
Abstract
Abstract
Cardiac-specific troponins are elevated in blood following cardiac injury and are the preferred diagnostic biomarkers when acute myocardial infarction is suspected clinically. Cardiac troponin (cTn) elevations are also observed in clinical conditions without obvious connection to cardiac injury. Irrespective of the underlying condition, cTn elevation is linked to a poor prognosis, even if the elevation is stable over time. Here, we explore mechanisms that may lead to cTn elevations, including necrosis, apoptosis, necroptosis, cell wounds and decreased clearance. The aim is to broaden the perspective of how we interpret unexpected cTn elevations in patients. The cTn elevations may not be able to serve as direct proof of myocardial necrosis especially in the absence of a clear-cut reason for its release.
Abbreviations:
AMI: acute myocardial infarction; cTn: cardiac troponin; cTnI: cardiac troponin I; cTnT: cardiac troponin T; MLKL: mixed lineage kinase domain-like; TUNEL: terminal deoxynucleotidyl transferase nick end labeling
Introduction
Cardiac troponin I (cTnI) and cardiac troponin T (cTnT)) are cardiac-specific proteins that are part of the troponin complex of thin filaments which form together with thick filaments the contractile apparatus, the sarcomere, of cardiomyocytes (). Cardiac troponin (cTn) is released and is transiently increased in the blood following an acute myocardial infarction (AMI) and other types of acute myocardial injury. The cTns are the preferred biomarkers for the laboratory diagnosis of AMI (Roffi et al. Citation2016), due to their excellent cardiac specificities, the fact that healthy individuals have only low levels (Cardinaels et al. Citation2012), and the fact that there are excellent assays available for clinical use.
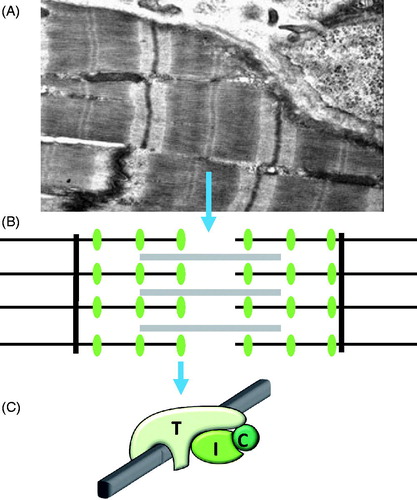
Figure 1. Cardiac troponin. (A) Electron microscope image of a cardiomyocyte showing the repeating sarcomeres in myofibrils. (B) The sarcomere contains thick filaments (grey lines), containing ATP consuming myosin interdigitated by thin filaments (black horizontal lines) that bind troponin (green ellipses), and is anchored in Z-bands (black vertical lines). (C) The cardiac troponin complex is composed of cTnT (T) that directly binds to thin filaments (grey bar), cTnI (I) and TnC (C), the Ca2+-binding subunit.
The cTn elevations are also found in patients with chronic cardiac comorbidities. Recently, increases have been observed in normal individuals in response to what might be considered physiological stress such as exercise, rapid atrial pacing and after the infusion of dobutamine. A large number of studies have shown in non-AMI populations that elevation of the cTn concentration, including some not above the 99th% upper reference limit, is a strong risk marker for death, the development of heart failure and ischaemic heart disease and other non-ischaemic cardiac diseases (Galvani et al. Citation1997, Aviles et al. Citation2002, Peacock et al. Citation2008, Omland et al. Citation2009, Kawahara et al. Citation2011, Aldous et al. Citation2015, Omland et al. Citation2015, Roos et al. Citation2017). At present, we only have evidence-based treatment for patients who have had AMI. For other conditions associated with cTn elevations, treatments are frequently advocated based on the assumed pathophysiology but data supporting their effectiveness are not available (Roos et al. Citation2017). Herein, we summarize what is known about the possible cellular mechanisms behind cTn release and clearance, which may broaden the perspective on how to interpret cTn values in clinical practice. In most cases, information is available for both cTnT and cTnI (cTn), but in some instances, information is only known for cTnT or cTnI and in those situations, that fact will be made clear.
cTn release by necrosis
The most obvious reason for cTn elevation in a patient, especially if the cTn elevation is transient, is necrosis. In fact, compared with other cell types, cardiomyocytes are more prone to undergo necrosis triggered by situations such as the Ca2+ paradox and/or the oxygen paradox (Piper Citation2000).
Myofibrils, which constitute 50–60% of the cardiomyocyte volume, are repeating units of Ca2+-activated, ATP-consuming sarcomeres (). Therefore, if Ca2+ leaks into the cardiomyocyte cytoplasm, the sarcomeres contract and quickly consume all the ATP (), resulting in rapid necrosis and release of cTn along with other cellular contents (Piper et al. Citation2003).
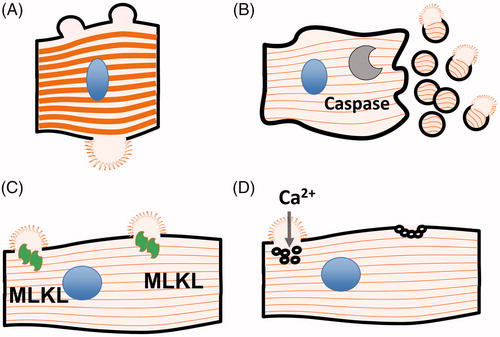
Figure 2. Possible release mechanisms for cTn discussed in this review (see text for details). (A) Necrosis (B) Apoptosis (C) Necroptosis (D) Cell wound.
A visual representation of the rapid nature of cardiac necrosis is the ‘stone heart syndrome’ after the Ca2+ paradox. If isolated hearts are first perfused with a buffer without, and then a buffer with Ca2+, the entire heart contracts tonically, turning the perfusion buffer red due to the release of myoglobin and the heart becomes a white hard contracting mass of necrotic cardiomyocytes called a ‘stone heart’ (Piper Citation2000).
Groundbreaking studies by Jennings showed that ischaemic cardiomyocytes quickly develop intracellular oedema when ATP levels drop (Jennings et al. Citation1978). As the sarcomere is anchored in the plasma membrane via the dystrophin protein complex, swelling does not result in uniform enlargement of the cardiomyocyte; rather, membrane ‘blebs’ (Schwartz et al. Citation1984) are formed between the dystrophin protein complex anchoring points (Jennings et al. Citation1978, Sage and Jennings Citation1988) (). In this situation, the cardiomyocyte plasma membrane becomes fragile but does not break immediately (Jennings et al. Citation1983), unless exposed to an acidic or hyposmotic environment (Jennings et al. Citation1983, Steenbergen et al. Citation1985). However, when contractility occurs after reoxygenation (Vander Heide and Ganote Citation1987), or when anoxic cardiomyocytes are forced to move by surrounding non-ischaemic cardiomyocytes (Steenbergen et al. Citation1985), or if the ischaemic heart is acutely dilated by a balloon in the ventricular chamber (Vander Heide and Ganote Citation1987), these cells will succumb and result in contraction band necrosis.
