
ABSTRACT
. COL18A1 gene mutations have been associated with Knobloch syndrome, which is characterized by ocular and brain abnormalities. Here we report a 4.5 years-old male child with autism and two novel COL18A1 mutations (NM_030582.4: c.1883_1891dup and c.1787C>T). Hypermetropic astigmatism, but not brain migration disorders, was observed. However, an asymmetric pattern of cerebellar perfusion and a smaller arcuate fascicle were found. Low levels of collagen XVIII were also observed in the patient´s serum. Thus, biallelic loss-of-function mutations in COL18A1 may be a new cause of autism without the brain malformations typically reported in patients with Knobloch syndrome.
Introduction
Knobloch syndrome (KS) -KNO1, MIM# 267,750- is an infrequent autosomal recessive syndrome characterized by retinal problems and midline occipital abnormalities (Knobloch & Layer, Citation1971). Encephalocele, meningocele and cutis aplasia were initially described as cardinal clinical features of KS (Knobloch & Layer, Citation1971). However, after initial descriptions, other cases with neuronal migration disorders and epilepsy have been reported (Caglayan et al., Citation2014; Charsar & Goldberg, Citation2017; Czeizel et al., Citation1992; Hull et al., Citation2016; Keren et al., Citation2007; Kliemann et al., Citation2003; Passos-Bueno et al., Citation1994; L. S. Zhang et al., Citation2018; Seaver et al., Citation1993; White et al., Citation2017; C. Wilson et al., Citation1998). The presence of different ocular abnormalities has been the rule in this syndrome; lens subluxation, cataracts, vitreoretinal degeneration, retinal detachment, and glaucoma have been observed in KS (AlBakri et al., Citation2017; Bongiovanni et al., Citation2011; Duh et al., Citation2004; Ebrahimiadib et al., Citation2017; Gradstein et al., Citation2017; Hull et al., Citation2016; Khan et al., Citation2012; Knobloch & Layer, Citation1971; C. Wilson et al., Citation1998).
COL18A1 was described as the KS disease-causing gene (Heljasvaara et al., Citation2017; Sertie et al., Citation1996, Citation2000). It encodes for the collagen type XVIII α1 chain, a ubiquitously expressed, non-fibrillar collagen that is found in association with vascular and epithelial basement membranes (BM) (Heljasvaara et al., Citation2017). Collagen XVIII is expressed as three isoforms, namely short, medium and long isoforms; all of them are encoded by the same gene, COL18A1, which localizes on chromosome 21 in humans (Oh et al., Citation1994; O. T. Suzuki et al., Citation2002). Although COL18A1 is widely expressed, its isoforms have different tissue distribution (O. T. Suzuki et al., Citation2002). Altogether, collagen XVIII is needed for normal brain development and it is essential for the formation of most eye structures (Heljasvaara et al., Citation2017) .
Here, we describe a patient with autism and global developmental delay. Whole-exome sequencing in trio, confirmed via Sanger sequencing, identified compound heterozygous COL18A1 mutations. At the age of 4 years, none of the two previously described hallmark features of KS were observed in this case. COL18A1 expression studies demonstrated very low expression of total collagen XVIII and its isoforms in the blood of the affected patient.
Case report
Clinical features of the patients and previous diagnostic studies
A 3 years-old male, a second child from non-consanguineous healthy parents of Spanish origin, was referred to our clinic. Partum occurred via uncomplicated vaginal delivery after 39-weeks of pregnancy by the 35-year-old mother. Apgar scores were 9–10 at 1 and 5 minutes, respectively. Birth weight was 2960 gr (15th centile), length 49 cm (30th centile), OFD 34 cm (30th centile). Family history was not relevant; his parents and his sister were healthy.
His initial developmental milestones were in the normal range; he walked unsupported at 12 months; the first bisyllabic words occurred at 12 months. At the age of 3 years, he uses several words but no sentences. He has no interest in other children; he always plays alone. His social eye contact is minimal.
The clinical examination disclosed a weight of 15.1 kg (50th centile), a height of 95 cm (40th centile), and an OFD of 50.5 cm (40th centile), without dysmorphic features. Observation of the patient revealed verbal and nonverbal communication deficits, echolalia, neologisms, a stereotyped and idiosyncratic language associated with severe hyperkinetic behavior.
Neuropsychological assessment to measure intellectual functioning and development, as well as language abilities, were conducted. Specifically, the following tests and scales were used: the Wechsler Preschool and Primary Scale of Intelligence, Fourth Edition – WPPSI-IV-, the Merrill Palmer-Revised scale of development (MP-R), the Peabody Picture Vocabulary Test, Third Edition -PPVT-III-, and the verbal ability test of the British Ability Scales, Second Edition -BAS II. Notably, WPPSI-IV revealed the presence of an IQ of 75 (5th centile); verbal comprehension was severely affected (score of 65; 1st centile). Global and separate (cognitive, communication, motor, social-emotion development, and adaptative behavior) MP-R scores were below normal range; adaptative behavior and expressive language were severely affected. Specific language evaluation revealed a psycholinguistic level below 5th centil according to PPVT-III and BAS II scores.
