated in the setting of ruxolitinib failure where we risk missing a small measurable benefit in the absence of a control arm. We need to adopt statistical designs that improve efficiency and shorten development time, such as randomization selection (pick-the-winner) and adaptive designs. A refined approach guiding MPN community is the patient-focused BEAT AML master trial where various stakeholders have collaborated to provide personalized treatment for patients with AML. This has ensured robust recruitment and a provision of control for every patient. Such productive alliances between multiple institutions, both academic and pharmaceutical based, can overcome the economic, intellectual, and logistical issues, and pave a way for future success in MF.
Article Highlights
Most non-JAKi agents evaluated so far have shown modest benefit in increasing the efficacy of ruxolitinib.
The BET inhibitor- CPI-0610 + ruxolitinib has shown early promise in reducing splenomegaly and transfusion dependency and is well tolerated.
The activin receptor ligand trap- luspatercept has been shown to mitigate ruxolitinib- and disease- induced anemia.
Telomerase inhibitor- imetelstat and recombinant pentraxin-PRM-151 have encouraging anti-fibrosis activity.
Early phase trials of new molecular targets (MDM2, p-selectin, TIM-3, bcl-2, TGF-β, aurora kinase, hsp-90) and immune-based strategies (CALR vaccine, anti-PD-1, allogeneic cord blood regulatory T cells) are ongoing.
Further translational studies targeting specific mutation and leukemic stem cells along with improvement in trial designs and patient-focused collaborations could pave a way for future success in MF drug development.
Declaration of interest
V Gupta received an honorarium and clinical trial funding through his institution and has served on an advisory board for Novartis, Celgene and Sierra Oncology. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
One reviewer has received research funding [via their institution] from CTI Biopharma, Constellation, Roche, Promedior, Janssen, Incyte, Novartis, Merck, Merus, Arog, Kartos, and Celgene. He/she has also been on the consulting, clinical trial and scientific advisory board for Roche, Constellation, Kartos, Incyte, and Celgene. One reviewer has received honoraria and research support from Incyte, Celgene (now BMS), Constellation, Kartos and CTI Biopharma. Peer reviewers on this manuscript have no other relevant financial or other relationships to disclose.
Additional information
Funding
References
- Tefferi A, Lasho TL, Jimma T, et al. One thousand patients with primary myelofibrosis: the mayo clinic experience. Mayo Clin Proc. 2012;87:25–33.
- Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the international working group for myelofibrosis research and treatment. Blood, J American Soc Hematol. 2009;113:2895–2901.
- Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (international working group for myeloproliferative neoplasms research and treatment). Blood,JAmerican Soc Hematol. 2010;115:1703–1708.
- Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined dynamic international prognostic scoring system for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J clin oncol. 2011;29:392–397.
- Tefferi A, Guglielmelli P, Lasho TL, et al. MIPSS70+ version 2.0: mutation and karyotype-enhanced international prognostic scoring system for primary myelofibrosis. J Clin Oncol. 2018;36:1769.
- Devlin R, Gupta V. Myelofibrosis: to transplant or not to transplant? Hematology Am Soc Hematol Educ Program. 2016;2016:543–551.
- Elliot Smith LL, Viswabandya A, Maze D, et al. Factors influencing selection of upfront hematopoietic stem cell transplantation versus best available non-transplant therapy in myelofibrosis. New York: 12th International Congress on Myeloproliferative Neoplasms; 2019.
- Blair HA. Fedratinib: first Approval. Drugs. 2019;79:1719–1725.
- Mascarenhas J, Hoffman R. Ruxolitinib: the first FDA approved therapy for the treatment of myelofibrosis. Clin Cancer Res. 2012;18:3008–3014.
- Palandri F, Breccia M, Bonifacio M, et al. Life after ruxolitinib: reasons for discontinuation, impact of disease phase, and outcomes in 218 patients with myelofibrosis. Cancer. 2020;126(6):1243-1252.