Figure 3. Cardiomyocyte necrosis. (A) Phase contrast microscopic images of isolated cardiomyocyte undergoing contraction band necrosis (reproduced from [Garcia-Dorado and Ruiz-Meana Citation2000] with permission). (B) Electron microscope image of an ischaemic cardiomyocyte where intracellular oedema forces the plasma membrane to form ‘blebs’ between dystrophin anchoring points in the Z-band of sarcomeres (reproduced from [Sage and Jennings Citation1988] with permission).
![Figure 3. Cardiomyocyte necrosis. (A) Phase contrast microscopic images of isolated cardiomyocyte undergoing contraction band necrosis (reproduced from [Garcia-Dorado and Ruiz-Meana Citation2000] with permission). (B) Electron microscope image of an ischaemic cardiomyocyte where intracellular oedema forces the plasma membrane to form ‘blebs’ between dystrophin anchoring points in the Z-band of sarcomeres (reproduced from [Sage and Jennings Citation1988] with permission).](/cms/asset/61b2dbeb-c979-4ccc-a2c9-a773333cc3a4/ibmk_a_1490969_f0003_b.jpg)
Some of this damage can be mitigated by a reduction in contractile performance, so called stunning. Myocyte contraction stops within a few heartbeats following total ischaemia in what is thought to be a mechanism that protects oedematous ischaemic cardiomyocytes from contraction-mediated cell damage, termed the ‘smart heart hypothesis’ (St Louis et al. Citation2000, Depre and Vatner Citation2005).
If the resulting membrane damage allows enough Ca2+ to enter the cardiomyocyte cytoplasm, the sarcomeres contract tonically and consume what little ATP is left, limiting the ability of the cardiomyocytes to pump out excess Ca2+. In the ATP-depleted and Ca2+-loaded state, myofibrillar shortening stays fixed in all the sarcomeres, as the cross-bridges between actin and myosin remain in an attached state (Piper'et al. Citation2003), resulting in contraction band necrosis where collapsed sarcomeres and large membrane defects are seen in histological preparations (; Jennings Citation2013). Unfortunately, much of the literature concerning cardiomyocyte necrosis is substantially dated and probably need to be revisited with newer techniques. In addition, during the past 15 years, this simple ‘necrosis by breaking’ picture has been complicated by the realization that programmed necrosis – necroptosis, described below – also operates in the ischaemically damaged heart (Kung et al. Citation2011). In essence, the possibility of programmed necrosis occurring at a later stage further complicates the interpretation of the cTn release kinetics following AMI and other myocardial injury. Nonetheless, in this acute situation, it is thought that most cTn release is due to irreversible cellular injury.
cTn release mechanisms after membrane damage
The cTn binds with finite affinity to thin filaments of the sarcomere (Shiraishi et al. Citation1992) (). Although release is immediate after membrane damage, the washout of cTn is slowed when compared to other cardiac injury biomarkers due to what has been called ‘the trapping effect’ (Starnberg et al. Citation2014). This is especially true when there are large volumes of the myocardium that become necrotic, such as after a transmural AMI. These types of insults result in sustained elevations of cTn for days and sometimes weeks (Katus et al. Citation1991, Laugaudin et al. Citation2016). The delayed washout of cTn is in contrast to that of cytoplasmic cardiac injury biomarkers (), such as lactate dehydrogenase (LDH), creatine kinase (CK) (Katus et al. Citation1989), CKMB (Katus et al. Citation1987) and myoglobin (Sylven and Bendz Citation1978), which have no affinity for cardiac tissue. Thus, if cardiac tissue is crushed in warm plasma, myoglobin and cTn are released immediately but the washout of cTn takes more time, due to its binding to the insoluble sarcomere. Starting within a few hours, the necrotic cardiac tissue induces local inflammation and accumulation of neutrophils and macrophages that digest damaged tissue, forming granulation tissue and then a stable scar (Pfeffer and Braunwald Citation1990). During this later process, cTn release is likely, largely due to degradation of myofibrils. Many of these other proteins, such as CK, are locally degraded and not detected by conventional clinical assays. Consequently, even once blood flow is re-established, they no longer can be appreciated (Clark et al. Citation1978).
Figure 4. Kinetics of cardiac troponin T (cTnT), creatine kinase (CK) and lactate dehydrogenase (LDH) from the first published patient with acute myocardial infarction where a cTnT assay was used by Katus [Katus et al. Citation1989].
![Figure 4. Kinetics of cardiac troponin T (cTnT), creatine kinase (CK) and lactate dehydrogenase (LDH) from the first published patient with acute myocardial infarction where a cTnT assay was used by Katus [Katus et al. Citation1989].](/cms/asset/23c41ee8-8032-41f4-b628-491938565464/ibmk_a_1490969_f0004_c.jpg)
As in all binding reactions, the washout of cTn is volume dependent and, hence, is faster and more complete if the plasma volume or blood flow is capacious (Starnberg et al. Citation2014). Increases in cTn levels hours or days after the initial non-reperfused AMI can, in addition to re-infarction, be due to partial restoration of flow (Hermens et al. Citation1990, Voss et al. Citation1995). Also, if cardiomyocyte necrosis is scattered, as observed after isoproterenol-induced type II infarctions in rats, the washout of cTn from necrotic myocytes could, in principle, be over within 24 hours (York et al. Citation2007, Zhang et al. Citation2008, Clements et al. Citation2010). The cTn elevations that are cleared within 24 hours thus still can be due to cardiomyocyte necrosis.
cTn release by apoptosis and other regulated cell death pathways
All cells, including cardiomyocytes, have built-in ATP-consuming death programmes that are activated when appropriate signals are present. This process is essential for the removal of interdigitate cells in the formation of fingers during'embryogenesis (Svandova et al. Citation2017). However, apoptosis and other death programmes are probably most important in the defence against intracellular parasites like viruses.
Mammalian cells have evolved an array of mechanisms to detect the presence of intracellular pathogens, with many of these pathways culminating in the activation of a cell death response. Some of these responses were found through studies of the cytomegalovirus (CMV) that harbours several genes essential for viral replication dedicated to block apoptosis and other cell death programmes. Inhibition of cell death not only affords CMV an opportunity to complete the replication cycle, but also restricts the release of cellular content and, hence, limits the inflammatory response required for the generation of the adaptive immune response (Brune and Andoniou Citation2017).
In the unlocking of the multitude of signals that can activate programmed cell death, including TNF receptor activation (Guo et al. Citation2017), it has become evident that our cells are like primed rat traps, eager to go off at any sign of invasion, but – perhaps not intentionally – also in response to oxidative overload, ischaemia and other stressors (Brune and Andoniou Citation2017).
In principle, using its original description by Kerr (Kerr et al. Citation1972) () and the latest definition of apoptosis by the Nomenclature Committee on Cell Death (NCCD) (Galluzzi et al. Citation2012), apoptosis should not result in cTn elevations. In fact, it has been suggested that apoptosis cannot be the culprit if the patient has cTn elevations (Takemura et al. Citation2013). This is because no intracellular content is expected to be released when the apoptotic cell is divided into membrane-enclosed apoptotic bodies with phosphatidylserine and other ‘eat-me’ signals exposed on the surface, resulting in the engulfment and lysosomal degradation of the apoptotic bodies by surrounding cells and sometimes by immune cells (Nagata et al. Citation2010).