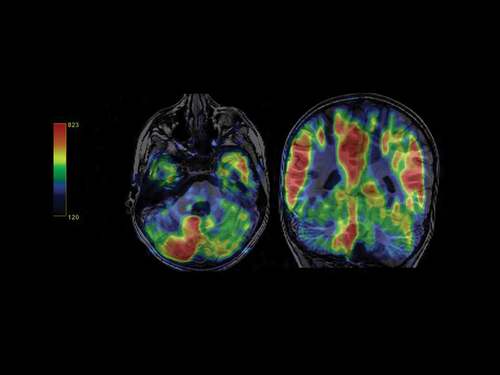
Routine laboratory screening including thyroid function and neurometabolic tests were within the normal range. Sleep video-EEG test and auditory evoked potentials displayed normal results. The ophthalmologic examination demonstrated hypermetropic astigmatism. Brain 3 T MRI did not reveal any significant structural malformations; diffusion tensor imaging with 3D-tractography reconstruction showed a smaller left arcuate fascicle with lower connections with the adjacent white matter (); besides, arterial spin-labeling (ASL) perfusion imaging evidenced marked cortical hypoperfusion in the left cerebellar hemisphere, with a normal pattern of the supratentorial cortical blood flow ().
Figure 1. Color orientation 3D-tractography reconstruction of the arcuate fascicle. The patent´s right-left asymmetry evidence a shorter tract and a lower amount of nerve fibers on the left side.
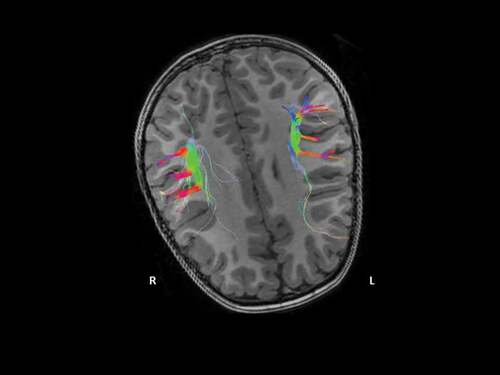
Figure 2. Color-code perfusion map with the pulsed-ASL sequence. Decreased cortical blood flow in the patient´s left cerebellar hemisphere.
Conventional genetic studies (karyotype and array-based comparative genomic hybridation with a 400k custom array) revealed no abnormalities.
At the age of 4.5 years, his neurological examination remained normal. He still had significant verbal and nonverbal communication deficits, associated with a severe hyperactivity behavior. At this age, because of additional concerns regarding social communication and repetitive behaviors, further information was obtained using the Autism Diagnostic Interview-Revised (ADI-R) and the Autism Diagnostic Observation Scale (ADOS). He met the criteria for autism spectrum disorder on both instruments. A new ophthalmological evaluation confirmed the presence of astigmatism, without lens- or retina-associated problems.
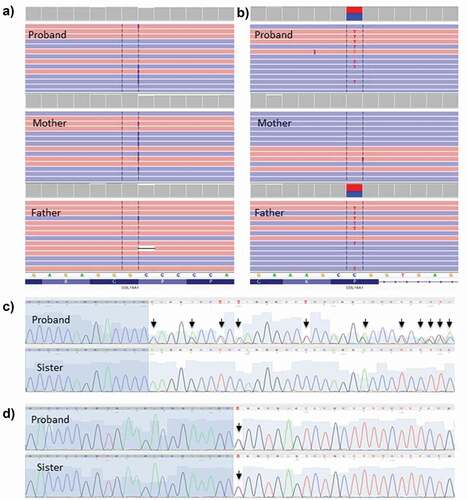
The study was completed by whole-exome sequencing; it revealed two genetic variants in the COL18A1 gene (Collagen Type XVIII Alpha 1 Chain). A maternally inherited non-frameshift insertion variant NM_030582.4: ca. 1883_1891dup; p.(Pro628_Pro630dup) and a paternally inherited missense variant NM_030582.4: ca. 1787 C > T,; p.(Pro596Leu) were identified. Both variants were classified as Variants of Uncertain Significance (VUS). The ca. 1883_1891dup categorization was supported by the effect on protein as length changes resulting from in-frame insertion in a non-repeat región (PM4). The population data (PM2), supported ca. 1787 C > T categorization, as an extremely low frequency in gnomAD population database has been reported (with a maximal non founder subpopulations frequency of 0.003%, and no homozygotes). On this variant in silico predictions criteria are controversial. Splicing tools did not predicted a clear splice-altering consequence. On the other hand, GnomAD missense Z-Score less than 0.647 does not support the pathogenicity of a missense variant (Z-score = −0.797). However, null variants are not the only known mechanism of variant pathogenicity in this gene, as three missense variants (NM_030582.4: ca. 2153 G > T, ca. 3559 G > A, ca. 4378 G > T) have been previously characterized as probably pathogenic (Clinvar database) and the CADD (Combined Annotation-Dependent Depletion) score of 18.20 may support a deleterious effect.
Segregation analysis confirmed a compound heterozygosity state, as both were identified in heterozygosity in the mother and father, consistent with an autosomal recessive inheritance. The missense mutation was also observed in his healthy sister. These mutations were confirmed by Sanger sequencing (). In the WES trio analysis, no other missense variants with a CADD score over 15 (Rentzsch et al., Citation2019) or LOF variants with a clear phenotypic association or a compatible segregation pattern were identified.