- Verstovsek S, Mesa RA, Gotlib J, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Newberry KJ, Patel K, Masarova L, et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood. 2017;130:1125–1131.
- Tefferi A, Lasho TL, Jimma T, et al. One thousand patients with primary myelofibrosis: the mayo clinic experience. Mayo Clin Proc. 2012;87:25–33.
- Mascarenhas J, Hoffman R, Talpaz M, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol. 2018;4:652–659.
- Xu L, Feng J, Gao G, et al. Momelotinib for the treatment of myelofibrosis. Expert Opin Pharmacother. 2019;20:1943–1951.
- Mullally A, Lane SW, Brumme K, et al. Myeloproliferative neoplasm animal models. Hematol/Oncol Clin. 2012;26:1065–1081.
- Jain T, Mesa R. The development, safety and efficacy of pacritinib for the treatment of myelofibrosis. Expert Rev Anticancer Ther. 2016;16:1101–1108.
- Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med. 1996;125:605–613.
- Mandrekar SJ, Sargent DJ. Randomized phase II trials: time for a new era in clinical trial design. J Thorac Oncol. 2010;5:932–934.
- Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood,JAmerican Soc Hematol. 2017;129:667–679.
- Zoi K, Cross N. Genomics of myeloproliferative neoplasms. J clin oncol. 2017;35:947–954.
- Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061.
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790.
- Levine RL, Loriaux M, Huntly BJ, et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood. 2005;106:3377–3379.
- Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–2405.
- Defour J-P, Itaya M, Gryshkova V, et al. Tryptophan at the transmembrane–cytosolic junction modulates thrombopoietin receptor dimerization and activation. Proceedings of the National Academy of Sciences 2013;110:2540–2545.
- Levine RL, Pardanani A, Tefferi A, et al. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673.
- Lundberg P, Karow A, Nienhold R, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123:2220–2228.
- Wagner-Ballon O, Chagraoui H, Prina E, et al. Monocyte/macrophage dysfunctions do not impair the promotion of myelofibrosis by high levels of thrombopoietin. J Immunol. 2006;176:6425–6433.
- Kleppe M, Koche R, Zou L, et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell. 2018;33:29–43.e27.
- Florence B, Faller DV. You bet-cha: a novel family of transcriptional regulators. Front Biosci. 2001;6:D1008–D1018.
- Mascarenhas J, Kremyanskaya M, Hoffman R, et al. MANIFEST, a phase 2 study of cpi-0610, a bromodomain and extraterminal domain inhibitor (BETi), as monotherapy or” add-on” to ruxolitinib, in patients with refractory or intolerant advanced myelofibrosis. Blood. 2019;134(Supplement_1):670-670.
- Harrison CN, Patriarca A, Mascarenhas J, et al. Preliminary Report of MANIFEST, a phase 2 study of cpi-0610, a bromodomain and extraterminal domain inhibitor (BETi), in combination with ruxolitinib, in JAK inhibitor (JAKi) treatment naïve myelofibrosis patients. Blood. 2019;134(Supplement_1):4164-4164.
- Mesa RA, Verstovsek S, Rivera C, et al. 5-Azacitidine has limited therapeutic activity in myelofibrosis. Leukemia. 2009;23:180–182.
- Masarova L, Verstovsek S, Hidalgo-Lopez JE, et al., A phase 2 study of ruxolitinib in combination with azacitidine in patients with myelofibrosis. Blood. 2018;132(16): 1664–1674. .
- Gao S-M, Chen C-Q, Wang L-Y, et al. Histone deacetylases inhibitor sodium butyrate inhibits JAK2/STAT signaling through upregulation of SOCS1 and SOCS3 mediated by HDAC8 inhibition in myeloproliferative neoplasms. Exp Hematol. 2013;41(261–270):e264. .
