
ABSTRACT
Introduction
Antisense oligonucleotides (ASOs) represent a class of drugs which can be rationally designed to complement the coding or non-coding regions of target RNA transcripts. They could modulate pre-messenger RNA splicing, induce mRNA knockdown, or block translation of disease-causing genes, thereby slowing disease progression. The pharmacokinetics of intravitreal delivery may enable ASOs to be effective in the treatment of inherited retinal diseases.
Areas covered
We review the current status of clinical trials of ASO therapies for inherited retinal diseases, which have demonstrated safety, viable durability, and early efficacy. Future applications are discussed in the context of alternative genetic approaches, including gene augmentation and gene editing.
Expert opinion
Early efficacy data suggest that the splicing-modulating ASO, sepofarsen, is a promising treatment for Leber congenital amaurosis associated with the common c.2991+1655A>G mutation in CEP290. However, potential variability in clinical response to ASO-mediated correction of splicing defect on one allele in patients who are compound heterozygotes needs to be assessed. ASOs hold great therapeutic potential for numerous other inherited retinal diseases with common deep-intronic and dominant gain-of-function mutations. These would complement viral vector-mediated gene augmentation which is generally limited by the size of the transgene and to the treatment of recessive diseases.
1. Introduction
Antisense oligonucleotides (ASOs) represent a class of genetic therapies which exert their action by directly modulating target gene expression or function, and could form an important therapeutic approach, alongside gene augmentation and gene editing, for the treatment of inherited retinal diseases [Citation1].
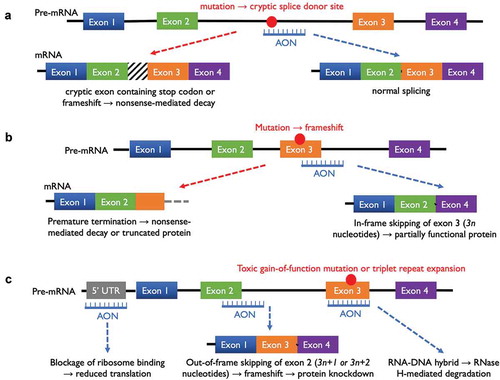
Synthetic ASOs are single-stranded oligodeoxynucleotides or oligoribonucleotides, typically 15–30 nucleotides (nt) in length, that can be rationally designed to hybridize with the target pre-messenger or messenger RNA (mRNA) of disease-related genes by Watson/Crick base pairing. A variety of effects on pre-mRNA processing or mRNA translation could result depending on where the ASO binds along the RNA transcript, as summarized in .
Figure 1. Mechanisms of action of antisense oligonucleotide (ASO) therapies. (a) An ASO hybridizing with a deep-intronic mutation could prevent aberrant splicing which would otherwise lead to the incorporation of a cryptic exon or frameshift. (b) An ASO could be designed to block a splice donor site in order to cause skipping of a mutation-containing exon, so that a shortened but partially functional protein may be produced. (c) ASOs could be designed to knockdown a dominant negative allele by direct binding to the mutant RNA sequence to generate a RNA-DNA hybrid, which would be recognized and degraded by RNase H. Alternatively, knockdown of both alleles may be achieved through ASO binding to the 5ʹ-untranslated region of a target mRNA or ASO-mediated exon skipping with frameshift.
The clinical viability of early ASOs was hampered by their poor stability in vivo due to the abundance of endonucleases which could degrade oligonucleotides with unmodified sugar phosphate backbone. Over the past two decades, three generations of synthetic ASOs have been developed with a range of chemical modifications to both the nucleic acid phosphate backbone and sugar rings. Particularly notable advances have been (first generation) ASOs with phosphorothioate (PS) backbone, (second generation) ASOs with PS backbone and a sugar modification such as 2ʹ-O-methoxyethyl (MOE) or 2ʹ-O-methyl (OMe), and (third generation) phosphorodiamidate morpholino oligonucleotides (PMO) [Citation2–Citation4]. The biochemical properties of these modifications and their effects have been reviewed in detail elsewhere [Citation5]. These developments have led to the emergence of clinical grade ASOs with increased resistance to endonuclease-mediated degradation while improving their ability to penetrate the cell membrane and affinity for hybridizing with pre-mRNA [Citation6–Citation8]. Moreover, the uniqueness of a 15 to 30-mer oligonucleotide sequence is generally associated with highly specific on-target hybridization and low levels of off-target effects [Citation9].