Figure 3. Illustration of the COL18A1 variants present in the proband by NGS and sanger sequencing and segregation studies.
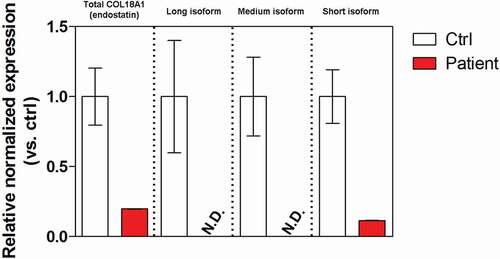
Real-time quantitative PCR (RT-qPCR) was used to evaluate the potential impact of the compound mutations on the COL18A1 gene expression. Peripheral blood leucocytes showed a mere 20–25% residual expression of both total COL18A1 (measured as C-terminal Endostatin domain expression) and the short isoform. Expression of the long and medium isoform was below the detection limit. Globally, decreased COL18A1 expression was consistent with a detrimental effect of the compound mutations on the gene expression ().
Figure 4. Expression of COL18A1 (total and isoforms) in peripheral blood leukocytes.
Materials and methods
The study was carried out in accordance with the Declaration of Helsinki of the World Medical Association and approved by the Local Ethics Committees. Informed consent was obtained from the family, after full explanation of the procedures.
Neuroimaging study: acquisition and analysis
Data were acquired using an 8-channel head coil with a 3 T system (Signa HDx, GE Medical System, Milwaukee, Wisconsin). Images were carefully inspected by an experienced neuroradiologist looking for structural defects; neuroimaging study and its analysis were performed before knowing the final clinical diagnosis and the result of the genetic study.
DTI images were obtained using a SS-SE echoplanar Diffusion weighted image (DWI) sequence (TR:12,000; FOV: 240 mm; sections thickness:3 mm, 0 spacing; matrix 128 × 128; bandwith: 250; 1 nex; diffusion encoding in 45 directions) with maximum b = 1000 sec/mm2.
3D-tractography was performed in an off-line workstation by using commercially available processing software as provided by the manufacturer (Functool 3D Fiber Tracking, GE, France) based on fiber assignment by contiguous tracking (FACT) method, achieved by connecting voxel to voxel. The threshold values were 0.3 for FA and 45° for the trajectory angles, between the regions of interest (ROIs). DTI tracts were also co-registered to the 3D-T1 weighted data set.
The Arterial Spin Labeling sequence (ASL) is a 3D pseudo-continuous ASL technique with a spiral readout with the following acquisition parameters: Field of view 24 cm, full brain coverage, slice thickness 4.0 mm, Arms 8, points per spiral 512, NEX 3, TDelay 1525 ms, TI 1525 ms, reconstructed matrix 128x128, resolution 1.875 × 1.875x4mm, TR 4.847s and TE 9.8 ms. The cerebral blood flow map was calculated using the same commercially available processing software (FuncTool perfussion, GE, France) which uses the pCASL CBF calculation equation (Alsop et al., Citation2015).
Genetic study
Exome sequencing was performed using genomic DNA isolated (MagnaPure, Roche) from whole blood from proband and parents. Libraries were prepared using the Ion AmpliSeq™ Exome Kit (Life Technologies) and quantified by qPCR. The enriched libraries were prepared using Ion Chef™ and sequenced on PI™ Chip in the Ion Proton™ System (Life Technologies) to provide >90% of amplicons covered with at least 20X. Signal processing, base calling, alignment, and variant calling were performed on a Proton™ Torrent Server using the Torrent Suite™ Software. Variants were annotated using Ion Reporter™ Software, and pedigree analysis was performed using the Genetic Disease Screen (GDS) trio workflow. Variant filtering and prioritization were performed with an in-house software program and a local database. The list of candidate variants was evaluated using previous published criteria (Eilbeck et al., Citation2017), Information from specific databases including variants already described in association with a known phenotype (ClinVar) and population frequency databases (dbSNP, gnomAD, 1000 Genome Project, or NHLBI-ESP 6500 exomes) to annotate variants that usually exist in the general population was used. We also estimated the pathogenicity of variants using CADD and a summation of selected prediction systems included in the dbNSFP database (SIFT, PolyPhen2, MutationTaster, MutationAssessor, LRT, FATHMM, and MetaSVM) for missense mutations. For mutations identified in splicing regions (including synonymous mutations), the effect on mRNA processing has been evaluated using the SpliceSiteFinder and MaxEntScan prediction systems, included in the SPiCE algorithm. Nucleotide position conservation has been evaluated according to the UCSC score ranges for the PhyloP tool. Finally, priorization variant was based on stringent assessments at both the gene and variant levels, and taking into consideration patient’s phenotype and the associated inheritance pattern. Candidate variants were visualized using IGV (Integrative Genomics Viewer). Candidate variants were evaluated based on stringent assessments at both the gene and variant levels, taking into consideration both the patient’s phenotype and the inheritance pattern. Variants were classified following the guidelines of the American College of Medical Genetics and Genomics (ACMG)(Richards et al., Citation2015). A board of molecular clinical geneticist evaluated each variant classified as pathogenic, likely pathogenic, or a variant of uncertain significance, and decided which, if any, had to be reported. In every case, causal variants were discussed with the referring physician and/or clinical geneticist. Identified variants were confirmed by Sanger sequencing.
