
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
We report a patient with autism-like deficits in emotional connectedness, executive dysfunction, and ataxia beginning at age 39. He had compound heterozygous variants in SPG7 (A510V and 1552+1 G>T substitutions), mutation of which is classically associated with spastic paraparesis. Diffusion MRI demonstrated abnormalities in the cerebellar outflow tracts. Transcranial magnetic stimulation showed a prolonged cortical silent period representing exaggerated cortical inhibition, as previously described with pure cerebellar degeneration. The acquired cerebellar cognitive affective syndrome in association with specific anatomic and neurophysiological abnormalities in the cerebellum expand the spectrum of SPG7-related neurodegeneration and support a role for cerebellar output in socio-emotional behavior.
Introduction
With our expanding knowledge of genetics, heritable causes of cerebellar degeneration continue to be identified and elucidated(Parodi et al., Citation2018). The hereditary ataxias can be divided into those with pure (featuring only ataxia) and complicated phenotypes which can be protean(Parodi et al., Citation2018). This can include decline in cognitive and social abilities which has been termed the “cerebellar cognitive affective syndrome” (CCAS)(Schmahmann, Citation2019). Deficits can include a dysexecutive syndrome(Schmahmann, Citation2019) and autism-like impairment of social skills with deficits in emotional attribution(Hoche et al., Citation2016). These deficits have been attributed to pathology in circuits including projections from the frontal lobes to the cerebellum via the pons and its connections back to the cortex via the thalamus(Schmahmann, Citation2019). In addition, abnormalities in cerebellar connectivity have been described in idiopathic autism without other overt cerebellar signs(Crippa et al., Citation2016).
Genetic mutations initially described in the context of other neurological manifestations have been found to include ataxia and other deficits attributable to the cerebellum. Here we describe a patient with cerebellar ataxia, a dysexecutive syndrome, and profound deficits in social behaviors associated with mutations in the SPG7 gene, originally described to cause spastic paraparesis(De Michele et al., Citation1998). He underwent a comprehensive clinical protocol, transcranial magnetic stimulation (TMS) and the Human Connectome imaging Protocol (HCP) to delineate abnormalities in connectivity underlying his symptoms.
Case report
The evaluation protocol was approved by the USC IRB and written informed consent was obtained from the patient. Our patient presented at age 47 with changes in emotional connectedness, cognition, and gait, which had begun insidiously at age 39. Previously healthy and described as having been intellectually gifted, he began to have difficulty maintaining employment as an accountant and financial analyst. He indicated that he had difficulty remembering things he needed to do and was described by others as distractible, lacking focus, thinking slowly, and having difficulty with decision making. He showed diminished judgment; for example, tipping an airport porter twenty dollars despite that being all the cash he had, and, when on vacation with his children to a mountain resort, he woke up early and fed the entire remaining supply of raw peanuts to squirrels before his children woke up–so they were not able to join him in this activity they would all have enjoyed. He developed slurred, rapid, and hypophonic speech such that he stopped his active participation in Toastmasters at age 41. Aphasia, disinhibition, and episodic memory deficits per se were denied. Over the same time period he had increasing difficulties with balance, finding it challenging to ride a bicycle.
The patient’s parents described him as always having been somewhat passive and a “follower.” As his symptoms developed they described him as even less confident, and more quiet and reserved. His conversation became superficial, often stating the obvious, with excessive focus on trivial details. They noted a decreased range of facial expression and that his voice became “flat” and “deadpan” and that he had lost “social niceties” to some extent. They were given questionnaires rating his personality retrospectively (before the onset of his condition) and currently. They indicated a decline from 30 to 25 (maximum score of 35) on the Empathic Concern Score on the Interpersonal Reactivity Index(Davis, Citation1983), supporting a measurable decline in expression of empathy. Nonetheless, they note that he could become irritable and angry such that his problems seemed to be more of expressing emotions than an inability to experience them.