One of the main modes of actions of ASOs is to block gene expression through targeted degradation of mRNA. For instance, if an ASO hybridizes with the complementary sequence of a target mRNA in the cytoplasm or nucleus, the resulting RNA-DNA hybrid would be recognized by the ubiquitous endonuclease, RNase H, for degradation, leading to reduced protein expression. The clinical viability of this gene knockdown approach was first demonstrated by fomivirsen, a 21-mer first-generation phosphorothioate ASO that binds to the mRNA encoding the major immediate-early region 2 protein of the cytomegalovirus (CMV), which can be administered via intravitreal injection for the treatment of CMV retinitis [Citation10]. While fomivirsen was the first ASO to receive FDA approval in 1998, it was subsequently withdrawn by the manufacturer due to low demand associated with reduced incidence of CMV retinitis following the introduction of highly active antiretroviral therapy (HAART) for HIV. Nevertheless, this proof-of-concept demonstrated the viable pharmacokinetics of an intravitreally delivered ASO and paved the path for subsequent development of a number of gene knockdown ASOs for life-threatening inherited neurodegenerations, including familial amyloid polyneuropathy, Huntington disease, SOD1 familial amyotrophic lateral sclerosis, and myotonic dystrophy 1 [Citation11,Citation12]. More recently, ASO-mediated gene knockdown therapy has been proposed as an antiviral mechanism to treat severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection; either by targeting the viral RNA directly, or by targeting mRNA for the host angiotensin converting enzyme 2 (ACE2) receptor for viral entry [Citation13].
An alternative mode of action of ASOs is through alteration of pre-mRNA splicing. By hybridizing with a nascent splice donor site during RNA polymerase II-mediated transcription, an ASO could prevent the normal recruitment of splicing factors, thus causing skipping of the targeted exon during RNA splicing. Depending on the length of the skipped exon and whether the alternatively spliced transcript remains in-frame or becomes frame-shifted, the ASO could lead to either a shortened (but potentially functional) protein lacking a possible domain, or gene knockdown through non-sense mediated decay of the mRNA, respectively. The exon-skipping approach is ideally suited to partially restoring protein function to a large gene containing many exons and harboring a mutation hotspot. It has already demonstrated clinical success in treating a number of neurodegenerations, in particular, eteplirsen – also known as Exondys 51 (Sarepta Therapeutics Inc, Cambridge, MA, USA) for Duchenne muscular dystrophy and nusinersen (Biogen Inc, Cambridge, MA, USA) for spinal muscular dystrophy [Citation14–Citation17].
In addition to the single-stranded ASOs, double-stranded small interfering RNAs (RNAi) have also been used to target mRNAs for degradation through the Dicer-RNA induced silencing complex (RISC) pathway [Citation18]. Partisiran (Alnylam Pharmaceuticals Inc, Cambridge, MA, USA) was the first-in-class RNAi therapy to be approved by the FDA. It is delivered by lipid nanoparticles into hepatocytes to suppress the production of both wild-type and mutant transthyretin (TTR), which can misfold and aggregate to cause debilitating amyloidosis with polyneuropathy [Citation19]. So far, however, RNAi therapeutics have seen limited clinical application in the treatment of retinal diseases, possibly due to the greater perceived challenge associated with their penetration of cell membrane compared with single-stranded ASOs. However, there are ongoing phase II trials of a chemically modified RNAi targeting the caspase 2 gene (QPI-1007, Quark Pharmaceuticals Inc, Newark, USA) with the aim of preventing neuronal apoptosis in non-arteritic ischemic optic neuropathy (NAION) and acute angle closure glaucoma (ClinicalTrials.gov ID: NCT02341560) [Citation20].
2. Antisense oligonucleotide therapy for CEP290-associated Leber congenital amaurosis
In a recent phase I/II clinical trial, a splice modifying ASO has demonstrated promising results in treating Leber congenital amaurosis 10 (LCA10) caused by a common mutation in the 290 kDa centrosomal protein gene (CEP290; OMIM 611,755) [Citation21]. The CEP290 protein normally plays a critical role in ciliogenesis, being located to the transition zone between the basal body and axoneme. Mutations in CEP290 are most commonly associated with childhood-onset blinding LCA but can also cause syndromic ciliopathies, including Senior-Loken syndrome, Joubert syndrome, nephronophthisis, Bardet-Biedl syndrome, and Meckel-Gruber syndrome [Citation22]. The most common mutation in CEP290 is a deep-intronic c.2991 + 1655A>G single nucleotide transition (also known as p.Cys998X) which is found in around 10% of patients with LCA [Citation23]. The mutation creates a cryptic splice donor site, which leads to the insertion of an aberrant pseudoexon (exon X) containing a premature stop-codon between exon 26 and 27. Rescue of aberrant splicing of the c.2991 + 1655A>G mutant gene and restoration of significant CEP290 protein levels using naked ASOs that block the cryptic splice donor site were first demonstrated in patient-derived fibroblasts and lymphoblastoid cells [Citation24,Citation25]. Subsequently, feasibility of this ASO-mediated splicing-modulation approach was further shown in a humanized mutant Cep290 mouse model (using naked 2ʹ-OMe/PS molecules and AAV delivery) [Citation26] and in-patient iPSC-derived retinal organoids (using an antisense morpholino) [Citation27]. One of the best performing ASOs to emerge from these pre-clinical studies, QR-110, was shown to restore pre-mRNA splicing and ciliogenesis in CEP290 c.2991 + 1655A>G homozygous iPSC-derived retinal organoids, reach the outer nuclear layer in rabbit retina after intravitreal delivery with a half-life of 58 days, as well as being well tolerated in non-human primates [Citation28].