- Calzada AA, Pedrini O, Finazzi G, et al. Givinostat and hydroxyurea synergize in vitro to induce apoptosis of cells from JAK2V617F myeloproliferative neoplasm patients. Exp Hematol. 2013;41(253–260):e252.
- Fiskus W, Verstovsek S, Manshouri T, et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin Cancer Res. 2011;17:7347–7358.
- Bose P, Swaminathan M, Pemmaraju N, et al. A phase 2 study of pracinostat combined with ruxolitinib in patients with myelofibrosis. Leuk Lymphoma. 2019;60:1767–1774.
- Mascarenhas J, Marcellino B, Lu M, et al. A phase I study of panobinostat and ruxolitinib in patients with primary myelofibrosis (PMF) and post-polycythemia vera/essential thrombocythemia myelofibrosis (post-PV/ET MF). Leuk Res. 2020;88:106272.
- Jean S, Kiger AA. Classes of phosphoinositide 3-kinases at a glance. Journal of Cell Science. 2014;127(5):923.
- Khan I, Huang Z, Wen Q, et al. AKT is a therapeutic target in myeloproliferative neoplasms. Leukemia. 2013;27:1882.
- Choong ML, Pecquet C, Pendharkar V, et al. Combination treatment for myeloproliferative neoplasms using JAK and pan-class I PI3K inhibitors. J Cell Mol Med. 2013;17:1397–1409.
- Vakkalanka S, Viswanadha S, Niecestro R, et al. TGR-1202 Suppresses AML and ALL Cells Via Selective Inhibition of PI3Kδ Kinase. Blood. 2012;120:2610.
- Moyo TK, Palmer J, Huang Y, et al. Resurrecting response to ruxolitinib: a phase I study of ruxolitinib and umbralisib (TGR-1202) in ruxolitinib-experienced myelofibrosis. HemaSphere. 2018;2(S1):19-20.
- Daver N, Dao K-H, Assad A, et al. 2 Study of the Safety and Efficacy of INCB050465 in Combination with Ruxolitinib in Patients with Myelofibrosis (MF). Clin Lymphoma Myeloma Leukemia. 2017;17:S351.
- Durrant ST, Nagler A, Guglielmelli P, et al. Results from HARMONY: an open-label, multicentre, 2-arm, phase 1b, dose-finding study assessing the safety and efficacy of the oral combination of ruxolitinib and buparlisib in patients with myelofibrosis. Haematologica. 2019:haematol.2018.209965.
- Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013;32:2601.
- Mazzacurati L, Collins RJ, Pandey G, et al. The pan-PIM inhibitor INCB053914 displays potent synergy in combination with ruxolitinib in models of MPN. Blood Adv. 2019;3:3503–3514.
- Bhagwat N, Keller MD, Rampal RK, et al. Improved efficacy of combination of JAK2 and hedgehog inhibitors in myelofibrosis. Blood. 2013;122(21):666-666.
- Couban S, Benevolo G, Donnellan W, et al. Phase 1b results of a study to assess the efficacy and safety of vismodegib in combination with ruxolitinib in patients with intermediate-or high-risk myelofibrosis. Blood. 2017;130(Supplement 1):4179-4179.
- Gupta V, Harrison CN, Hasselbalch H, et al. Phase 1b/2 study of the efficacy and safety of sonidegib (LDE225) in combination with ruxolitinib (INC424) in patients with myelofibrosis. Am Soc Hematology; 2015.
- Gerds AT, Tauchi T, Ritchie E, et al. Phase 1/2 trial of glasdegib in patients with primary or secondary myelofibrosis previously treated with ruxolitinib. Leuk Res. 2019;79:38–44.
- Tognon R, Gasparotto EPL, Neves RP, et al. Deregulation of apoptosis-related genes is associated with PRV1 overexpression and JAK2 V617F allele burden in essential thrombocythemia and myelofibrosis. J Hematol Oncol. 2012;5:2.
- Maiuri MC, Criollo A, Tasdemir E, et al. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-XL. Autophagy. 2007;3:374–376.