After the family’s relocation to a distant state (where no other family members lived), his wife described him as having lost communication skills and being “Asperger-like”, not expressing love or care for her. He would stay late at work, not calling home to say if and when he would be home for dinner. In addition to becoming detached from his wife and children, he became detached from new acquaintances. At one point he failed to pick up his son from school. His emotional detachment ultimately contributed to their divorce and he has had continued difficulty engaging with his teenage son and daughter. Per the patient’s report, he acknowledges feeling emotionally distant, but desires to socially connect with others. His social ability did improve somewhat upon his return to the state he grew up in. He began meeting with a couple of long-time friends every week, joined an adaptive rehabilitation physical activity group, and he began volunteering weekly at a local hospital. He verbally expresses distress about his strained relationships with his wife and children; however, his affect is blunted and prosody of speech and emotion are restricted. He reported that he cannot cry, although recognizes the emotional significance of his current circumstances. He has limited understanding and insight as to what led to his divorce and job losses.
There was no family history of any similar conditions, his parents being unrelated to each other and in their late 60’s without neurological symptoms. He has one healthy 44 year-old brother. The maternal grandfather had been diagnosed with Parkinson’s disease and the paternal grandfather had depression with a suicide attempt.
On the bedside mental status examination at age 47 he showed deficits in judgment and in abstraction and was circumlocutory in his replies. He scored 28/30 on the MMSE, making 2 errors on serial 7’s but could subtract 3 serially from 20. Digit span forwards was 6 and in reverse was 4. Episodic memory, language, and visuospatial function were grossly preserved.
On physical examination he had slightly dysarthric and hypophonic speech. There was no nystagmus. He had minimal dysmetria on finger-to-nose testing which was more apparent on heel-to-shin testing. He took two attempts to stand without the use of his hands and had a wide-based gait, slightly short stride, and slightly diminished arm swing bilaterally. He could not tandem walk. He had no truncal ataxia or pyramidal signs and the neurological examination was otherwise within normal limits. At age 51 he had no evidence of spasticity or hyperreflexia and muscle tone continued to be normal without Parkinsonism. He scored 85/120 on the Cerebellar Cognitive Affective/Schmahmann Syndrome Scale (CCAS-Scale), failing the category switching, verbal recall, and affect subscales, consistent with “definite” CCAS(Hoche et al., Citation2018). He was treated with amphetamines for presumed attention deficit disorder, carbidopa/levodopa, and with sertraline (see below) without improvement.
Neuropsychological testing at age 46 showed mild declines in attention, working memory, processing speed, verbal memory retrieval and executive functioning (specifically, sequencing and cognitive flexibility). In contrast, language, verbal comprehension, perceptual reasoning, visual memory and abstract reasoning were intact. At age 47, repeat neuropsychological testing again suggested slowed processing speed, poor cognitive flexibility and mild verbal memory retrieval difficulty. In addition, phonemic verbal fluency was impaired, while semantic verbal fluency and confrontation naming remained intact. Visual spatial skills, visual memory and sequencing were also within expected ranges. Testing at age 50 again showed executive deficits though with improvement in phonemic verbal fluency, verbal memory retrieval and attention. Cognitive decline had therefore not been progressive over a 4 − 5 year period.
Psychological history was remarkable for episodes of depression and anxiety. At age 46, depression and anxiety were “minimal” (Beck Depression Inventory-II: 8; Beck Anxiety Inventory: 3); however, he endorsed feeling social discomfort and isolation, low self-concept, and mild day-to-day stress on a self-reported personality measure. At that time, his cognitive symptoms were thought to be psychiatrically driven, because of reported stress related to marital discord, financial loss and difficulty maintaining a job. At age 49, depression and social isolation were heightened, which appeared to have been exacerbated by an impending divorce and separation from his children. At age 50, depression was moderate-severe (Patient Health Questionnaire (PHQ-9): 11–15), along with endorsement of social isolation and mistrust of others, and elevated anxiety related to cognition and interpersonal relationships (Symptom Checklist-90-R). Thus, mood symptoms have been persistent, with no reports of suicidal ideation. His mood has been treated with sertraline and individual psychotherapy.
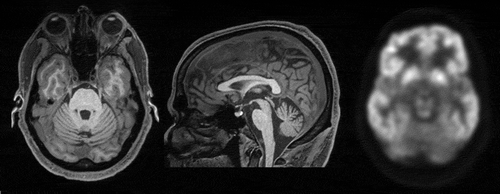
MRI at age 49 showed atrophy of the cerebellum in the midline (). A brain SPECT scan was thought to be within normal limits but a subsequent FDG-PET scan showed cerebellar hypometabolism (). Laboratory tests including a serum screen for heavy metals and a lumbar puncture were unremarkable except for a total protein level being elevated at 96 mg/dl. Electroencephalography was within normal limits. An autoimmune encephalopathy panel was negative as was a genetic panel for autosomal dominant ataxias. Specific genetic tests for Friedrich’s ataxia, Fragile X, and the C9orf72 repeat expansion were negative.