COL18A1 expression studies
RNA was isolated with Qiagen kits (QIAzol and RNeasy mini), and cDNA was produced with the iScript cDNA synthesis kit (Bio-Rad). RT-qPCR was performed with iTaq SYBR Green Supermix with ROX reagents (Bio-Rad). All assays were performed in duplicate using a CFX96 Real-Time System (Bio-Rad). Values in all samples were normalized to GAPDH and normalized relative expression (2Cq) was calculated using CFX Manager software (Bio-Rad). To rule out technical artifacts, parallel analyses were also conducted on cDNA isolated with Quanta kit (qScript cDNA Synthesis Kit, Quanta Biosciences), on different RNA input quantities (1 and 2 g) and on different reverse transcription (RT) products’ dilutions (1:10 and undiluted). The following primers were used:
Statistical significance between healthy donors (N = 10) and the proband was assessed by the Mann-Whitney U test, with a threshold for significance set at P < 0.05. The concordance of the different qPCR analyses was evaluated by the Bland and Altman’s limits of agreement (LOA) method. No differences were found to be significant in LOA evaluation.
Discussion
Here we describe the first case of autism spectrum disorder and developmental delay associated with compound heterozygous COL18A1 mutations. Our findings expand the clinical spectrum of mutations in the COL18A1located on the long arm of chromosome 21 (chr21q22.3) and composed of 43 exons, that has been related to KS (Oh et al., Citation1994; Rehn et al., Citation1996). Most cases have loss-of-function (LoF) mutations; missense mutations have been unfrequently described in this syndrome (Menzel et al., Citation2004; Passos-Bueno et al., Citation2006; O. Suzuki et al., Citation2009) . Interestingly, patients with KS and COL18A1 missense mutations in homozygous state or compound heterozygous state (with LoF mutations) also showed lack of type XVIII collagen (O. Suzuki et al., Citation2009). Regulation by micro RNAs might explain the impact of missense mutations on the COL18A1 gene expression; point mutations might increase the affinity for binding of the miRNA or create a new miRNA binding site (Valencia-Sanchez et al., Citation2006; Zeng et al., Citation2003). On the other hand, based on the expression study results, we can´t exclude that the splicing prediction for the missense variant (located at donor site 2 bps downstream) may not be accurate, and this really has an effect on the expression levels.
Ocular and brain defects are the two cardinal features of this syndrome (Caglayan et al., Citation2014; Kliemann et al., Citation2003; O. T. Suzuki et al., Citation2002). After initial descriptions, multisystemic manifestations have been described, namely renal abnormalities, leukemia, hypertriglyceridemia, lung hypoplasia and dysmorphic features (Heljasvaara et al., Citation2017).
Ocular abnormalities are often present in the first years of the life of KS patients and include progressive high myopia, lens subluxation, early-onset cataracts, vitreoretinal degeneration, macular abnormalities, retinal detachment, and glaucoma (AlBakri et al., Citation2017; Bongiovanni et al., Citation2011; Caglayan et al., Citation2014; Duh et al., Citation2004; Gradstein et al., Citation2017; Heljasvaara et al., Citation2017; Khan et al., Citation2012). These anomalies may lead to blindness at an early age. The presence of collagen XVIII in various BMs of the ocular structures (Bruch´s membrane, retina, and the lens capsule) explains the eye defects observed in patients with COL18A1 mutations (Heljasvaara et al., Citation2017). Notably, no evidence of ocular malformations could be inferred for the case reported. This suggests that the compound mutations do not completely abolish COL18A1 expression, in line with both previous reports (Duan et al., Citation2017; Karbassi et al., Citation2016) and the residual, though severely hampered, gene expression observed in leukocytes, and that such residual quantity might still be enough to sustain normal ocular development. Indeed, short isoform -partially conserved in our patient- is critical for eye development and maintenance, and for the normal closure of neural tube (O. Suzuki et al., Citation2009).
The initial neurological manifestations of KS were circumscribed to infratentorial malformations (encephalocele, meningocele and cutis aplasia) (Czeizel et al., Citation1992; Knobloch & Layer, Citation1971; Passos-Bueno et al., Citation1994; Seaver et al., Citation1993). The presence of collagen XVIII in the neuroectoderm, pial BM, blood vessels BM and epithelial BM of the choroid plexuses, as well as its role in neuronal migration, could also explain the neurological malformations in KS (Caglayan et al., Citation2014; Heljasvaara et al., Citation2017; Utriainen et al., Citation2004). In the last years, other neurological problems (functional or structural) have been described: brain migration disorders (polymicrogyria and heterotopic nodules; a review of previous neuroimaging findings can be found in ), ataxia, epilepsy, and cognitive delay (Charsar & Goldberg, Citation2017; Corbett et al., Citation2017; Kliemann et al., Citation2003; White et al., Citation2017) . The absence of structural anomalies in the MRI is an exceptional situation, with only 3 previously described cases (9%); in counterpart, 70% of the published cases with available MRI showed different neuronal migration disorders. Although psychomotor retardation is not a constant feature in this syndrome, cognitive regression or developmental delay have been previously described (Caglayan et al., Citation2014; Keren et al., Citation2007; Kliemann et al., Citation2003; Paisan-Ruiz et al., Citation2009); cognitive impairment is not clearly related to the presence of heterotopias or polymicrogyria (Caglayan et al., Citation2014; Kliemann et al., Citation2003). The roles of collagen XVIII in mitochondrial function, organization of brain synapses and cortical brain development may contribute to this feature (Heljasvaara et al., Citation2017; Kliemann et al., Citation2003; Momota et al., Citation2013; Su et al., Citation2012). In the case described here, the presence of an abnormal pattern of infratentorial perfusion as well as the marked asymmetry between the arcuate fascicles possibly reflects the presence of microstructural and/or vascular defects secondary to the observed low levels of collagen XVIII in vessels’ BM, in line with a previously reported role for collagen XVIII in brain and cerebellar organization (Su et al., Citation2012). Interestingly, perfusion impairments of cerebellar hemispheres have been described in autism (Pagani et al., Citation2012; Ryu et al., Citation1999); of particular interest is the poor development of the left arcuate fascicle as this is involved in the development of language, and similar findings have also been found in autistic patients or other syndromes (B. J. Wilson et al., Citation2011; Roberts et al., Citation2014; L. S. Zhang et al., Citation2018). The anomalies we describe might be caused by microstructural cortical defects or by the disturbance in both axonal and myelin development (Poretti et al., Citation2013).