- Waibel M, Solomon VS, Knight DA, et al. Combined targeting of JAK2 and Bcl-2/Bcl-xL to cure mutant JAK2-driven malignancies and overcome acquired resistance to JAK2 inhibitors. Cell Rep. 2013;5:1047–1059.
- Harrison CN, Garcia JS, Mesa RA, et al. Results from a phase 2 study of navitoclax in combination with ruxolitinib in patients with primary or secondary myelofibrosis. Blood. 2019;134:671.
- Quach H, Ritchie D, Stewart AK, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. 2010;24:22.
- Weinkove R, Reilly JT, McMullin MF, et al. Low-dose thalidomide in myelofibrosis. Haematologica. 2008;93:1100–1101.
- Mesa RA, Pardanani AD, Hussein K, et al. Phase1/‐2 study of Pomalidomide in myelofibrosis. Am J Hematol. 2010;85:129–130.
- Rampal RK, Verstovsek S, Devlin SM, et al. Safety and efficacy of combined ruxolitinib and thalidomide in patients with myelofibrosis: a phase II study. Blood. 2019;134(Supplement_1):4163-4163.
- Tefferi A, Al-Ali H, Barosi G, et al. A randomized study of pomalidomide vs placebo in persons with myeloproliferative neoplasm-associated myelofibrosis and RBC-transfusion dependence. Leukemia. 2017;31:896.
- Stegelmann F, Koschmieder S, Isfort S, et al. Updated results from the german mpnsg-0212 combination trial: ruxolitinib plus pomalidomide in Myelofibrosis with Anemia. Blood. 2019;134(Supplement_1):672-672.
- Ceglia I, Dueck AC, Masiello F, et al. Preclinical rationale for TGF-β inhibition as a therapeutic target for the treatment of myelofibrosis. Exp Hematol. 2016;44(1138–1155):e1134. .
- Suragani RN, Cadena SM, Cawley SM, et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20:408.
- Gerds AT, Vannucchi AM, Passamonti F, et al. A phase 2 study of luspatercept in patients with Myelofibrosis-Associated Anemia. Blood. 2019;134(Supplement_1):557-557.
- Bose P, Pemmaraju N, Daver N, et al. Sotatercept (ace-011) in subjects with mpn-associated myelofibrosis and anemia: S829. Hemasphere. 2019;3:367–368.
- Cappellini MD, Porter J, Origa R, et al. Sotatercept, a novel transforming growth factor β ligand trap, improves anemia in β-thalassemia: a phase II, open-label, dose-finding study. Haematologica. 2019;104:477.
- Xia Y, Schneyer AL. The biology of activin: recent advances in structure, regulation and function. J Endocrinol. 2009;202:1–12.
- Ianotto JC, Kiladjian JJ, Demory JL, et al. PEG‐IFN‐α‐2a therapy in patients with myelofibrosis: A study of the French groupe d’etudes des myelofibroses (GEM) and France intergroupe des syndromes myéloprolifératifs (FIM). Br J Haematol. 2009;146:223–225.
- Silver RT, Vandris K, Goldman JJ. Recombinant interferon-α may retard progression of early primary myelofibrosis: a preliminary report. Blood. 2011;117:6669–6672.
- Mikkelsen SU, Kjær L, Bjørn ME, et al. Safety and efficacy of combination therapy of interferon-α2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med. 2018;7:3571–3581.
- Kiladjian -J-J, Soret-Dulphy J, Resche-Rigon M, et al. Ruxopeg, a multi-center bayesian phase 1/2 adaptive randomized trial of the combination of ruxolitinib and pegylated interferon alpha 2a in patients with myeloproliferative neoplasm (MPN)-associated myelofibrosis. Blood. 2018;132:581.
- Nakatake M, Monte-Mor B, Debili N, et al. JAK2V617F negatively regulates p53 stabilization by enhancing MDM2 via La expression in myeloproliferative neoplasms. Oncogene. 2012;31:1323–1333.