Figure 1. T1-weighted MRI at age 49. Cerebellar atrophy is appreciable in the axial (left) and midline sagittal plane (middle). FDG-PET scan revealed cerebellar hypometabolism.
Clinical Exome Sequencing revealed compound heterozygosity for variants in the SPG7 gene. Specifically, he was found to have one copy of the C to T substitution at position 1529 (ca. 1529 C > T) causing an alanine to valine substitution at codon 510 (A510V) and a G to T substitution at position 1552 + 1 (ca. 1552 + 1 G > T) resulting in loss of function of a splice donor. Compound heterozygosity was confirmed, as we observed that one mutation was inherited from the patient’s mother and the other from his father.
Using the high-resolution diffusion MRI protocol of the HCP(Sotiropoulos et al., Citation2013) and an atlas of brainstem pathways derived using it(Tang et al., Citation2017), diffusion parameters were determined in multiple brainstem and other white matter pathways. High-resolution diffusion MRI (dMRI) was acquired using the HCP protocol (8) and preprocessed using the HCP-Pipeline(Makropoulos et al., Citation2018). After distortion due to susceptibility and eddy current was corrected, the dMRI image was warped to the native T1-weighted MRI space. The diffusion tensor model and associated eigenvalues () were estimated with the MRtrix3 software(Tournier et al., Citation2019). Axial diffusivity (
), radial diffusivity (
) and mean diffusivity (
)) were then computed. To analyze the diffusivity in the brainstem region, we used the brainstem atlas from our previous work (Tang et al., Citation2017). With the non-linear warping, the brainstem pathways were warped to the native subject space. To measure the diffusivity in whole brain white matter regions, we segmented the brain using the Desikan-Killiany atlas such that each hemisphere is segmented into 34 white matter regions(Desikan et al., Citation2006). The mean values of diffusivity measures were calculated in all white matter pathways and compared to that of 11 healthy controls (6 males, mean age = 54.2, range 40–71) by two-sided t-tests.
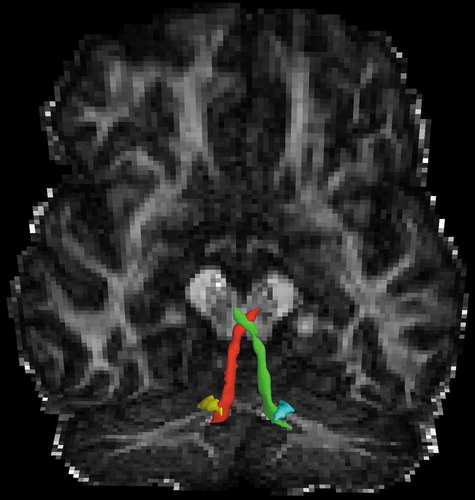
In our patient, axial diffusivity (AD), radial diffusivity (RD), and mean diffusivity was greater in the bilateral cerebellorubral tracts (p’s </ = 0.003) and cerebellothalamic tracts (p’s </ = 0.005) of the superior cerebellar peduncles (SCP) () as well as in the tracts from the medulla oblongata to the cerebellum (p’s </ = 0.01) and in the vestibulocerebellar tract of the inferior cerebellar peduncles (ICP, p’s </ = 0.006). No differences were seen in the adjacent anterior spinocerebellar tract of the SCP nor in other brainstem tracts or corticospinal tracts as delineated in our brainstem atlas(Tang et al., Citation2017) nor in any of white matter tracts elsewhere in the cerebrum.
Figure 2. Outflow pathways of the superior cerebellar peduncle (left: red; right: green) and inferior cerebellar peduncle (left: yellow; right: cyan), superimposed on the fractional anisotropy map, in which there was significantly elevated diffusion parameters in the patient with the SPG7 mutation relative to controls.