It is, however, possible that the myofibrils in cardiomyocytes interfere with the formation of tight membrane-enclosed apoptotic bodies, or that apoptotic bodies break and expel their content before they are cleared (). It is also possible that other forms of programmed cell death operate, like necroptosis, which ultimately results in lysis of the cell after the formation of multimers of the protein mixed lineage kinase domain-like (MLKL) that bind to and make holes in cellular membranes (Kung et al. Citation2011) (). Although never reported, cardiomyocytes undergoing necroptosis most likely also release cTn as the nuclear protein HMGB1 is released in the process. Our experience is that cTn release from damaged primary cardiomyocytes always overlaps with the release of other intracellular proteins, like LDH and myoglobin (Starnberg et al. Citation2014).
Unfortunately, many studies of apoptosis in the human heart following AMI have methodological problems (Takemura et al. Citation2013). In addition, most studies are performed on hearts from dead patients, possibly adding a selection bias and effects secondary to the cause of death (Abbate et al. Citation2003). Sometimes, studies have neglected the fact that only 30% of the cells in the normal heart are cardiomyocytes and even less in granulation tissue, because of accumulation of apoptosis-prone immune cells (Saraste et al. Citation1997, Wakabayashi et al. Citation2015, Jose Corbalan et al. Citation2016). In addition, no attempt was made to relate the extent of apparent apoptosis to the cTn levels in these human studies. Consequently, we do not know if, or to what extent, apoptosis contributes to acute or stable cTn elevations in humans. However, increased levels of caspase-3, thought to be a mediator of apoptosis, are found systemically in patients with AMI (Agosto et al. Citation2011).
What is clear, however, is that the cells in human hearts seem to undergo some kind of apoptosis-like process during an extended period following AMI, also in areas not affected by ischaemia (Abbate et al. Citation2008), possibly involved in the thinning of the remote myocardium that is often observed after a large MI (Pfeffer and Braunwald Citation1990). Similar apoptosis-like processes have been found in human biopsy studies of other cardiac pathologies like heart failure (Narula et al. Citation1999). In these studies, cardiac cells display several hallmarks of apoptosis, including terminal deoxynucleotidyl transferase nick end labeling (TUNEL)-positive nuclei and caspase activation co-localizing in cells expressing myocyte-specific proteins that correlate with cardiac disease and outcome (Abbate et al. Citation2003, Biondi-Zoccai et al. Citation2004). Some authors have faith in methods to detect apoptosis not involving electron microscopy (Saraste and Pulkki Citation2000). However, according to other authors, TUNEL and other DNA-end labelling methods cannot unequivocally prove that apoptosis has occurred. This controversy has led to some cases where the interpretation of the published results has been questioned because of discrepancies between the methods (Takemura et al. Citation2013).
There is a large body of studies in mice where cardiomyocyte apoptosis or necroptosis can be induced by genetic manipulation, like limited induction of the apoptosis-inducing protease caspase-8, which has been shown to result in the development of heart failure (Wencker et al. Citation2003) and larger infarcts after coronary occlusion. If the apoptotic or necroptic programme is inhibited by genetic or pharmacological means, the almost uniform finding is that the size of experimental infarcts is reduced (Yaoita et al. Citation1998, Holly et al. Citation1999, Hochhauser et al. Citation2003, Abbate et al. Citation2008) and mice are protected from the development of heart failure (Wencker et al. Citation2003, Kitamura et al. Citation2014, Wang et al. Citation2014). There are a few studies reporting apoptosis following ischaemic challenges in larger animals (Goussev et al. Citation1998). One study has shown a correlation between sustained cTnI elevations and the amount of TUNEL-positive cells in pigs after brief periods of ischaemia (Weil et al. Citation2017).
At this stage, it is safe to conclude that some type of programmed cell death operates in the heart and that necroptosis could contribute to cTn elevations in patients; however, the current data are not sufficient to make strong statements about how programmed cell death influences cTn elevations more chronically in patients. However, it is fair to speculate that increases in myocardial stress due to volume expansion (Feng et al. Citation2001) and potentially subendocardial ischaemia due to supply–demand imbalance or transient increases in pulmonary pressures could result in activation of these pathways with cTn release. Such speculation would fit with the release of cTn shown to occur in normal subjects who are rapidly atrially paced (Turer et al. Citation2011) and/or exposed to dobutamine (Siriwardena et al. Citation2012). However, other non-apoptotic mechanisms also could explain these findings.
cTn release by cell wounds
Apparently viable cardiomyocytes in Petri dishes or in animal models exchange macromolecules over their plasma membranes (Cooper and McNeil Citation2015, Demonbreun and McNally Citation2016). The evidence that macromolecular exchange occurs in living cardiomyocytes is based on the detection of albumin (McNeil and Khakee Citation1992, Clarke et al. Citation1995) and other extracellular'macromolecules (Hoffstein et al. Citation1975) in the cytoplasm of apparently normal cardiomyocytes. This macromolecular exchange over the plasma membrane is higher if the cardiomyocytes are stressed either by contraction, are stretched by external forces (Page et al. Citation1992, Swildens et al. Citation2010), are challenged by beta-adrenergic stimulation (Boutet et al. Citation1976, Clarke et al. Citation1995) or after limited ischaemia. The cellular uptake of albumin and other large molecules in these studies seems to occur through transient disruptions in the plasma membrane and is not due to endocytosis or albumin entering T tubules (Clarke et al. Citation1995). Macromolecular exchange via holes in the plasma membrane without cardiomyocyte death is possible, in part, because the cytoplasm is a macromolecular gel with restricted diffusion (Ellis Citation2001), and because dystrophin complexes stabilize the membrane by forming links between the contracting sarcomere and the extracellular matrix (Allen et al. Citation2016) ().
However, the most important protection against cell death when the plasma membrane is injured is a Ca2+-dependent repair system, thoroughly examined by McNeil (Terasaki et al. Citation1997) and discovered by Heilbrunn almost 90 years ago during his studies of echinoderm oocytes (Heilbrunn Citation1930). This is why cells do not expel the cytoplasm like a balloon bursting if poked with a micropipette (Terasaki et al. Citation1997) or if the plasma membrane is injured by a localized laser burn (Demonbreun et al. Citation2016). Cells are surprisingly resilient and repair membrane holes larger than 10 μm2 within seconds, in a process called cell wound repair (Terasaki et al. Citation1997) (). If the same insults are induced in Ca2+-free culture media, the membrane holes persist, eventually leading to cell death (McNeil and Kirchhausen Citation2005). The Ca2+ concentration required to elicit a cell wound response is 100 times higher than the Ca2+ concentration seen during muscle contraction in most experimental systems (Steinhardt et al. Citation1994, Terasaki et al. Citation1997). The result of the cell wound response is the formation of a Ca2+-impermeable patch within a few seconds.
The molecular details of cell wound repair are still unclear but most data indicate that the Ca2+-impermeable patch is formed by intracellular membrane vesicles fusing with the plasma membrane (Miyake and McNeil Citation1995, Terasaki et al. Citation1997, Andrews et al. Citation2015, Davenport et al. Citation2016), aided by MG53 (Cai et al. Citation2009) and dysferlin (Han and Campbell Citation2007), proteins able to bind to and promote the fusion of two membranes () (McNeil Citation2014).