- Spangrude GJ, Lewandowski D, Martelli F, et al. P-Selectin Sustains Extramedullary Hematopoiesis in the Gata1 low Model of Myelofibrosis. Stem Cells. 2016;34:67–82.
- Kikushige Y, Shima T, Takayanagi S, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7:708–717.
- Kikushige Y, Miyamoto T, Yuda J, et al. A TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell. 2015;17:341–352.
- Ndhlovu LC, Lopez-Vergès S, Barbour JD, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. 2012;119:3734–3743.
- Wang JC, Sindhu H, Chen C, et al. Immune derangements in patients with myelofibrosis: the role of Treg, Th17, and sIL2Rα. PloS One. 2015;10:e0116723.
- Hasselbalch HC, Holmström MO. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: minimal residual disease and cure? Semin Immunopathol. 2019 Jan;41(1):5-19.
- Kadia TM, Ma H, Zeng K, et al. Phase I clinical trial of CK0801 (cord blood regulatory T cells) in patients with bone marrow failure syndrome (BMF) including aplastic anemia, myelodysplasia and myelofibrosis. Blood. 2019;134(Supplement_1):1221-1221.
- Sprüssel A, Schulte J, Weber S, et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia. 2012;26:2039.
- Jutzi JS, Kleppe M, Dias J, et al. LSD1 Inhibition prolongs survival in mouse models of MPN by selectively targeting the disease clone. Hemasphere. 2018;2:e54.
- Pettit K, Gerds AT, Yacoub A, et al. A phase 2a study of the LSD1 inhibitor img-7289 (bomedemstat) for the treatment of myelofibrosis. Blood. 2019;134(Supplement_1):556-556. .
- Bernard L, Belisle C, Mollica L, et al. Telomere length is severely and similarly reduced in JAK2V617F-positive and-negative myeloproliferative neoplasms. Leukemia. 2009;23:287.
- Wang X, Hu CS, Petersen B, et al. Imetelstat, a telomerase inhibitor, is capable of depleting myelofibrosis stem and progenitor cells. Blood Adv. 2018;2:2378–2388.
- Mascarenhas J, Komrokji RS, Cavo M, et al. imetelstat is effective treatment for patients with intermediate-2 or high-risk myelofibrosis who have relapsed on or are refractory to janus kinase inhibitor therapy: results of a phase 2 randomized study of two dose levels. Blood. 2018;132:685.
- Tefferi A, Lasho TL, Begna KH, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015;373:908–919.
- Heaton WL, Senina AV, Pomicter AD, et al. Autocrine Tnf signaling favors malignant cells in myelofibrosis in a Tnfr2-dependent fashion. Leukemia. 2018;32:2399–2411.
- Deveraux QL, Reed JC. IAP family proteins–suppressors of apoptosis. Genes Dev. 1999;13:239–252.
- Du C, Fang M, Li Y, et al. Smac, a mitochondrial protein that promotes cytochrome c–dependent caspase activation by eliminating IAP Inhibition. Cell. 2000;102:33–42.
- Pemmaraju N, Carter BZ, Kantarjian HM, et al. Final results of phase 2 clinical trial of LCL161, a novel oral SMAC mimetic/IAP antagonist, for patients with intermediate to high risk myelofibrosis. Blood. 2019;134:555.
- Pastore F, Bhagwat N, Krishnan A, et al. PRMT5 inhibition modulates E2F1 methylation and gene regulatory networks leading to therapeutic efficacy in JAK2VF mutant MPN. Blood. 2019;134(Supplement_1):473-473.
- Yan D, Pomicter AD, Tantravahi S, et al. Nuclear–Cytoplasmic Transport Is a Therapeutic Target in Myelofibrosis. Clin Cancer Res. 2019;25:2323–2335.
- Marubayashi S, Koppikar P, Taldone T, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010;120:3578–3593.