In addition, the physiology of the corticospinal tracts was interrogated using TMS. Multiple TMS measurements were compared between our patient and 5 healthy age-matched controls including resting motor threshold (rMT), interhemispheric inhibition (IHI), transcallosal conduction time (TCT), central conduction time (CCT), and cortical silent period (CSP). TMS assessments were performed using a single-pulse magnetic stimulator (Magstim 2002; Magstim Company, Ltd., United Kingdom) with a figure-of-8 and double-cone coil to target bilateral primary motor cortical representational areas of abductor pollicis brevis (APB) and tibialis anterior (TA), respectively. Brainsight Frameless (Rogue Research Inc, Montreal, Canada), a stereotactic image guidance system, was used to mark the APB and TA hotspots on a 3-D reconstruction of a participant-specific magnetic resonance image scan of the brain. The use of Brainsight allows the TMS coil to be accurately positioned over the muscle specific hotspots for subsequent stimulations, minimizing variability in TMS measures.
rMT was determined as the lowest TMS intensity required to elicit a 50 μV motor evoked potential at least 50% of the time (5 out of 10 trials) at each hotspot. IHI was quantified as the normalized ipsilateral silent period of APB and TA muscle activity following a TMS pulse(Kuo et al., Citation2017). Participants were instructed to activate each muscle to 50% of their maximal voluntary contraction as a TMS pulse was delivered to the ipsilateral hemisphere (intensity: 130% rMT for APB and 110% rMT for TA). TCT was then calculated by subtracting the motor evoked potential onset latency from the ipsilateral silent period onset latency. Both IHI and TCT provide insight on the neurophysiological function of transcallosal fibers. CCT was calculated by first determining the onset latency of an F-wave, a muscle action potential following an antidromic stimulus on a peripheral nerve. The F-wave latency represents the time required for an antidromic signal to travel to the spinal cord from a peripheral stimulation site and an orthodromic signal to travel from the spinal cord to the targeted nerve’s associated muscle following activation of an anterior horn cell. Dividing the F-wave latency by 2 reveals the conduction time of the peripheral nerve. F-wave latency was determined by delivering a TMS stimulation to the median nerve (for APB) and peroneal nerve (for TA). CCT was computed by subtracting half of the F-wave latency from the MEP onset latency (representing the conduction time of both the central and peripheral system). CSP, a measure of cortical inhibition, values were obtained by averaging 10 trials consisting of a stimulation at 100% of each individual’s rMT while participants maintained a moderate contraction of the target muscle (APB or TA). CSP was quantified as the time between onset of the TMS pulse and CSP offset (return of EMG activity to voluntary contraction level).
No significant differences were found between our patient and controls in threshold to elicit responses in the APB or TA, central conduction time to APB or TA, transcallosal conduction time, or in a measure of interhemispheric inhibition. However, a significantly prolonged CSP was found when eliciting a response in the left (156 ms vs. 105 ms, p = 0.0016) and right TA (271 vs. 110 ms, p < 0.001).
Discussion
Here we document neuroanatomical and neurophysiological changes indicating cerebellar dysfunction in a patient with emotional detachment and ataxia associated with compound heterozygosity for mutations in the SPG7 gene. Our patient had two mutations previously described in patients with spastic paraparesis but this feature and its underlying neuroanatomical and neurophysiological substrate have been notably absent. One mutation, the A510V substitution, has been repeatedly reported in association with spastic paraparesis(Van Gassen et al., Citation2012) and ataxia, presenting between childhood and age 54(Pfeffer et al., Citation2015). The global frequency of the minor allele is reported to be 0.2% (https://www.ncbi.nlm.nih.gov/clinvar/variation/42016/). Polyphen predicts this substitution to be probably damaging. The 1552 + 1 G > T substitution, a splice site mutation causing omission of exon 11 and loss of part of the paraplegin AAA-domain(Karlberg et al., Citation2009; Warnecke et al., Citation2010), has been reported in association with a complicated phenotype including spastic paraparesis, ptosis, horizontal nystagmus, and subtle executive dysfunction when in its homozygous state(Warnecke et al., Citation2010). It was absent in 202 controls(Warnecke et al., Citation2010).
The SPG7 gene encodes the paraplegin protein, an ATPase located in the mitochondrial inner membrane. As such, a complex phenotype with features of other mitochondrial encephalopathies might be anticipated. Though mutations in SPG7 are classically associated with progressive spastic paraparesis, ataxia occurred in > 50% of cases in a European cohort(Van Gassen et al., Citation2012) with a trend for it to be more common in persons with at least one A510V mutation as in our patient(Coarelli et al., Citation2019). Though ataxia was often the presenting symptom in this series of patients, it appears that spastic paraparesis usually, if not always, develops. Our patient, when assessed at ages 49 through 51 did not demonstrate any evidence of hyperreflexia or spasticity, indicating a relatively pure ataxia motor phenotype thus far.