Mice (Wenzel et al. Citation2007) and humans (Han and Campbell Citation2007) with mutations in genes involved in cell wound repair develop dystrophies, that is contraction-induced accumulation of muscle injury that ultimately leads to muscle weakness. In addition, dystrophies with mutations in other muscle cell-stabilizing functions, such as Duchenne muscular dystrophy, show up to 16-fold upregulation of dysferlin, MG53 and other cell wound repair genes (Waddell et al. Citation2011). Mice with MG53 mutations show impaired cell wound repair in skeletal muscle (Cai et al. Citation2009) and are more sensitive to cardiac ischaemia than normal mice (Cao et al. Citation2010). Dystrophies caused by dysferlin mutations sometimes lead to cardiomyopathy and heart failure in humans (Kuru et al. Citation2004). Mice with dysferlin mutations display inefficient cell wound repair in cardiomyocytes and are susceptible to pressure overload-induced cardiomyopathy. In addition, dysferlin-mutated mice have stable cTnI elevations that are aggravated by exercise (Han et al. Citation2007). These findings indicate that cell wound repair is important in cardiomyocytes and, if impaired, can result in stable cTnI elevations, at least in mice.
Cell wounds induced by contraction likely increase under ischaemic conditions. However, if the plasma membrane injury is limited, the cardiomyocytes survive after reoxygenation, likely as a result of cell wound repair with accumulation of extracellular macromolecules in the cytoplasm as the only evidence of transient plasma membrane injury (Boutet et al. Citation1976). This can be shown by resident colloidal lanthanum in apparently healthy cardiomyocytes surrounding necrotic cardiomyocytes after experimental ischaemic injury and i.v. injection of colloidal lanthanum complexes with a median diameter of 40 Å (Hoffstein et al. Citation1975, Burton et al. Citation1977), roughly half the size of albumin.
Under limited ischaemic conditions or limited beta-adrenergic stimulation, elevated serum levels of myocardial injury biomarkers such as LDH, CK and cTn are observed without obvious cell death and sometimes with a reciprocal lowering of these markers in apparently living cardiomyocytes (Chiong et al. Citation1974, Zhang et al. Citation2008, Hickman et al. Citation2010). Although these studies did not detect cardiomyocyte death, it remains possible that areas of cell death and in particular apoptosis could have been missed by their histological examinations. These and similar studies of elevated cardiac injury biomarkers are therefore unable to prove that the leakage occurred from viable cardiomyocytes. However, Spieckermann group showed that 35% of all LDH can be expelled from resting cardiomyocytes under ischaemic challenge without finding cell death. Taken at face value, this finding would indicate that extensive leakage of the cellular content can result from ischaemically stressed but living cardiomyocytes (Piper et al. Citation1984, Schwartz et al. Citation1984). However, how cytoplasmic proteins could escape in such an abundance and still allow the cardiomyocytes to survive is unclear and have, to the best of our knowledge, never been replicated. If the above is correct, then increases in cTn elevation should not be considered proof of cardiomyocyte cell death (Kawahara et al. Citation2011).
In summary, it is safe to conclude that there are mechanisms that could allow cTn release from living cardiomyocytes. The extent to which cell wounds contribute to the cTn elevations we encounter in the clinic remains to be shown.
cTn release from skeletal muscle
The clinical cTn assays all use antibodies that bind epitopes specific to cardiac isoforms of TnI and TnT. The Roche high-sensitive cTnT assay does, however, react with something in skeletal muscle, not yet observed with the cTnI assays. This was first observed in patients with neuromuscular disorders and patients with myositis (Botta et al. Citation2008, Jaffe et al. Citation2011, Rittoo et al. Citation2014, Valaperta et al. Citation2016, Wens et al. Citation2016).
A cross-reactivity with skeletal muscle extracts from control subjects has also been found. When serum was supplemented in vitro with extracts from normal human skeletal muscle such that myoglobin levels were within what can be observed after rhabdomyolysis, cTnT levels, but not cTnI levels, increased above the current cut-off for MI (Schmid et al. Citation2018).
We (unpublished) and others (Valaperta et al. Citation2016) have observed a similar reactivity using the Roche high-sensitive cTnT assay in skeletal muscle extracts from healthy humans, pigs and rats, while the Abbott high-sensitive cTnI assay was negative in the same extracts. Therefore, the Roche high-sensitive cTnT assay could in theory become positive in those with chronic skeletal muscle disease or in response to rhabdomyolysis. In these instances, as suggested by the Biomarker Group of the European Society of Cardiology (Thygesen et al. Citation2010), an alternative cTnI assay could be used to check whether the heart is involved, since most of these assays have the same ability to diagnose and exclude myocardial infarction and to define prognosis as the Roche high-sensitive cTnT assay (Shah et al. Citation2015, Pickering et al. Citation2016). Whether this strategy it is safe in these conditions has not been examined in a prospective study, but it is in use in several clinics.
cTn elevation due to decreased cTn clearance
Once cTnT and cTnI reach the circulation, they are initially cleared with a half-life of 0.5 hours in dogs and rats (Dunn et al. Citation2011), followed by what we have shown is a slower, apparently kidney-dependent clearance when cTnT drops to concentrations often observed in patients with stable cTn elevations (Friden et al. Citation2017) ().
It may be that this phenomenon is similar to myoglobin clearance, thoroughly studied by Sylven (). At high levels, clearance of radioactively labelled myoglobin injected in healthy controls or in patients with acute MI has a half-life of roughly 1 h, followed by a much slower phase at low myoglobin levels (Sylven Citation1978, Groth and Sylven Citation1981) (). As myoglobin is a small protein with a molecular weight of 17 kDa, it can pass through the glomerular membrane and is cleared in part by kidney function, as found in patients with rhabdomyolysis and myoglobin-induced acute tubular renal failure.
Figure 5. Clearance of myoglobin in humans and cTnT in rats. (A) Concentrations of 125I-labeled myoglobin during 6 hours following i.v. injection in humans. Circles represent 125I levels in serum and triangles represent 125I levels in urine. Dotted lines are projections of the two kinetic profiles found (reproduced from [Sylven Citation1978] with permission). Most of the 125I in urine did not precipitate in acid, indicating that 125I in the urine was not linked to myoglobin. (B) Relative concentrations of cTnT in rats during 2 hours following i.v. injection of rat cardiac extracts. Dotted lines are projections of two kinetic profiles found (reproduced from [Friden et al. Citation2017] with permission).
![Figure 5. Clearance of myoglobin in humans and cTnT in rats. (A) Concentrations of 125I-labeled myoglobin during 6 hours following i.v. injection in humans. Circles represent 125I levels in serum and triangles represent 125I levels in urine. Dotted lines are projections of the two kinetic profiles found (reproduced from [Sylven Citation1978] with permission). Most of the 125I in urine did not precipitate in acid, indicating that 125I in the urine was not linked to myoglobin. (B) Relative concentrations of cTnT in rats during 2 hours following i.v. injection of rat cardiac extracts. Dotted lines are projections of two kinetic profiles found (reproduced from [Friden et al. Citation2017] with permission).](/cms/asset/44a0b441-530c-4be4-8c55-534b5ce1dc9c/ibmk_a_1490969_f0005_b.jpg)
Despite its ability to pass through the glomerular membrane, clearance of myoglobin at high levels observed after MI or rhabdomyolysis is not influenced by renal function (Wakabayashi et al. Citation1994, Lappalainen et al. Citation2002), whereas the low levels found in normal subjects are strongly related to renal function (Hallgren et al. Citation1978). Myoglobin apparently undergoes predominant extra-renal clearance at high levels () and its renal clearance () becomes apparent only at lower levels.