QR-110 or sepofarsen (ProQR Therapeutics NV, Leiden, Netherlands) is a 17-mer RNA with 2ʹ-O-methyl modified bases and phosphorothioated backbone. In the phase I/II trial of sepofarsen (ClinicalTrials.gov ID: NCT03140969), 10 LCA10 patients with a mean age of 22.5 yr (SD 12.1) who were all compound heterozygotes for the c.2991 + 1655A>G mutation and another mutant allele in CEP290 received unilateral intravitreal injections of a loading dose (either 160 or 320 µg in 50 µl) of ASO followed by 3 monthly injections of a maintenance dose (either 80 or 160 µg). The best-corrected visual acuities (BCVA) of the study eyes ranged from 1.1 logMAR to light perception at baseline. By month 3 post-treatment, the BCVA asymmetry between treated and untreated eyes shifted from a mean of 0.12 logMAR in favor of the untreated eyes at baseline to 0.54 logMAR in favor of the treated eyes. The overall improvement in visual acuity of the treated eyes over the untreated eyes was 0.67 logMAR at 3 months and shown to be statistically significant by linear mixed-effect model (p = 0.022). The improvements in visual acuity corresponded to similar trends in red and blue full-field stimulus testing (FST) thresholds (which are indicative of improved cone function) and quantified improvement in fixation stability. In particular, one patient (a 41-year-old male who has a c.4723A>T transversion on the other allele) was an exceptional responder who reported subjective visual improvements in the treated eye from 6 weeks onwards. By 2 months after initiation of ASO therapy, the acuity in the treated eye improved from perception of light to 1.46 logMAR (20/580 Snellen equivalent). Further improvement to 1.28 logMAR was observed by month 4, which was sustained up to 6 months. FST results at 6 months were consistent with improved cone-mediated visual responses with threshold changes of 0.71 and 0.55 log units for red flash FST under dark and light-adapted conditions, respectively, and 1.21 and 0.77 log units for blue flash FST. With up to 9 months of follow-up, the trial encountered no serious adverse events, and in particular no signs of intraocular inflammation.
The finding of significant visual acuity improvements from 6 weeks onward in the CEP290-LCA trial provides strong evidence that a degree of outer segment regeneration from ‘dormant’ photoreceptors is possible following correction of the underlying genetic defect in primary ciliopathies. Indeed, two of the ten patients in the CEP290-LCA trial showed improved reflectance of the inner segment/outer segment line on optical coherence tomography (OCT) in the treated eyes, which was not seen in the untreated eyes. This observation is consistent with recent report of the first-in-human trial of gene replacement therapy for RPGR-associated retinitis pigmentosa, another ciliopathy, in which visual acuity and microperimetry improvements were accompanied by OCT evidence of outer segment regeneration in at least one individual [Citation29]. Altogether, these trial results would indicate that visual functional endpoints (e.g. BCVA and threshold testing) in retinal ciliopathies are likely to be achievable in a much shorter time frame compared with anatomical endpoints to demonstrate slowing of retinal degeneration in other forms of inherited retinal diseases.
Based on the results of the phase I/II trial, a multicentre phase II/III double-masked, randomized, sham-controlled clinical trial of sepofarsen (ClinicalTrials.gov ID: NCT03913143) has been approved with plans to recruit 30 patients. The patients will be randomized into three groups to receive (i) low-dose ASO, (ii) high-dose ASO, or (iii) sham intravitreal injections. At 12 months, the second eye may also be treated, or in the case of the sham group, ASO treatment may be started in one eye. In addition, an extension of the phase I/II study of sepofarsen (ClinicalTrials.gov ID: NCT03913130) is also under way which enables continued dosing of the first-treated eye, initiation of dosing in the contralateral eye, and monitoring of long-term therapeutic effects. The results of these studies are anticipated in the next 2 years.
3. Antisense oligonucleotide therapy for RHO-associated autosomal dominant retinitis pigmentosa
Using a similar approach to sepofarsen for CEP290-LCA, ASO therapies are in early clinical trials for a number of other common mutations found in inherited retinal diseases ().
Table 1. Current clinical trials of antisense oligonucleotides (ASOs) in inherited retinal diseases. All three ASOs are by ProQR Therapeutics NV, Leiden, the Netherlands. LCA.
Mutations in the rhodopsin gene (RHO) account for over 25% of autosomal dominant retinitis pigmentosa (adRP). Due to a suspected founder effect, around 10% of RHO-associated adRP are due to a C-to-A transversion in codon 23 of RHO, which causes a Pro-23-His single amino acid change [Citation30,Citation31]. The misfolded RHO proteins expressed from the mutant allele appear to accumulate in the endoplasmic reticulum, leading to pro-apoptotic stress responses and a toxic dominant negative effect on rod photoreceptors [Citation32]. Due to the dominant nature of most RHO mutations, the disease is not amenable to standard adeno-associated viral (AAV) vector-mediated gene augmentation therapy even though the RHO gene is under 1.5 kb. However, a ‘block and replace’ therapeutic strategy involving co-expression of an inhibitory shRNA against RHO and a codon-modified cDNA of RHO resistant to the shRNA has shown early success in a canine model of RHO-adRP [Citation33].