TMS measures of cortical excitability and motor conduction times did not differ between our patient and controls. However, we did find a longer CSP duration in our patient relative to controls. A longer CSP duration to low threshold stimulation has been previously reported in patients with pure cerebellar degeneration(Tamburin et al., Citation2004) and in particular cerebellar degeneration due to SCA6 repeat expansion(Teo et al., Citation2008). Such prolongation is thought to represent exaggerated cortical inhibition, potentially as a result of reduced cerebello-thalamic-cortical input. This observation in a patient with ataxia and cerebellar degeneration associated with SPG7 mutations provides support for prolonged CSP representing a general effect of cerebellar dysfunction rather than a gene-specific effect.
The limited neuropathological data regarding SPG7 mutations that are available have demonstrated spongiosis and swollen axons of the spinocerebellar tracts in a homozygote for the R470Q substitution(Van Gassen et al., Citation2012). Neuropathology in a homozygote for the A510V mutation showed neuronal loss in the inferior olivary nucleus, the dentate nucleus, substantia nigra, and basal nucleus of Meynert. In this case, neurofibrillary tangles composed of 4 repeat (4 R) tau, neuropil threads, and Lewy Bodies were found in diverse brain areas, particularly in the brainstem and cerebellum including in the dentate nucleus. The cerebellorubral and cerebellothalamic tracts are composed in part of outputs from the dentate nucleus and neuronal loss there would be expected to manifest in deterioration of their axonal projections. Our case supports the utility of diffusion MRI in identifying pathology potentially attributable to neuronal loss. Parenthetically, as part of our protocol, we obtained a flortaucipir PET scan sensitive to the mixed 3 R and 4 R tau pathology characteristic of Alzheimer’s disease. Though showing some increased signal in the cerebellar dentate nucleus in our patient, the signal was not elevated above age-matched controls (data not shown). Whether such tau pathology was absent in our patient or if flortaucipir is not sensitive to the subtype of tau pathology seen in association with SPG7 mutations cannot be determined.
Our patient’s most clinically relevant deficit was an autism-like impairment of social skills. These are characteristic of CCAS(Hoche et al., Citation2016; Schmahmann & Sherman, Citation1998) as has been previously described in cerebellar degeneration(Heilman et al., Citation2014) and specifically in association with a compound heterozygote for SPG7 mutations (the A510V substitution and a ca. 2271delG, pMet757fs*65 mutation)(Zhang et al., Citation2017). Diffusion abnormalities were restricted to pathways in the ICP and SCP; specifically in the cerebellorubral and cerebellothalamic tracts. These include outputs from the limbic cerebellum (vermis, adjacent regions of the posterior cerebellar hemispheres, and the associated fastigial nuclei(Schmahmann et al., Citation2007)) which are connected to the amygdala, septum and hippocampus. Patients with midline lesions, as evident in the vermal atrophy of our patient, have been shown to have aberrant socio-emotional behaviors(Levisohn et al., Citation2000; Tavano et al., Citation2007). Consistent with our patient’s phenotype of ataxia and emotional detachment is the finding that abnormalities in the cerebellar-rubral-thalamic tracts are the most consistently reported abnormalities in developmental autism(Crippa et al., Citation2016). This observation provides evidence for a direct neuroanatomical substrate for socio-emotional behavior as abnormalities in these pathways, whether acquired or developmental, can cause a similar autism-like state.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Coarelli, G., Schule, R., van de Warrenburg, B. P. C., De Jonghe, P., Ewenczyk, C., Martinuzzi, A., Synofzik, M., Hamer, E. G., Baets, J., Anheim, M., Schöls, L., Deconinck, T., Masrori, P., Fontaine, B., Klockgether, T., D’Angelo, M. G., Monin, M.-L., De Bleecker, J., Migeotte, I., Bassi, M. T., … Durr, A. (2019). Loss of paraplegin drives spasticity rather than ataxia in a cohort of 241 patients with SPG7. Neurology, 92(23), e2679–e2690. https://doi.org/10.1212/WNL.0000000000007606