Figure 6. Possible clearance mechanisms for cTn discussed in this review (see text for details). (A) Endocytosis (B) Filtration (C) Degradation.
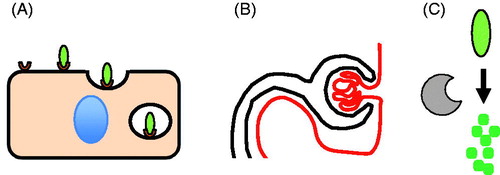
Myoglobin shares this pharmacokinetic behaviour with low molecular weight heparin cleared by a fast saturable receptor-mediated endocytosis-based system () and slow kidney-dependent clearance (Boneu et al. Citation1990) (). Early studies have shown that myoglobin accumulates in the liver and spleen (Amako et al. Citation1963), followed by bilirubinaemia (Bywaters and Beall Citation1998) in patients who die from massive rhabdomyolysis, indicating that receptor-mediated endocytosis in the reticuloendothelial system is responsible for the extra-renal clearance of myoglobin (). This dual clearance system is likely similar to how cTnT is cleared, at least in rats ().
For unknown reasons, most of the cTnT measured with clinical high-sensitive cTnT assays are degradation products in patients with stable cTnT elevations (Diris et al. Citation2003, Friden et al. Citation2017), and a few hours after the ischaemic event in patients with acute MI (Cardinaels et al. Citation2013) (). The major degradation products are <20 kDa (Cardinaels et al. Citation2013, Streng et al. Citation2016, Friden et al. Citation2017), expected to undergo free passage over the glomerular membrane. Therefore, cTnT measured by the current high-sensitive cTnT assay could be, to some extent, cleared by the kidneys. We have shown this in experiments where we block renal blood flow in rats. In this context, it is important to note that very little radioactive myoglobin reached the urine in Sylven’s subjects mentioned above, likely because tubular resorption prevented myoglobin from reaching the bladder (Sylven Citation1978). Likewise, tubular resorption of cTn degradation products may explain the fact that, despite apparent renal clearance, cTn levels are low in urine (Ziebig et al. Citation2003).
Assuming the above, it appears that at low cTnT levels, often found in patients with stable elevations, kidney clearance appears to dominate. Therefore, at steady state and everything else being the same, cTnT levels are roughly twice as high if kidney function is reduced by 50% (Bjurman et al. Citation2015). As many patients with kidney failure often have stable cTnT levels 10–100 times above the 99th% upper reference limit (Bjurman et al. Citation2015), other mechanisms must also contribute to the cTnT elevations found in this patient group (Waldum and Os Citation2013).
However, if the cTnT levels are adjusted for kidney function, the clinical high-sensitive cTnT assay becomes slightly better at distinguishing between patients with and without AMI in the emergency room (Friden et al. Citation2017). However, cTnT levels adjusted for kidney function do not improve prognostication (Friden et al. Citation2017).
At the higher cTnT levels often found after large AMIs, extra-renal clearance of cTnT dominates in rats and is not influenced by renal function (Friden et al. Citation2017), potentially explaining why clearance of cTn is similar among AMI patients with or without renal function (Ellis et al. Citation2001). The nature of this extra-renal clearance of cTnT has not been studied but likely occurs via scavenger receptors, a loosely defined group of receptors that are able to bind to a plethora of proteins and other molecules and direct them to intracellular degradation (Prabhudas et al. Citation2014) (). Scavenger receptor clearance of LDH has been studied by Gruber in rats and rabbits (Smit et al. Citation1987, Smit et al. Citation1988). Radioactive LDH is taken up by macrophages (Hayashi and Notkins Citation1994) in the liver and spleen after i.v. injection. If macrophages are specifically destroyed by the LDH virus, by silica nanoparticles or by other means, mice and monkeys develop stable LDH and CK elevations (Hayashi et al. Citation1988, Radi et al. Citation2011), likely due to decreased clearance. An indication that this mechanism may be relevant with regard to stable elevations in humans is a recent finding, showing that stable elevations of CK and LDH correlate with genetic mutations in scavenger receptors (Kristjansson et al. Citation2016). It is therefore formally possible that stable cTnT elevations could be due to inefficient renal or extra-renal clearance and this is therefore an area of intense study.
Conclusions
Unfortunately, although current knowledge might broaden the perspective, the information in this review makes interpretation of cTn elevations no less complicated. First, because of the possibility that cell wounds could explain some of the release and given the data concerning altered renal clearance, cTn elevations may not be able to serve as direct proof of myocardial necrosis especially in the absence of a clear-cut reason for its release. However, early timely limited cTn increases may be the result of scattered cardiomyocyte necrosis and/or apoptosis, or theoretically even reversible cardiomyocyte injury. Prolonged cTn elevations (>24 h), however, may be due to slow washout from local myocardial injury or the delayed necroptosis following an ischaemic event in both granulation tissue and in the remote myocardium undergoing remodelling.
Disclosure statement
JM none to declare concerning the content of the manuscript. BL has received consulting fees from Roche Diagnostics, bioMérieux Clinical Diagnostics, Philips Healthcare, ThermoFischer and Fiomi Diagnostics; and has received a research grant from bioMérieux Clinical Diagnostics and Fiomi Diagnostics. ASJ presently or in the past has consulted for most of the major diagnostic companies.
Additional information
Funding
Related Research Data
References
- Abbate, A., et al., 2003. Increased myocardial apoptosis in patients with unfavorable left ventricular remodeling and early symptomatic post-infarction heart failure. Journal of the American college of cardiology, 41 (5), 753–760.
- Abbate, A., et al., 2008. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation, 117 (20), 2670–2683.
- Abbate, A., et al., 2008. Right ventricular cardiomyocyte apoptosis in patients with acute myocardial infarction of the left ventricular wall. American journal of cardiology, 102 (6), 658–662.
- Agosto, M., et al., 2011. Serum caspase-3 p17 fragment is elevated in patients with ST-segment elevation myocardial infarction: a novel observation. Journal of the American college of cardiology, 57 (2), 220–221.
- Aldous, S., et al., 2015. Outcomes in patients presenting with symptoms suggestive of acute coronary syndrome with elevated cardiac troponin but non-obstructive coronary disease on angiography. Heart, lung and circulation, 24 (9), 869–878.
- Allen, D.G., Whitehead, N.P., and Froehner, S.C., 2016. Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiological reviews, 96 (1), 253–305.
- Amako, T., et al., 1963. Experimental investigation of the metabolism of myoglobin. Kyushu journal of medical science, 14, 277–287.
- Andrews, N.W., Corrotte, M., and Castro-Gomes, T., 2015. Above the fray: surface remodeling by secreted lysosomal enzymes leads to endocytosis-mediated plasma membrane repair. Seminars in cell and developmental biology, 45, 10–17.