An ASO therapy targeting the P23 H mutation in RHO-adRP (QR-1123, ProQR Therapeutics) is currently under evaluation through a multi-center phase I/II clinical trial (ClinicalTrials.gov ID: NCT04123626). QR-1123 has been designed to hybridize with the mutant P23 H mRNA and induce allele-selective knockdown via RNase H-mediated cleavage while sparing the normal RHO transcripts. The molecule is a gapmer ASO containing a central sequence of DNA nucleotides flanked by RNA residues on either side, which facilitates RNase H-mediated cleavage of the target mRNA across the DNA gap. It was first developed by Ionis Pharmaceuticals Inc (Carlsbad, California, USA) with proof-of-concept studies performed in a P23 H rat model, demonstrating slowing of photoreceptor degeneration and preservation of visual function [Citation34]. The license was subsequently acquired by ProQR Therapeutics NV, who is conducting the current phase I/II trial. The study design includes an open-label group which will receive single dose-escalation therapy with the ASO, and a double-masked randomized group which will receive 3 monthly intravitreal injections of either ASO or sham. In total, 35 adult patients are being recruited and the planned follow-up is 12 months. The main objectives are to assess dose-related safety, and the effects on visual function (visual acuity, visual field, and FST) as well as retinal structure (OCT). Secondary outcomes also include changes in quality-of-life. Interim results may be expected within the next year.
4. Antisense oligonucleotide therapy for usher syndrome type 2
Usher syndrome type 2 (USH2A; OMIM 608,400) is an autosomal recessive ciliopathy and the most common subtype of Usher syndrome, which is characterized by a combination of retinitis pigmentosa and moderate congenital neurosensory hearing impairment. Most patients develop early rod-related peripheral visual field loss but preservation of cone-mediated central visual acuity until the end stage of degeneration [Citation35–Citation37]. Over 75% of Usher type 2 are associated with mutations in USH2A, encoding two spliceoforms of usherin consisting of 21 and 72 exons, respectively [Citation38–Citation41]. USH2A has been localized to the connecting cilium where the protein is believed to play an important role in the development and maintenance of cilia function [Citation42]. The size of the USH2A cDNA (15.6 kb) far exceeds the cargo capacity of AAV vectors commonly used in gene augmentation therapy; hence, alternative therapeutic approaches are required. While a large number mutations have been identified in patients with USH2A, the mutations are generally heterogeneous with the exception of the c.2299delG (p.Glu767fs) mutation in exon 13 [Citation41,Citation43].
A splicing-modulating ASO therapy (QR-421a, ProQR Therapeutics NV) for the subgroup of Usher 2A patients harboring mutations within exon 13 has been in phase I/II clinical trial since March 2019 (ClinicalTrials.gov ID: NCT03780257). QR-421a is a 21-mer single-stranded second-generation ASO designed to cause in-frame skipping of exon 13 during USH2A pre-mRNA splicing. Production of a shortened version of usherin (without exon 13) has been demonstrated in patient iPSC-derived retinal organoids. Moreover, this shortened protein is also able to provide rescue of visual function in an ush2a mutant zebrafish model [Citation44]. The QR-421 clinical trial involves intravitreal injections of three doses of the ASO (50, 100, or 200 µg) or sham in 18 adult participants with the objective of assessing safety, effects on visual function (visual acuity, FST, and dark-adapted perimetry) and retinal structure (photoreceptor ellipsoid zone on OCT). So far, ProQR Therapeutics has announced non-peer-reviewed interim results at 3 months suggesting concordant improvements in visual function across multiple modalities in the treated eyes of two out of eight subjects who received low and middle doses of the ASO, respectively. In addition, no serious adverse event was reported and no consistent visual changes were seen in six sham-treated eyes. Given the visual function improvements seen in therapeutic rescue of primary ciliopathies to date, more clinical efficacy data could be anticipated in the near future.
5. Conclusion
Antisense oligonucleotides could modulate pre-mRNA splicing or downregulate mRNA expression in vivo and are being investigated as therapeutics for a number of common mutations in inherited retinal diseases, including CEP290-associated Leber congenital amaurosis, USH2A-associated Usher syndrome, and RHO-associated autosomal dominant retinitis pigmentosa. Phase I/II clinical trials have demonstrated good safety profile and viable pharmacokinetics of intravitreally delivered ASOs. While the mechanism of how the ASOs reach the nuclei of outer retinal cells remains poorly defined, the promising signs of clinical efficacy seen are consistent with observations from intrathecal delivery of ASOs for inherited neurodegenerations and intra-otic delivery of ASOs for inherited hearing loss [Citation11,Citation45]. If early efficacy data were confirmed in phase II/III trials, the splicing-modulating ASO, sepofarsen, may become the first available treatment for LCA10 associated with the common c.2991 + 1655A>G mutation in the CEP290 gene.