Table 1. Neuroimaging findings in patients with biallelic COL18A1 mutations. * Cases with anomalies in brain MRI (only cases with performed and available results)
The presence of other co-existing genetic disorders (CNVs or SNVs) responsible for his neurodevelopmental disorder may occur. However, described genetic results, COL18A1 expression studies and cognitive impairment in patients with KS support this relationship. The case described here extends the clinical spectrum of mutations of COL18A1 and calls for further studies of this gene in autism.
Author Contributions
Díez and S. Alvarez provided molecular genetic data; M. Jiménez provided neuroimaging data; S. López-Martín & J. Albert: neuropsychological assessment and study supervision. D. Martín, B. Calleja and A. Fernández-Jaén provided clinical data and reviewed the manuscript; M.T. Gómez provided the ophthalmological reports; T. Pihlajaniemi and V. Izzi provided expression studies; T. Pihlajaniemi, V. Izzi, S. López-Martín, J. Albert and A. Fernández-Jaén supervised the study and wrote the paper.
Statement of Ethics
This study complied with the guidelines for human studies and was conducted in accordance with the World Medical Association Declaration of Helsinki. Written consent was obtained from the family to publish this case (including publication of images).
Acknowledgments
Authors would like to thank Jaana Peters for technical assistance.
Disclosure Statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- AlBakri, A., Ghazi, N. G., & Khan, A. O. (2017). Biometry, optical coherence tomography, and further clinical observations in knobloch syndrome. Ophthalmic Genetics, 38 (2), 138–142. https://doi.org/https://doi.org/10.3109/13816810.2016.1164197
- Alsop, D. C., Detre, J. A., Golay, X., Gunther, M., Hendrikse, J., Hernandez-Garcia, L., Lu, H., MacIntosh, B. J., Parkes, L. M., Smits, M., Van Osch, M. J., Wang, D. J., Wong, E. C., & Zaharchuk, G. (2015). Recommended implementation of arterial spin-labeled perfusion MRI for clinical applications: A consensus of the ISMRM perfusion study group and the European consortium for ASL in dementia. Magnetic Resonance In Medicine : Official Journal Of The Society Of Magnetic Resonance In Medicine / Society of Magnetic Resonance in Medicine, 73 (1), 102–116. https://doi.org/https://doi.org/10.1002/mrm.25197
- Bongiovanni, C. S., Ferreira, C. C., Rodrigues, A. P., Fortes Filho, J. B., & Tartarella, M. B. (2011). Cataract surgery in knobloch syndrome: A case report. Clin Ophthalmol, 5, 735–737. https://doi.org/https://doi.org/10.2147/OPTH.S18989
- Caglayan, A. O., Baranoski, J. F., Aktar, F., Han, W., Tuysuz, B., Guzel, A., Guclu, B., Kaymakcalan, H., Aktekin, B., Akgumus, G. T., Murray, P. B., Erson-Omay, E. Z., Caglar, C., Bakircioglu, M., Sakalar, Y. B., Guzel, E., Demir, N., Tuncer, O., Senturk, S., Ekici, B., Gunel, M. (2014). Brain malformations associated with knobloch syndrome–review of literature, expanding clinical spectrum, and identification of novel mutations. Pediatric Neurology, 51 (6), 806–813 e808. https://doi.org/https://doi.org/10.1016/j.pediatrneurol.2014.08.025
- Charsar, B. A., & Goldberg, E. M. (2017). Polymicrogyria and intractable epilepsy in siblings with knobloch syndrome and homozygous mutation of COL18A1. Pediatric Neurology, 76, 91–92. https://doi.org/https://doi.org/10.1016/j.pediatrneurol.2017.08.003
- Corbett, M. A., Turner, S. J., Gardner, A., Silver, J., Stankovich, J., Leventer, R. J., Derry, C. P., Carroll, R., Ha, T., Scheffer, I. E., Bahlo, M., Jackson, G. D., Mackey, D. A., Berkovic, S. F., & Gecz, J. (2017). Familial epilepsy with anterior polymicrogyria as a presentation of COL18A1 mutations. European Journal of Medical Genetics, 60 (8), 437–443. https://doi.org/https://doi.org/10.1016/j.ejmg.2017.06.002
- Czeizel, A. E., Goblyos, P., Kustos, G., Mester, E., & Paraicz, E. (1992). The second report of knobloch syndrome. American Journal of Medical Genetics, 42 (6), 777–779. https://doi.org/https://doi.org/10.1002/ajmg.1320420605