- Aviles, R.J., et al., 2002. Long-term prognosis of patients with clinical unstable angina pectoris without elevation of creatine kinase but with elevation of cardiac troponin i levels. American journal of cardiology, 90 (8), 875–878.
- Biondi-Zoccai, G.G., et al., 2004. Increased apoptosis in remote non-infarcted myocardium in multivessel coronary disease. International journal of cardiology, 94 (1), 105–110.
- Bjurman, C., et al., 2015. High-sensitive cardiac troponin, NT-proBNP, hFABP and copeptin levels in relation to glomerular filtration rates and a medical record of cardiovascular disease. Clinical biochemistry, 48 (4–5), 302–307.
- Boneu, B., Caranobe, C., and Sie, P., 1990. Pharmacokinetics of heparin and low molecular weight heparin. Bailliere's clinical haematology, 3 (3), 531–544.
- Botta, A., et al., 2008. The CTG repeat expansion size correlates with the splicing defects observed in muscles from myotonic dystrophy type 1 patients. Journal of medical genetics, 45 (10), 639–646.
- Boutet, M., Huttner, I., and Rona, G., 1976. Permeability alteration of sarcolemmal membrane in catecholamine-induced cardiac muscle cell injury. In vivo studies with fine structural diffusion tracer horse radish peroxidase. Laboratory investigation, 34 (5), 482–488.
- Brune, W., and Andoniou, C., 2017. Die another day: inhibition of cell death pathways by cytomegalovirus. Viruses, 9 (9), 249.
- Burton, K.P., et al., 1977. Lanthanum probe studies of cellular pathophysiology induced by hypoxia in isolated cardiac muscle. Journal of clinical investigation, 60 (6), 1289–1302.
- Bywaters, E.G., and Beall, D., 1998. Crush injuries with impairment of renal function. Journal of the American society of nephrology, 9 (2), 322–332.
- Cai, C., et al., 2009. MG53 nucleates assembly of cell membrane repair machinery. Nature cell biology, 11 (1), 56–64.
- Cao, C.M., et al., 2010. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation, 121 (23), 2565–2574.
- Cardinaels, E.P., et al., 2012. A comprehensive review of upper reference limits reported for (high-)sensitivity cardiac troponin assays: the challenges that lie ahead. Clinical chemistry and laboratory medicine, 50 (5), 791–806.
- Cardinaels, E.P., et al., 2013. Time-dependent degradation pattern of cardiac troponin T following myocardial infarction. Clinical chemistry, 59 (7), 1083–1090.
- Chiong, M.A., West, R., and Parker, J.O., 1974. Myocardial balance of inorganic phosphate and enzymes in man. Effects of tachycardia and ischemia. Circulation, 49 (2), 283–290.
- Clark, G.L., et al., 1978. Effects of lymphatic transport of enzyme on plasma creatine kinase time-activity curves after myocardial infarction in dogs. Circulation research, 43 (2), 162–169.
- Clarke, M.S., et al., 1995. Contraction-induced cell wounding and release of fibroblast growth factor in heart. Circulation research, 76 (6), 927–934.
- Clements, P., et al., 2010. Time course characterization of serum cardiac troponins, heart fatty acid-binding protein, and morphologic findings with isoproterenol-induced myocardial injury in the rat. Toxicologic pathology, 38 (5), 703–714.
- Cooper, S.T., and McNeil, P.L., 2015. Membrane repair: mechanisms and pathophysiology. Physiological reviews, 95 (4), 1205–1240.
- Davenport, N.R., et al., 2016. Membrane dynamics during cellular wound repair. Molecular biology of the cell, 27 (14), 2272–2285.
- Demonbreun, A.R., and McNally, E.M., 2016. Plasma membrane repair in health and disease. Current topics in membranes, 77, 67–96.
- Demonbreun, A.R., et al., 2016. An actin-dependent annexin complex mediates plasma membrane repair in muscle. The journal of experimental medicine, 213 (7), 2137OIA58–2137OIA18.
- Depre, C., and Vatner, S.F., 2005. Mechanisms of cell survival in myocardial hibernation. Trends in cardiovascular medicine, 15 (3), 101–110.
- Diris, J.H., et al., 2003. Impaired renal clearance explains elevated troponin T fragments in hemodialysis patients. Circulation, 109 (1), 23–25.
- Dunn, M.E., et al., 2011. The complete pharmacokinetic profile of serum cardiac troponin I in the rat and the dog. Toxicological sciences: an official journal of the society of toxicology, 123 (2), 368–373.
- Ellis, K., Dreisbach, A.W., and Lertora, J.L., 2001. Plasma elimination of cardiac troponin I in end-stage renal disease. Southern medical journal, 94 (10), 993–996.
- Ellis, R.J., 2001. Macromolecular crowding: obvious but underappreciated. Trends in biochemical sciences, 26 (10), 597–604.
- Feng, J., et al., 2001. Preload induces troponin I degradation independently of myocardial ischemia. Circulation, 103 (16), 2035–2037.
- Friden, V., et al., 2017. Clearance of cardiac troponin T with and without kidney function. Clinical biochemistry, 50 (9), 468–474.
- Galluzzi, L., et al., 2012. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell death and differentiation, 19 (1), 107–120.
- Galvani, M., et al., 1997. Prognostic influence of elevated values of cardiac troponin I in patients with unstable angina. Circulation, 95 (8), 2053–2059.
- Garcia-Dorado, D., and Ruiz-Meana, M., 2000. Propagation of cell death during myocardial reperfusion. Physiology, 15 (6), 326–330.
- Goussev, A., et al., 1998. Effects of ACE inhibition on cardiomyocyte apoptosis in dogs with heart failure. The American journal of physiology, 275 (2), H626–H631.
- Groth, T., and Sylven, C., 1981. Myoglobin kinetics in patients suffering from acute myocardial infarction in its early phase—as studied by the single injection method. Scandinavian journal of clinical and laboratory investigation, 41 (1), 79–85.
- Guo, X., et al., 2017. Cardioprotective role of tumor necrosis factor receptor-associated factor 2 by suppressing apoptosis and necroptosis. Circulation, 136 (8), 729–742.
- Hallgren, R., et al., 1978. Myoglobin turnover-influence of renal and extrarenal factors. Journal of laboratory and clinical medicine, 91 (2), 246–254.
- Han, R., and Campbell, K.P., 2007. Dysferlin and muscle membrane repair. Current opinion in cell biology, 19 (4), 409–416.
- Han, R., et al., 2007. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. Journal of clinical investigation, 117 (7), 1805–1813.
- Hayashi, T., and Notkins, A.L., 1994. Clearance of LDH-5 from the circulation of inbred mice correlates with binding to macrophages. International journal of experimental pathology, 75 (3), 165–168.
- Hayashi, T., et al., 1988. Regulation of enzyme levels in the blood. Influence of environmental and genetic factors on enzyme clearance. The American journal of pathology, 132 (3), 503–511.,
- Heilbrunn, L.V., 1930. The surface precipitation reaction of living cells, Proc. American philosophical society, 419, 295–301.
- Hermens, W.T., et al., 1990. Complete recovery in plasma of enzymes lost from the heart after permanent coronary artery occlusion in the dog. Circulation, 81 (2), 649–659.