6. Expert opinion
AAV-vector-based gene augmentation therapies for blinding inherited retinal diseases, such as RPE65-associated LCA, choroideremia (CHM), and RPGR-associated X-linked retinitis pigmentosa, are rapidly approaching clinical deployment [Citation29,Citation46,Citation47]. However, AAV gene augmentation remains constrained by the size of the AAV genome (4.7 kb) and has seen limited application in autosomal dominant disorders. Splicing-modulating ASOs provide a particularly elegant and safe mode of treatment for diseases caused by deep-intronic mutations. Particularly when clinical observations suggest that less than 1% of correctly spliced mRNA transcripts may be sufficient to ameliorate the disease significantly [Citation48]. Moreover, gene knockdown ASOs can be clinically effective in slowing retinal degeneration when the mutation exerts a dominant negative effect. Both these modes of actions of ASOs fulfil a major therapeutic void left by standard AAV-mediated gene augmentation. The main advantages of ASOs lie in their simple rational design, ease of chemical synthesis, and limited off-target and inflammatory effects. ASO-mediated splicing modulation or mRNA knockdown could also be validated relatively easily in vitro using cell lines or patient-derived iPSC retinal organoids without the need for protein functional assays. Subsequent pre-clinical testing in animal models may be limited to an assessment of safety, except for when residual protein function is sought from exon-skipping to generate a shortened protein. Therefore, compared with the preclinical development pathway for viral vector-based gene augmentation or gene editing, the development of ASOs may be more predictable, less dependent on animal models or functional assays, and associated with lower costs. In addition, while most current AAV gene therapies for RPE or photoreceptor degenerations are delivered surgically via subretinal injections, ASOs can be given via repeated intravitreal injections, further enhancing their relative safety profile.
One of the limitations of ASO therapies is the need to generate a unique oligonucleotide for each specific disease-causing mutation. While current clinical trials have focused on a number of mutation hotspots, the majority of mutations in inherited retinal diseases are heterogeneous and spread out across the entire gene. Therefore, a potentially large number of mRNA-knockdown ASO molecules will need to be produced and tested to cover the multitude of mutations in adRP. Clinical trial may also need to be designed to enable testing of a variety of mutation-specific ASO molecules for an inherited retinal degeneration in a single study once the general safety of intravitreal administration of oligonucleotides has been established. Although it might be possible to develop a cocktail of mRNA-knockdown ASOs as a single investigational medicinal product (IMP) that covers a variety of common mutant alleles in an adRP, there is a theoretical risk that different ASOs could interfere with each other’s actions or adversely affect the wildtype mRNA in vivo.
A noteworthy feature of ASO-based splicing-modulating therapy is that the clinical benefits may vary between patients with compound heterozygous mutations. For instance, most LCA10 patients are compound heterozygotes for the common c.2991 + 1655A>G mutation, and the clinical effect of ASO-mediated correction of this splicing defect may vary depending on the severity of the mutation on the second allele (e.g. whether it is a missense or nonsense mutation). A potential alternative therapeutic strategy, which could target both alleles, is to utilize spliceosome-mediated pre-mRNA trans-splicing. In this approach, an AAV vector is used to deliver a 5ʹ partial CEP290-coding DNA sequence containing a binding domain to intron 26–27 (between exon X and 27). Trans-splicing between the exogenous 5ʹ partial CEP290 pre-mRNA and the endogenous mutant pre-mRNA has been shown to produce hybrid full-length CEP290 transcripts in a mini-gene mouse model [Citation49]. A similar trans-splicing therapeutic strategy for RHO-associated adRP has also been demonstrated in vitro using a 3ʹ partial RHO-coding sequence packaged within an AAV transgene construct [Citation50].
Even within the restricted niches of treating known mutation hotspots, ASO therapies are expected to encounter strong competition from emerging viral vector-based gene-editing approaches. For instance, pre-clinical studies of a CRISPR/Cas9-based gene-editing vector (EDIT-101, Editas Medicine, Cambridge, USA) which could target and ‘cut out’ the c.2991 + 1665A>G mutation-containing intronic DNA segment in CEP290-associated LCA has achieved levels of genome editing that met the therapeutic threshold in non-human primates without evidence of significant off-target effects [Citation51]. EDIT-101 has now entered phase I clinical trial (ClinicalTrials.gov ID: NCT03872479). In addition, CRISPR/Cas9-mediated correction of the P23 H mutant RHO allele without disruption of the wildtype allele has been shown in the Rho+/P23 H mouse model of adRP [Citation52]. Potentially therapeutic level of genome editing was achieved via intravitreal delivery of the gene-editing components packaged within an AAV serotype 9 vector, further demonstrating the translational potential of this approach in the near future. While many technical and ethical issues surrounding CRISPR/Cas9-based genome editing remain to be resolved, the technology is rapidly evolving to create safer editing tools, including self-inactivating Cas9, CRISPRi, and prime editing [Citation53]. Compared with ASOs, in the long-term, CRISPR-based genome editing techniques are likely to offer greater versatility for mutation-independent correction of genetic defects.
The advent of ASO therapeutics targeting specific mutations in inherited retinal diseases compels us to re-consider these diseases by focusing on common mutational hotspots. In addition, there is a greater-than-ever need to gather data on the mutation spectra of each retinal dystrophy. This information, combined with our knowledge of the size of each exon and the locations of key functional protein domains, would facilitate the evaluation of potential splicing-modulation or exon-skipping ASO therapeutic strategies. Currently, a number of pathogenic deep-intronic mutations associated with inherited retinal diseases are known which are theoretically amenable to ASO-mediated therapy to restore normal splicing. These include the deep-intronic mutations found in ABCA4-associated Stargardt disease [Citation54–Citation57], CHM-associated choroideremia [Citation58,Citation59], OFD1-associated X-linked retinitis pigmentosa [Citation60], USH2A-associated Usher syndrome type 2 [Citation61], and CLRN1-associated Usher syndrome type 3 [Citation62]. Previously, whole-exome sequencing of patients with Stargardt disease, the most common macular dystrophy, detected pathogenic mutations in around 70% of cases. However, full sequencing of the entire ABCA4 locus has uncovered a large number of the missing deep-intronic mutations. This highlights the importance of deep genotyping to reveal the intronic mutations which may be treatable using the ASO approach.