- Duan, Y., Lin, S., Xie, L., Zheng, K., Chen, S., Song, H., Zeng, X., Gu, X., Wang, H., Zhang, L., Shao, H., Hong, W., Zhang, L., & Duan, S. (2017). Exome sequencing identifies a novel mutation of the GDI1 gene in a Chinese non-syndromic X-linked intellectual disability family. Genetics and Molecular Biology, 40 (3), 591–596. https://doi.org/https://doi.org/10.1590/1678-4685-GMB-2016-0249
- Duh, E. J., Yao, Y. G., Dagli, M., & Goldberg, M. F. (2004). Persistence of fetal vasculature in a patient with knobloch syndrome: Potential role for endostatin in fetal vascular remodeling of the eye. Ophthalmology, 111 (10), 1885–1888. https://doi.org/https://doi.org/10.1016/j.ophtha.2004.03.030
- Ebrahimiadib, N., Modjtahedi, B. S., Ferenchak, K., Papakostas, T. D., Mantagos, J. S., & Vavvas, D. G. (2017). Optical coherence tomography findings and successful repair of retina detachment in knobloch syndrome. Digital Journal Of Ophthalmology : Djo / sponsored by Massachusetts Eye and Ear Infirmary, 23 (1), 29–32. https://doi.org/https://doi.org/10.5693/djo.02.2017.01.002
- Eilbeck, K., Quinlan, A., & Yandell, M. (2017). Settling the score: Variant prioritization and mendelian disease. Nature Reviews. Genetics, 18 (10), 599–612. https://doi.org/https://doi.org/10.1038/nrg.2017.52
- Gradstein, L., Hansen, R. M., Cox, G. F., Altschwager, P., & Fulton, A. B. (2017). Progressive retinal degeneration in a girl with knobloch syndrome who presented with signs of ocular albinism. Documenta Ophthalmologica. Advances in Ophthalmology, 134 (2), 135–140. https://doi.org/https://doi.org/10.1007/s10633-017-9574-1
- Heljasvaara, R., Aikio, M., Ruotsalainen, H., & Pihlajaniemi, T. (2017). Collagen XVIII in tissue homeostasis and dysregulation - lessons learned from model organisms and human patients. Matrix Biology : Journal of the International Society for Matrix Biology, 57-58, 55–75. https://doi.org/https://doi.org/10.1016/j.matbio.2016.10.002
- Hull, S., Arno, G., Ku, C. A., Ge, Z., Waseem, N., Chandra, A., Webster, A. R., Robson, A. G., Michaelides, M., Weleber, R. G., Davagnanam, I., Chen, R., Holder, G. E., Pennesi, M. E., & Moore, A. T. (2016). Molecular and clinical findings in patients with knobloch syndrome. JAMA Ophthalmology, 134 (7), 753–762. https://doi.org/https://doi.org/10.1001/jamaophthalmol.2016.1073
- Karbassi, I., Maston, G. A., Love, A., DiVincenzo, C., Braastad, C. D., Elzinga, C. D., Bright, A. R., Previte, D., Zhang, K., Rowland, C. M., McCarthy, M., Lapierre, J. L., Dubois, F., Medeiros, K. A., Batish, S. D., Jones, J., Liaquat, K., Hoffman, C. A., Jaremko, M., Wang, Z., … Higgins, J. J. (2016). A standardized DNA variant scoring system for pathogenicity assessments in mendelian disorders. Human Mutation, 37 (1), 127–134. https://doi.org/https://doi.org/10.1002/humu.22918
- Keren, B., Suzuki, O. T., Gerard-Blanluet, M., Bremond-Gignac, D., Elmaleh, M., Titomanlio, L., Delezoide, A. L., Passos-Bueno, M. R., & Verloes, A. (2007). CNS malformations in knobloch syndrome with splice mutation in COL18A1 gene. American Journal of Medical Genetics. Part A, 143A (13), 1514–1518. https://doi.org/https://doi.org/10.1002/ajmg.a.31784
- Khan, A. O., Aldahmesh, M. A., Mohamed, J. Y., Al-Mesfer, S., & Alkuraya, F. S. (2012). The distinct ophthalmic phenotype of knobloch syndrome in children. The British Journal of Ophthalmology, 96 (6), 890–895. https://doi.org/https://doi.org/10.1136/bjophthalmol-2011-301396
- Kliemann, S. E., Waetge, R. T., Suzuki, O. T., Passos-Bueno, M. R., & Rosemberg, S. (2003). Evidence of neuronal migration disorders in knobloch syndrome: Clinical and molecular analysis of two novel families. American Journal of Medical Genetics. Part A, 119A (1), 15–19. https://doi.org/https://doi.org/10.1002/ajmg.a.20070
- Knobloch, W., & Layer, J. (1971). Retinal detachment and encephalocele. Journal of Pediatric Ophthalmology, 8, 181–184. https://doi.org/https://doi.org/10.3928/0191-3913-19710801-11
- Menzel, O., Bekkeheien, R. C., Reymond, A., Fukai, N., Boye, E., Kosztolanyi, G., Aftimos, S., Deutsch, S., Scott, H. S., Olsen, B. R., Antonarakis, S. E., & Guipponi, M. (2004). Knobloch syndrome: Novel mutations in COL18A1, evidence for genetic heterogeneity, and a functionally impaired polymorphism in endostatin. Human Mutation, 23 (1), 77–84. https://doi.org/https://doi.org/10.1002/humu.10284