- Hickman, P.E., et al., 2010. Cardiac troponin may be released by ischemia alone, without necrosis. Clinica chimica acta, 411 (5–6), 318–323.
- Hochhauser, E., et al., 2003. Bax ablation protects against myocardial ischemia-reperfusion injury in transgenic mice. American journal of physiology: heart and circulatory physiology, 284 (6), H2351–H2359.
- Hoffstein, S., et al., 1975. Colloidal lanthanum as a marker for impaired plasma membrane permeability in ischemic dog myocardium. The American journal of pathology, 79 (2), 207–218.
- Holly, T.A., et al., 1999. Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. Journal of molecular and cellular cardiology, 31 (9), 1709–1715.
- Jaffe, A.S., et al., 2011. Diseased skeletal muscle: a noncardiac source of increased circulating concentrations of cardiac troponin T. Journal of the American college of cardiology, 58 (17), 1819–1824.
- Jennings, R.B., 2013. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circulation research, 113 (4), 428–438.
- Jennings, R.B., et al., 1978. Relation between high energy phosphate and lethal injury in myocardial ischemia in the dog. The American journal of pathology, 92 (1), 187–214.
- Jennings, R.B., et al., 1983. Comparison of the effect of ischaemia and anoxia on the sarcolemma of the dog heart. European heart journal, 4 (Suppl H), 123–137.
- Jose Corbalan, J., Vatner, D.E., and Vatner, S.F., 2016. Myocardial apoptosis in heart disease: does the emperor have clothes? Basic research in cardiology, 111 (3), 31.
- Katus, H.A., et al., 1987. Influence of reperfusion on serum concentrations of cytosolic creatine kinase and structural myosin light chains in acute myocardial infarction. American journal of cardiology, 60 (7), 440–445.
- Katus, H.A., et al., 1989. Enzyme linked immuno assay of cardiac troponin T for the detection of acute myocardial infarction in patients. Journal of molecular and cellular cardiology, 21 (12), 1349–1353.
- Katus, H.A., et al., 1991. Intracellular compartmentation of cardiac troponin T and its release kinetics in patients with reperfused and nonreperfused myocardial infarction. American journal of cardiology, 67 (16), 1360–1367.
- Kawahara, C., et al., 2011. Prognostic role of high-sensitivity cardiac troponin T in patients with nonischemic dilated cardiomyopathy. Circulation journal, 75 (3), 656–661.
- Kerr, J.F., Wyllie, A.H., and Currie, A.R., 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British journal of cancer, 26 (4), 239–257.
- Kitamura, Y., et al., 2014. Manipulation of cardiac phosphatidylinositol 3-kinase (PI3K)/Akt signaling by apoptosis regulator through modulating IAP expression (ARIA) regulates cardiomyocyte death during doxorubicin-induced cardiomyopathy. Journal of biological chemistry, 289 (5), 2788–2800.
- Kristjansson, R.P., et al., 2016. Common and rare variants associating with serum levels of creatine kinase and lactate dehydrogenase. Nature communications, 7, 10572.
- Kung, G., Konstantinidis, K., and Kitsis, R.N., 2011. Programmed necrosis, not apoptosis, in the heart. Circulation research, 108 (8), 1017–1036.
- Kuru, S., et al., 2004. [A patient with limb girdle muscular dystrophy type 2B (LGMD2B) manifesting cardiomyopathy]. Rinsho shinkeigaku, 44 (6), 375–378.
- Lappalainen, H., et al., 2002. Elimination kinetics of myoglobin and creatine kinase in rhabdomyolysis: implications for follow-up. Critical care medicine, 30 (10), 2212–2215.
- Laugaudin, G., et al., 2016. Kinetics of high-sensitivity cardiac troponin T and I differ in patients with ST-segment elevation myocardial infarction treated by primary coronary intervention. European heart journal: acute cardiovascular care, 5 (4), 310–354.
- McNeil, P.L., 2014. Cell biology: ESCRTing trouble out! Current biology, 24 (9), R370–R372.
- McNeil, P.L., and Khakee, R., 1992. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. The American journal of pathology, 140 (5), 1097–1109.
- McNeil, P.L., and Kirchhausen, T., 2005. An emergency response team for membrane repair. Nature reviews molecular cell biology, 6 (6), 499–505.
- Miyake, K., and McNeil, P.L., 1995. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. The journal of cell biology, 131 (6), 1737–1745.
- Nagata, S., Hanayama, R., and Kawane, K., 2010. Autoimmunity and the clearance of dead cells. Cell, 140 (5), 619–630.
- Narula, J., et al., 1999. Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proceedings of the national academy of sciences of the United States of America, 96 (14), 8144–8149.
- Omland, T., et al., 2009. A sensitive cardiac troponin T assay in stable coronary artery disease. The New England journal of medicine, 361 (26), 2538–2547.
- Omland, T., et al., 2015. Troponins in heart failure. Clinica chimica acta, 443, 78–84.
- Page, E., Upshaw-Earley, J., and Goings, G., 1992. Permeability of rat atrial endocardium, epicardium, and myocardium to large molecules. Stretch-dependent effects. Circulation research, 71 (1), 159–173.
- Peacock, WF., et al., 2008. Cardiac troponin and outcome in acute heart failure. The New England journal of medicine, 358 (20), 2117–2126.
- Pfeffer, M.A., and Braunwald, E., 1990. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation, 81 (4), 1161–1172.
- Pickering, J.W., et al., 2016. Assessment of the European society of cardiology 0-hour/1-hour algorithm to rule-out and rule-in acute myocardial infarction. Circulation, 134 (20), 1532–1541.
- Piper, H.M., 2000. The calcium paradox revisited: an artefact of great heuristic value. Cardiovascular research, 45 (1), 123–127.
- Piper, H.M., et al., 1984. Energy metabolism and enzyme release of cultured adult rat heart muscle cells during anoxia. Journal of molecular and cellular cardiology, 16 (11), 995–1007.
- Piper, H.M., Meuter, K., and Schäfer, C., 2003. Cellular mechanisms of ischemia-reperfusion injury. The annals of thoracic surgery, 75 (2), S644–S648.
- Prabhudas, M., et al., 2014. Standardizing scavenger receptor nomenclature. Journal of immunology, 192 (5), 1997–2006.
- Radi, Z.A., et al., 2011. Increased serum enzyme levels associated with kupffer cell reduction with no signs of hepatic or skeletal muscle injury. The American journal of pathology, 179 (1), 240–247.
- Rittoo, D., et al., 2014. Elevation of cardiac troponin T, but not cardiac troponin I, in patients with neuromuscular diseases: implications for the diagnosis of myocardial infarction. Journal of the American college of cardiology, 63 (22), 2411–2420.
- Roffi, M., et al., 2016. 2015 ESC guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: task force for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). European heart journal, 37 (3), 267–315.
- Roos, A., et al., 2017. Stable high-sensitivity cardiac troponin T levels and outcomes in patients with chest pain. Journal of the American college of cardiology, 70 (18), 2226–2236.
- Sage, M.D., and Jennings, R.B., 1988. Cytoskeletal injury and subsarcolemmal bleb formation in dog heart during in vitro total ischemia. The American journal of pathology, 133 (2), 327–337.