- Bhagwat N, Koppikar P, Keller M, et al. Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms. Blood. 2014;123:2075–2083.
- Bagley CJ, Woodcock JM, Stomski FC, et al. The structural and functional basis of cytokine receptor activation: lessons from the common β subunit of the granulocyte-macrophage colony-stimulating factor, interleukin-3 (IL-3), and IL-5 receptors. Blood,JAmerican Soc Hematol. 1997;89:1471–1482.
- Jin L, Lee EM, Ramshaw HS, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor α chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5:31–42.
- Lasho T, Finke C, Kimlinger TK, et al. Expression of CD123 (IL-3R-alpha), a therapeutic target of SL-401, on myeloproliferative neoplasms. Am Soc Hematology; 2014.
- Testa U, Riccioni R, Biffoni M, et al. Diphtheria toxin fused to variant human interleukin-3 induces cytotoxicity of blasts from patients with acute myeloid leukemia according to the level of interleukin-3 receptor expression. Blood. 2005;106:2527–2529.
- Pemmaraju N, Gupta V, Ali H, et al. Results from a phase 1/2 clinical trial of tagraxofusp (SL-401) in patients with intermediate, or high risk, relapsed/refractory myelofibrosis. Blood. 2019;134(Supplement_1):558-558.
- Zhang Y, Nagata Y, Yu G, et al. Aberrant quantity and localization of Aurora-B/AIM-1 and survivin during megakaryocyte polyploidization and the consequences of Aurora-B/AIM-1–deregulated expression. Blood. 2004;103:3717–3726.
- Wen Q, Goldenson B, Silver SJ, et al. Identification of regulators of polyploidization presents therapeutic targets for treatment of AMKL. Cell. 2012;150:575–589.
- Gangat N, Marinaccio C, Swords R, et al. Aurora kinase A inhibition provides clinical benefit, normalizes megakaryocytes and reduces bone marrow fibrosis in patients with myelofibrosis. Clin Cancer Res. 2019 Aug 15;25(16):4898-4906.
- Castaño AP, Lin S-L, Surowy T, et al. Serum amyloid P inhibits fibrosis through FcγR-dependent monocyte-macrophage regulation in vivo. Sci Transl Med. 2009;1:5ra13–15ra13.
- Verstovsek S, Talpaz M, Wadleigh M, et al. A randomized, double blind phase 2 study of 3 different doses of prm-151 in patients with myelofibrosis who were previously treated with or ineligible for ruxolitinib: S828. Hemasphere. 2019;3:367.
- Vannucchi AM, Bianchi L, Paoletti F, et al. A pathobiologic pathway linking thrombopoietin, GATA-1, and TGF-beta1 in the development of myelofibrosis. Blood. 2005;105:3493–3501.
- Drexler B, Passweg JR, Tzankov A, et al. The sympathomimetic agonist mirabegron did not lower JAK2-V617F allele burden, but restored nestin-positive cells and reduced reticulin fibrosis in patients with myeloproliferative neoplasms: results of phase II study SAKK 33/14. Haematologica. 2019;104:710–716.
- Arranz L, Sanchez-Aguilera A, Martin-Perez D, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014;512:78–81.
- Prestipino A, Emhardt AJ, Aumann K, et al. Oncogenic JAK2(V617F) causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Science Translational Medicine. 2018;10(429):eaam7729.
- Holmström MO, Ahmad SM, Klausen U, et al. High frequencies of circulating memory T cells specific for calreticulin exon 9 mutations in healthy individuals. Blood Cancer J. 2019;9:8.
- Spiegel JY, McNamara C, Kennedy JA, et al. Impact of genomic alterations on outcomes in myelofibrosis patients undergoing JAK1/2 inhibitor therapy. Blood Adv. 2017;1:1729–1738.
- Patel KP, Newberry KJ, Luthra R, et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood. 2015;126:790–797.