- Momota, R., Narasaki, M., Komiyama, T., Naito, I., Ninomiya, Y., & Ohtsuka, A. (2013). Drosophila type XV/XVIII collagen mutants manifest integrin mediated mitochondrial dysfunction, which is improved by cyclosporin A and losartan. The International Journal of Biochemistry & Cell Biology, 45 (5), 1003–1011. https://doi.org/https://doi.org/10.1016/j.biocel.2013.02.001
- Oh, S. P., Warman, M. L., Seldin, M. F., Cheng, S. D., Knoll, J. H., Timmons, S., & Olsen, B. R. (1994). Cloning of cDNA and genomic DNA encoding human type XVIII collagen and localization of the alpha 1(XVIII) collagen gene to mouse chromosome 10 and human chromosome 21. Genomics, 19 (3), 494–499. https://doi.org/https://doi.org/10.1006/geno.1994.1098
- Pagani, M., Manouilenko, I., Stone-Elander, S., Odh, R., Salmaso, D., Hatherly, R., Brolin, F., Jacobsson, H., Larsson, S. A., & Bejerot, S. (2012). Brief report: Alterations in cerebral blood flow as assessed by PET/CT in adults with autism spectrum disorder with normal IQ. Journal of Autism and Developmental Disorders, 42 (2), 313–318. https://doi.org/https://doi.org/10.1007/s10803-011-1240-y
- Paisan-Ruiz, C., Scopes, G., Lee, P., & Houlden, H. (2009). Homozygosity mapping through whole genome analysis identifies a COL18A1 mutation in an Indian family presenting with an autosomal recessive neurological disorder. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics : The Official Publication of the International Society of Psychiatric Genetics, 150B (7), 993–997. https://doi.org/https://doi.org/10.1002/ajmg.b.30929
- Passos-Bueno, M. R., Marie, S. K., Monteiro, M., Neustein, I., Whittle, M. R., Vainzof, M., & Zatz, M. (1994). Knobloch syndrome in a large Brazilian consanguineous family: Confirmation of autosomal recessive inheritance. American Journal of Medical Genetics, 52 (2), 170–173. https://doi.org/https://doi.org/10.1002/ajmg.1320520209
- Passos-Bueno, M. R., Suzuki, O. T., Armelin-Correa, L. M., Sertié, A. L., Errera, F. I., Bagatini, K., Kok, F., & Leite, K. R. (2006). Mutations in collagen 18A1 (COL18A1) and their relevance to the human phenotype. Anais Da Academia Brasileira De Ciencias, 78 (1), 123–131. https://doi.org/https://doi.org/10.1590/S0001-37652006000100012
- Poretti, A., Meoded, A., Rossi, A., Raybaud, C., & Huisman, T. A. (2013). Diffusion tensor imaging and fiber tractography in brain malformations. Pediatric Radiology, 43 (1), 28–54. https://doi.org/https://doi.org/10.1007/s00247-012-2428-9
- Rehn, M., Hintikka, E., & Pihlajaniemi, T. (1996). Characterization of the mouse gene for the alpha 1 chain of type XVIII collagen (Col18a1) reveals that the three variant N-terminal polypeptide forms are transcribed from two widely separated promoters. Genomics, 32 (3), 436–446. https://doi.org/https://doi.org/10.1006/geno.1996.0139
- Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., & Kircher, M. (2019). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47 (D1), D886–D894. https://doi.org/https://doi.org/10.1093/nar/gky1016
- Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W. W., Hegde, M., Lyon, E., & Spector, E. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine, 17 (5), 405–423. https://doi.org/https://doi.org/10.1038/gim.2015.30
- Roberts, T. P., Heiken, K., Zarnow, D., Dell, J., Nagae, L., Blaskey, L., Solot, C., Levy, S. E., Berman, J. I., & Edgar, J. C. (2014). Left hemisphere diffusivity of the arcuate fasciculus: Influences of autism spectrum disorder and language impairment. AJNR. American Journal of Neuroradiology, 35 (3), 587–592. https://doi.org/https://doi.org/10.3174/ajnr.A3754
- Ryu, Y. H., Lee, J. D., Yoon, P. H., Kim, D. I., Lee, H. B., & Shin, Y. J. (1999). Perfusion impairments in infantile autism on technetium-99m ethyl cysteinate dimer brain single-photon emission tomography: Comparison with findings on magnetic resonance imaging. European Journal of Nuclear Medicine, 26 (3), 253–259. https://doi.org/https://doi.org/10.1007/s002590050385