- Saraste, A., and Pulkki, K., 2000. Morphologic and biochemical hallmarks of apoptosis. Cardiovascular research, 45 (3), 528–537.
- Saraste, A., et al., 1997. Apoptosis in human acute myocardial infarction. Circulation, 95 (2), 320–323.
- Schmid, J., et al., 2018. Elevated cardiac troponin T in skeletal myopathies: new evidence for a skeletal muscle origin. Journal of the American college of cardiology, 71, 1540–1549.
- Schwartz, P., et al., 1984. Ultrastructure of cultured adult myocardial cells during anoxia and reoxygenation. The American journal of pathology, 115 (3), 349–361.,
- Shah, A.S., et al., 2015. High-sensitivity cardiac troponin I at presentation in patients with suspected acute coronary syndrome: a cohort study. Lancet, 386 (10012), 2481–2488.
- Shiraishi, F., Kambara, M., and Ohtsuki, I., 1992. Replacement of troponin components in myofibrils. Journal of biochemistry, 111 (1), 61–65.
- Siriwardena, M., et al., 2012. Cardiac biomarker responses to dobutamine stress echocardiography in healthy volunteers and patients with coronary artery disease. Clinical chemistry, 58 (10), 1492–1494.
- Smit, M.J., et al., 1987. Receptor-mediated endocytosis of lactate dehydrogenase M4 by liver macrophages: a mechanism for elimination of enzymes from plasma. Evidence for competition by creatine kinase MM, adenylate kinase, malate, and alcohol dehydrogenase. Journal of biological chemistry, 262 (27), 13020–13026.
- Smit, M.J., et al., 1988. Catabolism of circulating enzymes: plasma clearance, endocytosis, and breakdown of lactate dehydrogenase-1 in rabbits. Clinical chemistry, 34 (12), 2475–2480.
- St Louis, J.D., et al., 2000. An experimental model of chronic myocardial hibernation. The annals of thoracic surgery, 69 (5), 1351–1357.
- Starnberg, K., et al., 2014. Revision of the troponin T release mechanism from a damaged human myocardium. Clinical chemistry, 60 (8), 1098–1104.
- Steenbergen, C., Hill, M.L., and Jennings, R.B., 1985. Volume regulation and plasma membrane injury in aerobic, anaerobic, and ischemic myocardium in vitro. Effects of osmotic cell swelling on plasma membrane integrity. Circulation research, 57 (6), 864–875.
- Steinhardt, R.A., Bi, G., and Alderton, J.M., 1994. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science, 263 (5145), 390–393.
- Streng, A.S., et al., 2016. Development of a targeted selected ion monitoring assay for the elucidation of protease induced structural changes in cardiac troponin T. Journal of proteomics, 136, 123–132.
- Svandova, E.B., et al., 2017. Expression of Fas, FasL, caspase-8 and other factors of the extrinsic apoptotic pathway during the onset of interdigital tissue elimination. Histochemistry and cell biology, 147 (4), 497–510.
- Swildens, J., et al., 2010. Integrin stimulation favors uptake of macromolecules by cardiomyocytes in vitro. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology, 26 (6), 999–1010.
- Sylven, C., 1978. The kinetics of myoglobin in old volunteers and in patients with acute myocardial infarction. Scandinavian journal of clinical and laboratory investigation, 38 (6), 561–565.
- Sylven, C., and Bendz, R., 1978. Myoglobin, creatine kinase and its isoenzyme MB in serum after acute myocardial infarction. European journal of cardiology, 8 (4-5), 515–521.
- Takemura, G., et al., 2013. Cardiomyocyte apoptosis in the failing heart–a critical review from definition and classification of cell death. International journal of cardiology, 167 (6), 2373–2386.
- Terasaki, M., Miyake, K., and McNeil, P.L., 1997. Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. The journal of cell biology, 139 (1), 63–74.
- Thygesen, K., et al., 2010. Recommendations for the use of cardiac troponin measurement in acute cardiac care. European heart journal, 31 (18), 2197–2204.
- Turer, A.T., et al., 2011. Myocardial ischemia induced by rapid atrial pacing causes troponin T release detectable by a highly sensitive assay: insights from a coronary sinus sampling study. Journal of the American college of cardiology, 57 (24), 2398–2405.
- Valaperta, R., et al., 2016. High-sensitive cardiac troponin T (hs-cTnT) assay as serum biomarker to predict cardiac risk in myotonic dystrophy: a case-control study. Clinica chimica acta, 463, 122–128.
- Vander Heide, R.S., and Ganote, C.E., 1987. Increased myocyte fragility following anoxic injury. Journal of molecular and cellular cardiology, 19 (11), 1085–1103.
- Voss, E.M., et al., 1995. Human and canine cardiac troponin T and creatine kinase-MB distribution in normal and diseased myocardium. Infarct sizing using serum profiles. Archives of pathology and laboratory medicine, 119 (9), 799–806.,
- Waddell, L.B., et al., 2011. Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tubules of the T-system with stretch. Journal of neuropathology and experimental neurology, 70 (4), 302–313.
- Wakabayashi, H., et al., 2015. Correlation between apoptosis and left ventricular remodeling in subacute phase of myocardial ischemia and reperfusion. EJNMMI research, 5 (1), 72.
- Wakabayashi, Y., et al., 1994. Rapid fall in blood myoglobin in massive rhabdomyolysis and acute renal failure. Intensive care medicine, 20 (2), 109–112.
- Waldum, B., and Os, I., 2013. The cardiorenal syndrome: what the cardiologist needs to know. Cardiology, 126 (3), 175–186.
- Wang, H., et al., 2014. Double-stranded RNA-dependent protein kinase deficiency protects the heart from systolic overload-induced congestive heart failure. Circulation, 129 (13), 1397–1406.
- Weil, B.R., et al., 2017. Brief myocardial ischemia produces cardiac troponin I release and focal myocyte apoptosis in the absence of pathological infarction in swine. JACC basic to translational science, 2 (2), 105–114.
- Wencker, D., et al., 2003. A mechanistic role for cardiac myocyte apoptosis in heart failure. The journal of clinical investigation, 111 (10), 1497–1504.
- Wens, S.C., et al., 2016. Elevated plasma cardiac troponin T levels caused by skeletal muscle damage in Pompe disease. Circulation: cardiovascular genetics, 9 (1), 6–13.
- Wenzel, K., et al., 2007. Dysfunction of dysferlin-deficient hearts. Journal of molecular medicine (Berlin, Germany)), 85 (11), 1203–1214.
- Yaoita, H., et al., 1998. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation, 97 (3), 276–281.
- York, M., et al., 2007. Characterization of troponin responses in isoproterenol-induced cardiac injury in the Hanover Wistar rat. Toxicologic pathology, 35 (4), 606–617.
- Zhang, J., et al., 2008. Isoproterenol-induced cardiotoxicity in Sprague-Dawley rats: correlation of reversible and irreversible myocardial injury with release of cardiac troponin T and roles of iNOS in myocardial injury. Toxicologic pathology, 36 (2), 277–278.
- Ziebig, R., et al., 2003. Renal elimination of troponin T and troponin I. Clinical chemistry, 49 (7), 1191–1193.