- Crippa, A., Del Vecchio, G., Busti Ceccarelli, S., Nobile, M., Arrigoni, F., & Brambilla, P. (2016). Cortico-cerebellar connectivity in autism spectrum disorder: What do we know so far? Frontiers in Psychiatry, 7(20). https://doi.org/10.3389/fpsyt.2016.00020
- Davis, M. H. (1983). Measuring individual differences in empathy: Evidence for a multidemensional approach. Journal of Personality and Social Psychology, 44(1), 113–126. https://doi.org/10.1037/0022-3514.44.1.113
- De Michele, G., De Fusco, M., Cavalcanti, F., Filla, A., Marconi, R., Volpe, G., Monticelli, A., Ballabio, A., Casari, G., & Cocozza, S. (1998). A new locus for autosomal recessive hereditary spastic paraplegia maps to chromosome 16q24.3. The American Journal of Human Genetics, 63(1), 135–139. https://doi.org/10.1086/301930
- Desikan, R. S., Segonne, F., Fischl, B., Quinn, B. T., Dickerson, B. C., Blacker, D., Buckner, R. L., Dale, A. M., Maguire, R. P., Hyman, B. T., Albert, M. S., & Killiany, R. J. (2006). An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage, 31(3), 968–980. https://doi.org/10.1016/j.neuroimage.2006.01.021
- Heilman, K. M., Leon, S. A., Burtis, D. B., Ashizawa, T., & Subramony, S. H. (2014). Affective communication deficits associated with cerebellar degeneration. Neurocase, 20(1), 18–26. https://doi.org/10.1080/13554794.2012.713496
- Hoche, F., Guell, X., Sherman, J. C., Vangel, M. G., & Schmahmann, J. D. (2016). Cerebellar contribution to social cognition. The Cerebellum, 15(6), 732–743. https://doi.org/10.1007/s12311-015-0746-9
- Hoche, F., Guell, X., Vangel, M. G., Sherman, J. C., & Schmahmann, J. D. (2018). The cerebellar cognitive affective/Schmahmann syndrome scale. Brain: A Journal of Neurology, 141(1), 248–270. https://doi.org/10.1093/brain/awx317
- Karlberg, T., van den Berg, S., Hammarstrom, M., Sagemark, J., Johansson, I., Holmberg-Schiavone, L., & Schuler, H. (2009). Crystal structure of the ATPase domain of the human AAA+ protein paraplegin/SPG7. PloS One, 4(10), e6975. https://doi.org/10.1371/journal.pone.0006975
- Kuo, Y. L., Dubuc, T., Boufadel, D. F., & Fisher, B. E. (2017). Measuring ipsilateral silent period: Effects of muscle contraction levels and quantification methods. Brain Research, 1674, 77–83. https://doi.org/10.1016/j.brainres.2017.08.015
- Levisohn, L., Cronin-Golomb, A., & Schmahmann, J. D. (2000). Neuropsychological consequences of cerebellar tumour resection in children: Cerebellar cognitive affective syndrome in a paediatric population. Brain: A Journal of Neurology, 123(Pt 5), 1041–1050. https://doi.org/10.1093/brain/123.5.1041
- Makropoulos, A., Robinson, E. C., Schuh, A., Wright, R., Fitzgibbon, S., Bozek, J., Counsell, S. J., Steinweg, J., Vecchiato, K., Passerat-Palmbach, J., Lenz, G., Mortari, F., Tenev, T., Duff, E. P., Bastiani, M., Cordero-Grande, L., Hughes, E., Tusor, N., Tournier, J.-D., Price, A. N., … Rueckert, D. (2018). The developing human connectome project: A minimal processing pipeline for neonatal cortical surface reconstruction. NeuroImage, 173, 88–112. https://doi.org/10.1016/j.neuroimage.2018.01.054
- Parodi, L., Coarelli, G., Stevanin, G., Brice, A., & Durr, A. (2018). Hereditary ataxias and paraparesias: Clinical and genetic update. Current Opinion in Neurology, 31(4), 462–471. https://doi.org/10.1097/WCO.0000000000000585