One of the limitations of ASO therapies for genetic retinal diseases is the need for repeated intravitreal injections, which could cumulatively become a large therapeutic burden on the patients, especially in children who would require repeated general anesthesia. In this regard, a ‘one-off’ virus-based approach to delivering ASOs to retinal cells may be appealing. For instance, it is possible to use AAV to deliver a stably expressed ASO as part of a modified U7 small nuclear RNA (snRNA) transgene [Citation63]. The U7 snRNA is normally involved in 3ʹ-end processing of histone pre-mRNA, thus fusing U7 to the ASO improves its recruitment to the pre-mRNA processing machinery such as the spliceosome. This method has been shown to be effective in cellular and animal models of Duchenne muscular dystrophy as well as CEP290-associated LCA and could give rise to a one-off dose ASO-based gene therapy [Citation26]. Interestingly, however, in the CEP290-LCA mouse model, naked ASOs delivered via intravitreal injection were more effective at redirecting splicing compared with AAV-packaged ASOs delivered by subretinal injection. The reason for this remains uncertain, but may be partly due to the fact that intravitreal administration of ASOs allowed the small molecules to penetrate through the retina to reach the photoreceptors, whereas subretinal delivery of the AAV vectors led to restricted viral transduction within the area of the subretinal bleb.
With rapid development of gene-editing tools, additional hybrid technologies will likely arise at the intersection between ASO therapeutics and viral vector-mediated gene therapy. For example, mRNA-editing oligonucleotides have been engineered which combine an antisense oligonucleotide domain with an additional recruitment domain for the endogenous RNA-editing enzyme, adenosine deaminase acting on RNA (ADAR), to bring about A-to-I (equivalent of G when translated) transitions within the target mRNA transcript [Citation64]. Protein engineering has produced ADAR variants with altered target specificity, including C-to-U editing capability, and a variety of guide RNA recruiting domains [Citation65]. AAV-mediated delivery of engineered RNA-editing enzymes together with mRNA-targeting ASOs offers the potential to correct single nucleotide mutations in a wide range of inherited retinal diseases without the risks associated with genome editing [Citation66].
The retina is ideally suited to a wide range of genetic therapies. Over the next decade, we are likely to see parallel maturation of ASO, gene therapy, and gene-editing technologies. Alongside this, the availability of next-generation sequencing will enable more patients with inherited retinal diseases to be tested, genotype–phenotype relationships established, and mutation spectra of each disease characterized. If early efficacy results were confirmed in phase II/III clinical trials, it is likely that ASOs may gain traction as first-line therapy for a number of specific indications, in particular, deep-intronic mutations affecting splicing and slow-progressing autosomal dominant degenerations with long therapeutic windows, owing to their favorable safety profile, ease of administration, and lower cost. In contrast, gene augmentation therapies will likely remain the treatment-of-choice for severe recessive LCA or retinitis pigmentosa needing early intervention if the therapeutic transgene falls within the packaging capacity of single or dual vectors [Citation67,Citation68]. Finally, DNA or RNA-editing approaches will most likely advance both in terms of efficacy and safety to provide the broadest therapeutic possibility for correcting other individual retinal disease-causing mutations.
Article highlights
Therapeutic antisense oligonucleotides are synthetic endonuclease-resistant single-stranded oligodeoxynucleotides or oligoribonucleotides that can be designed to specifically knockdown the level of a disease-causing messenger RNA, alter its pattern of splicing, or block translation.
Phase I/II clinical trial of an intravitreally administered antisense oligonucleotide to correct a splicing defect associated with the common deep-intronic mutation in CEP290-Leber congenital amaurosis has demonstrated safety and early efficacy in terms of improved visual function.
Antisense oligonucleotide therapies are also in early clinical trials for common mutations found in RHO-associated autosomal dominant retinitis pigmentosa and Usher syndrome type 2.
In the future, antisense oligonucleotides may be designed to correct the splicing defects found in a number of other inherited retinal diseases, including Stargardt disease (ABCA4) and retinitis pigmentosa caused by the large EYS gene.
While the therapeutic potential of antisense oligonucleotides is restricted to a select group of genetic mutations, they complement gene augmentation and gene editing approaches, and represent an important modality in the treatment of inherited retinal diseases.
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
One reviewer works in an institution that received funding from ProQR.
Peer reviewers on this manuscript have no other relevant financial or other relationships to disclose.
Additional information
Funding
References
- Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. 1978;75:285–288.