- Seaver, L. H., Joffe, L., Spark, R. P., Smith, B. L., & Hoyme, H. E. (1993). Congenital scalp defects and vitreoretinal degeneration: Redefining the knobloch syndrome. American Journal of Medical Genetics, 46 (2), 203–208. https://doi.org/https://doi.org/10.1002/ajmg.1320460221
- Sertie, A. L., Quimby, M., Moreira, E. S., Murray, J., Zatz, M., Antonarakis, S. E., & Passos-Bueno, M. R. (1996). A gene which causes severe ocular alterations and occipital encephalocele (knobloch syndrome) is mapped to 21q22.3. Human Molecular Genetics, 5 (6), 843–847. https://www.ncbi.nlm.nih.gov/pubmed/8776601 . https://doi.org/https://doi.org/10.1093/hmg/5.6.843
- Sertie, A. L., Sossi, V., Camargo, A. A., Zatz, M., Brahe, C., & Passos-Bueno, M. R. (2000). Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (knobloch syndrome). Human Molecular Genetics, 9 (13), 2051–2058. https://www.ncbi.nlm.nih.gov/pubmed/10942434 . https://doi.org/https://doi.org/10.1093/hmg/9.13.2051
- Su, J., Stenbjorn, R. S., Gorse, K., Su, K., Hauser, K. F., Ricard-Blum, S., Pihlajaniemi, T., & Fox, M. A. (2012). Target-derived matricryptins organize cerebellar synapse formation through alpha3beta1 integrins. Cell Reports, 2 (2), 223–230. https://doi.org/https://doi.org/10.1016/j.celrep.2012.07.001
- Suzuki, O., Kague, E., Bagatini, K., Tu, H., Heljasvaara, R., Carvalhaes, L., Gava, E., De Oliveira, G., Godoi, P., Oliva, G., Kitten, G., Pihlajaniemi, T., & Passos-Bueno, M. R. (2009). Novel pathogenic mutations and skin biopsy analysis in knobloch syndrome. Molecular Vision, 15, 801–809. https://www.ncbi.nlm.nih.gov/pubmed/19390655 .
- Suzuki, O. T., Sertie, A. L., Der Kaloustian, V. M., Kok, F., Carpenter, M., Murray, J., Czeizel, A. E., Kliemann, S. E., Rosemberg, S., Monteiro, M., Olsen, B. R., & Passos-Bueno, M. R. (2002). Molecular analysis of collagen XVIII reveals novel mutations, presence of a third isoform, and possible genetic heterogeneity in knobloch syndrome. American Journal of Human Genetics, 71 (6), 1320–1329. https://doi.org/https://doi.org/10.1086/344695
- Utriainen, A., Sormunen, R., Kettunen, M., Carvalhaes, L. S., Sajanti, E., Eklund, L., Kauppinen, R., Kitten, G. T., & Pihlajaniemi, T. (2004). Structurally altered basement membranes and hydrocephalus in a type XVIII collagen deficient mouse line. Human Molecular Genetics, 13 (18), 2089–2099. https://doi.org/https://doi.org/10.1093/hmg/ddh213
- Valencia-Sanchez, M. A., Liu, J., Hannon, G. J., & Parker, R. (2006). Control of translation and mRNA degradation by miRNAs and siRNAs. Genes & Development, 20 (5), 515–524. https://doi.org/https://doi.org/10.1101/gad.1399806
- White, R. J., Wang, Y., Tang, P., & Montezuma, S. R. (2017). Knobloch syndrome associated with polymicrogyria and early onset of retinal detachment: Two case reports. BMC Ophthalmology, 17 (1), 214. https://doi.org/https://doi.org/10.1186/s12886-017-0615-z
- Wilson, B. J., Sundaram, S. K., Huq, A. H., Jeong, J. W., Halverson, S. R., Behen, M. E., Bui, D. Q., & Chugani, H. T. (2011). Abnormal language pathway in children with angelman syndrome. Pediatric Neurology, 44 (5), 350–356. https://doi.org/https://doi.org/10.1016/j.pediatrneurol.2010.12.002
- Wilson, C., Aftimos, S., Pereira, A., & McKay, R. (1998). Report of two sibs with knobloch syndrome (encephalocoele and viteroretinal degeneration) and other anomalies. American Journal of Medical Genetics, 78 (3), 286–290. https://www.ncbi.nlm.nih.gov/pubmed/9677068. https://doi.org/https://doi.org/10.1002/(SICI)1096-8628(19980707)78:3<286::AID-AJMG16>3.0.CO;2-B“3.0.CO;2-B
- Zeng, Y., Yi, R., & Cullen, B. R. (2003). MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proceedings of the National Academy of Sciences, 100 (17), 9779–9784. https://doi.org/https://doi.org/10.1073/pnas.1630797100
- Zhang, L., Li, K., Zhang, C., Qi, X., Zheng, N., & Wang, G. (2018). Arcuate fasciculus in autism spectrum disorder toddlers with language regression. Open Med (Wars), 13, 90–95. https://doi.org/https://doi.org/10.1515/med-2018-0014
- Zhang, L. S., Li, H. B., Zeng, J., Yang, Y., & Ding, C. (2018). Knobloch syndrome caused by homozygous frameshift mutation of the COL18A1 gene in a Chinese pedigree. International Journal of Ophthalmology, 11 (6), 918–922. https://doi.org/https://doi.org/10.18240/ijo.2018.06.04