- Pfeffer, G., Pyle, A., Griffin, H., Miller, J., Wilson, V., Turnbull, L., Fawcett, K., Sims, D., Eglon, G., Hadjivassiliou, M., Horvath, R., Németh, A., & Chinnery, P. F. (2015). SPG7 mutations are a common cause of undiagnosed ataxia. Neurology, 84(11), 1174–1176. https://doi.org/10.1212/WNL.0000000000001369
- Schmahmann, J. D. (2019). The cerebellum and cognition. Neuroscience Letters, 688, 62–75. https://doi.org/10.1016/j.neulet.2018.07.005
- Schmahmann, J. D., & Sherman, J. C. (1998). The cerebellar cognitive affective syndrome. Brain: A Journal of Neurology, 121(Pt 4), 561–579. https://doi.org/10.1093/brain/121.4.561
- Schmahmann, J. D., Weilburg, J. B., & Sherman, J. C. (2007). The neuropsychiatry of the cerebellum - insights from the clinic. The Cerebellum, 6(3), 254–267. https://doi.org/10.1080/14734220701490995
- Sotiropoulos, S. N., Jbabdi, S., Xu, J., Andersson, J. L., Moeller, S., Auerbach, E. J., Glasser, M. F., Hernandez, M., Sapiro, G., Jenkinson, M., Feinberg, D. A., Yacoub, E., Lenglet, C., Van Essen, D. C., Ugurbil, K., & Behrens, T. E. (2013). Advances in diffusion MRI acquisition and processing in the human connectome project. NeuroImage, 80, 125–143. https://doi.org/10.1016/j.neuroimage.2013.05.057
- Tamburin, S., Fiaschi, A., Andreoli, A., Marani, S., Manganotti, P., & Zanette, G. (2004). Stimulus-response properties of motor system in patients with cerebellar ataxia. Clinical Neurophysiology, 115(2), 348–355. https://doi.org/10.1016/s1388-2457(03)00357-2
- Tang, Y., Sun, W., Toga, A. W., Ringman, J. M., & Shi, Y. (2017). A probabilistic atlas of human brainstem pathways based on connectome imaging data. NeuroImage, 169, 227–239. https://doi.org/10.1016/j.neuroimage.2017.12.042
- Tavano, A., Grasso, R., Gagliardi, C., Triulzi, F., Bresolin, N., Fabbro, F., & Borgatti, R. (2007). Disorders of cognitive and affective development in cerebellar malformations. Brain: A Journal of Neurology, 130(Pt. 10), 2646–2660. https://doi.org/10.1093/brain/awm201
- Teo, J. T., Schneider, S. A., Cheeran, B. J., Fernandez-del-Olmo, M., Giunti, P., Rothwell, J. C., & Bhatia, K. P. (2008). Prolonged cortical silent period but normal sensorimotor plasticity in spinocerebellar ataxia 6. Movement Disorders: Official Journal of the Movement Disorder Society, 23(3), 378–385. https://doi.org/10.1002/mds.21847
- Tournier, J. D., Smith, R., Raffelt, D., Tabbara, R., Dhollander, T., Pietsch, M., Christiaens, D., Jeurissen, B., Yeh, C.-H., & Connelly, A. (2019). MRtrix3: A fast, flexible and open software framework for medical image processing and visualisation. NeuroImage, 202, 116137. https://doi.org/10.1016/j.neuroimage.2019.116137
- van Gassen, K. L., van der Heijden, C. D., de Bot, S. T., den Dunnen, W. F., van den Berg, L. H., Verschuuren-Bemelmans, C. C., Kremer, H. P. H., Veldink, J. H., Kamsteeg, E.-J., Scheffer, H., & van de Warrenburg, B. P. (2012). Genotype-phenotype correlations in spastic paraplegia type 7: A study in a large Dutch cohort. Brain: A Journal of Neurology, 135(Pt. 10), 2994–3004. https://doi.org/10.1093/brain/aws224
- Warnecke, T., Duning, T., Schirmacher, A., Mohammadi, S., Schwindt, W., Lohmann, H., Dziewas, R., Deppe, M., Ringelstein, E. B., & Young, P. (2010). A novel splice site mutation in the SPG7 gene causing widespread fiber damage in homozygous and heterozygous subjects. Movement Disorders: Official Journal of the Movement Disorder Society, 25(4), 413–420. https://doi.org/10.1002/mds.22949
- Zhang, L., McFarland, K. N., Subramony, S. H., Heilman, K. M., & Ashizawa, T. (2017). SPG7 and Impaired Emotional Communication. The Cerebellum, 16(2) 595–598. https://doi.org/10.1007/s12311-016-0818-5