- Lubini P, Zürcher W, Egli M. Stabilizing effects of the RNA 2ʹ-substituent: crystal structure of an oligodeoxynucleotide duplex containing 2ʹ-O-methylated adenosines. Chem Biol. 1994;1:39–45.
- Hudziak RM, Barofsky E, Barofsky DF, et al. Resistance of morpholino phosphorodiamidate oligomers to enzymatic degradation. Antisense Nucleic Acid Drug Dev. 1996;6:267–272.
- Geary RS, Yu RZ, Levin AA. Pharmacokinetics of phosphorothioate antisense oligodeoxynucleotides. Curr Opin Invest Drugs. 2001;2:562–573.
- Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259‐293.
- Yu RZ, Grundy JS, Geary RS. Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin Drug Metab Toxicol. 2013;9:169–182.
- Juliano RL, Carver K. Cellular uptake and intracellular trafficking of oligonucleotides. Adv Drug Deliv Rev. 2015;87:35–45.
- Juliano RL. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016;44:6518–6548.
- Lindow M, Vornlocher HP, Riley D, et al. Assessing unintended hybridization-induced biological effects of oligonucleotides. Nat Biotechnol. 2012;30:920–923.
- Vitravene Study Group. A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. Am J Ophthalmol. 2002;133:467–474.
- Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018;14:9–21.
- Scoles DR, Minikel EV, Stefan MP. Antisense oligonucleotides. A primer. Neurol Genet. 2019;5(2):e323.
- Rossi JJ, Rossi D. Oligonucleotides and the COVID-19 pandemic: A perspective. Nucleic Acid Ther. 2020;30:129–132.
- Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605.
- Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74:637–647.
- Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388(10063):3017–3026.
- Hammond SM, Hazell G, Shabanpoor F, et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc Natl Acad Sci U S A. 2016;113(39):10962–10967.
- Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in caenorhabditis elegans. Nature. 1998;391:806–811.
- Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:11–21.
- Solano EC, Kornbrust DJ, Beaudry A, et al. Toxicological and pharmacokinetic properties of QPI-1007, a chemically modified synthetic siRNA targeting caspase 2 mRNA, following intravitreal injection. Nucleic Acid Ther. 2014;24:258–266.
- Cideciyan AV, Jacobson SG, Drack AV, et al., Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med. 25(2): 225–228. 2019.
- Coppieters F, Lefever S, Leroy BP, et al. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat. 2010;31:1097–1108.
- den Hollander AI, Koenekoop RK, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79:556‐561.
- Collin RW, den Hollander AI, van der Velde-visser SD, et al. Antisense oligonucleotide (AON)-based therapy for Leber congenital amaurosis caused by a frequent mutation in CEP290. Mol Ther Nucleic Acids. 2012;1:e14.
- Gerard X, Perrault I, Hanein S, et al. AON-mediated exon skipping restores ciliation in fibroblasts harboring the common Leber congenital amaurosis CEP290 mutation. Mol Ther Nucleic Acids. 2012;1:e29.
- Garanto A, Chung DC, Duijkers L, et al. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum Mol Genet. 2016;25:2552‐2563.
- Parfitt DA, Lane A, Ramsden CM, et al. Identification and correction of mechanisms underlying inherited blindness in human iPSC-derived optic cups. Cell Stem Cell. 2016;18:769‐781.
- Dulla K, Aguila M, Lane A, et al. Splice-modulating oligonucleotide QR-110 restores CEP290 mRNA and function in human c.2991+1655A>G LCA10 models. Mol Ther Nucleic Acids. 2018;12:730‐740.
- Cehajic-Kapetanovic J, Xue K, Martinez-fernandez de la Camara C, et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med. 2020;26:354–359.
- Dryja TP, McGee TL, Hahn LB, et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med. 1990;323:1302–1307.
- Sung CH, Davenport CM, Hennessey JC, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88:6481–6485.
- Athanasiou D, Aguila M, Bellingham J, et al. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog Retin Eye Res. 2018;62:1–23.
- Cideciyan AV, Sudharsan R, Dufour VL, et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc Natl Acad Sci U S A. 2018;115:E8547–E8556.
- Murray SF, Jazayeri A, Matthes MT, et al. Allele-specific inhibition of rhodopsin with an antisense oligonucleotide slows photoreceptor cell degeneration. Invest Ophthalmol Vis Sci. 2015;56:6362‐6375.
- Edwards A, Fishman GA, Anderson RJ, et al. Visual acuity and visual field impairment in Usher syndrome. Arch Ophthalmol. 1998;116:165–168.
- Iannaccone A, Kritchevsky SB, Ciccarelli ML, et al. Kinetics of visual field loss in Usher syndrome Type II. Invest Ophthalmol Vis Sci. 2004;45:784–792.
- Fishman GA, Bozbeyoglu S, Massof RW, et al. Natural course of visual field loss in patients with Type 2 Usher syndrome. Retina. 2007;27:601–608.
- Espinos C, Millan JM, Beneyto M, et al. Epidemiology of usher syndrome in Valencia and Spain. Community Genet. 1998;1:223–228.
- van Wijk E, Pennings RJ, Te Brinke H, et al. Identification of 51 novel exons of the usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with usher syndrome type II. Am J Hum Genet. 2004;74:738–744.
- Reiners J, Nagel-Wolfrum K, Jürgens K, et al. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp Eye Res. 2006;83:97–119.
- Garcia-Garcia G, Aparisi MJ, Jaijo T, et al. Mutational screening of the USH2A gene in Spanish USH patients reveals 23 novel pathogenic mutations. Orphanet J Rare Dis. 2011;6:65.
- Bhattacharya G, Miller C, Kimberling WJ, et al. Localization and expression of usherin: a novel basement membrane protein defective in people with Usher’s syndrome type IIa. Hear Res. 2002;163:1–11.
- Eudy JD, Weston MD, Yao S, et al. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998;280:1753–1757.
- van Diepen H, Dulla K, Chan HL, et al. QR-421a, an antisense oligonucleotide, for the treatment of retinitis pigmentosa due to USH2A exon 13 mutations. IOVS. 2019;60(ARVO abstract). https://iovs.arvojournals.org/article.aspx?articleid=2743135.
- Hastings ML, Jones TA. Antisense oligonucleotides for the treatment of inner ear dysfunction. Neurotherapeutics. 2019;16(2):348‐359.
- Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860.
- Xue K, Jolly JK, Barnard AR, et al. Beneficial effects on vision in patients undergoing retinal gene therapy for choroideremia. Nat Med. 2018;24:1507–1512.
- Fry LE, Patrício MI, Williams J, et al. Association of messenger RNA level with phenotype in patients with choroideremia: potential implications for gene therapy dose. JAMA Ophthalmol. 2019;138:128‐135.
- Dooley SJ, McDougald DS, Fisher KJ, et al. Spliceosome-mediated Pre-mRNA trans-splicing can repair CEP290 mRNA. Mol Ther Nucleic Acids. 2018;12:294‐308.
- Berger A, Lorain S, Joséphine C, et al. Repair of rhodopsin mRNA by spliceosome-mediated RNA trans-splicing: a new approach for autosomal dominant retinitis pigmentosa. Mol Ther. 2015;23:918‐930.
- Maeder ML, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. 2019;25:229–233.
- Giannelli SG, Luoni M, Castoldi V, et al. Cas9/sgRNA selective targeting of the P23H Rhodopsin mutant allele for treating retinitis pigmentosa by intravitreal AAV9.PHP.B-based delivery. Hum Mol Genet. 2018;27:761–779.
- Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578:229–236.
- Bauwens M, De Zaeytijd J, Weisschuh N, et al. An augmented ABCA4 screen targeting noncoding regions reveals a deep-intronic founder variant in Belgian Stargardt patients. Hum Mutat. 2015;36:39‐42.
- Albert S, Garanto A, Sangermano R, et al. Identification and rescue of splice defects caused by two neighboring deep-intronic ABCA4 mutations underlying stargardt disease. Am J Hum Genet. 2018;102:517‐527.
- Zernant J, Lee W, Nagasaki T, et al. Extremely hypomorphic and severe deep-intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb Mol Case Stud. 2018;4:a002733.
- Sangermano R, Garanto A, Khan M, et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med. 2019;21:1751–1760.
- van den Hurk JA, van de Pol DJ, Wissinger B, et al. Novel types of mutation in the choroideremia (CHM) gene: a full-length L1 insertion and an intronic mutation activating a cryptic exon. Hum Genet. 2003;113:268–275.
- Garanto A, van der Velde-visser SD, Cremers FPM, et al. Antisense oligonucleotide-based splice correction of a deep-intronic mutation in CHM underlying choroideremia. Adv Exp Med Biol. 2018;1074:83–89.
- Webb TR, Parfitt DA, Gardner JC, et al. Deep-intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum Mol Genet. 2012;21:3647–3654.
- Slijkerman RW, Vaché C, Dona M, et al. Antisense oligonucleotide-based splice correction for USH2A-associated retinal degeneration caused by a frequent deep-intronic mutation. Mol Ther Nucleic Acids. 2016;5:e381.
- Khan AO, Becirovic E, Betz C, et al. A deep-intronic CLRN1 (USH3A) founder mutation generates an aberrant exon and underlies severe Usher syndrome on the Arabian Peninsula. Sci Rep. 2017;7:1411.
- Goyenvalle A, Vulin A, Fougerousse F, et al. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science. 2004;306:1796–1799.
- Merkle T, Merz S, Reautschnig P, et al. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat Biotechnol. 2019;37:133–138.
- Vogel P, Stafforst T. Critical review on engineering deaminases for site-directed RNA editing. Curr Opin Biotechnol. 2019;55:74‐80.
- Fry LE, Peddle CF, Barnard AR, et al. RNA editing as a therapeutic approach for retinal gene therapy requiring long coding sequences. Int J Mol Sci. 2020;21:777.
- Trapani I, Toriello E, de Simone S, et al. Improved dual AAV vectors with reduced expression of truncated proteins are safe and effective in the retina of a mouse model of Stargardt disease. Hum Mol Genet. 2015;24:6811–6825.
- McClements ME, Barnard AR, Singh MS, et al. An AAV dual vector strategy ameliorates the stargardt phenotype in adult Abca4-/- mice. Hum Gene Ther. 2019;30:590–600.