Abstract
The hierarchical self-assembly of disc-shaped molecules leads to the formation of discotic liquid crystals. These intriguing materials are of fundamental importance not only as models for the study of energy and charge migration in self-organised systems but also as functional materials for various device applications. This has generated tremendous interest in this field. In this review article, we have summarised our synthetic work on discotic liquid crystals.
1. Preamble
At the outset, let me express my sincere thanks to the International Liquid Crystal Society for the inaugural LG Philips Display Mid Carrier Award bestowed on me at the 22nd International Liquid Crystal Conference during 29 June to 4 July 2008 at Jeju Island, Korea. At this occasion, Professor Gleeson advised me to briefly summarise my research work on discotic liquid crystals and submit it to Dr Dierking who kindly agreed to consider its publication in Liquid Crystals Today. I would also like to thank all my collaborators for supporting my research work. This article summarises our efforts to prepare various novel discotic liquid crystals. Physical studies carried out on these materials have not been included in this article.
2. Philosophy and mission
Life on Earth begins with the self-organisation of molecules. No life would be possible without the self-assembly of lipids into bilayers within the cell membrane. Numerous biological supramolecular structures result from self-assembly of organic molecules (Citation1–7). In materials science, non-covalent interactions have been used to obtain well-defined self-assembled architectures in neat systems as well as in solvents (Citation1–7). Liquid crystals (LCs) belong to one of such systems. Supramolecular interactions such as van der Waals forces, dipolar or quadrupolar interactions, charge-transfer interactions and hydrogen bonding play a crucial role in the formation of LCs and in the determination of their mesomorphic properties. The liquid crystalline state, the fascinating, intriguing, beautiful, mysterious, delicate fourth state of matter, is an intermediate state between the solid and the liquid. Liquid crystals are unique nanostructures with remarkable electronic and optoelectronic properties (Citation 8 ).
The research in my group is concerned with the design and synthesis of various liquid crystalline materials. We are interested in understanding the structure–property relationships in these intriguing self-assembling supramolecular architectures. Our present objectives are: design and synthesis of novel liquid crystalline materials; development of new synthetic routes for the synthesis of LCs; search for new reagents for the synthesis of LCs; incorporation of nanoparticles in the supramolecular order of LCs to hybridise their properties; and green chemistry approaches to the synthesis of soft condensed matter.
3. Discotic liquid crystals
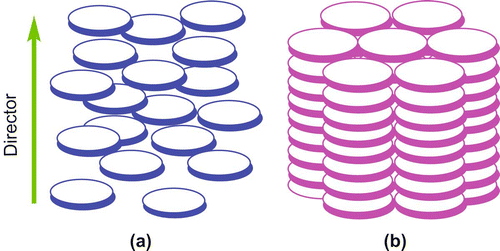
Shape anisotropy is the primary factor for any organic molecule to display mesomorphism. Until 1977, it was the belief that only rod-like molecules having much higher length than width could show liquid crystalline properties, commonly known as calamitic LCs. However, in 1977 Chandrasekhar and his colleagues at the Raman Research Institute realised that not only rod-like molecules, but also compounds with disc-like molecular shape are able to form mesophases (Citation 9 ). These are referred to as discotic liquid crystals (DLCs). Generally these compounds have flat or nearly flat cores surrounded by a plural number of aliphatic chains (Citation10–17). Mesophases formed by disc-shaped molecules are primarily of two types: nematic and columnar. In the discotic nematic phase, there is an orientationally ordered arrangement of discs with no long-range translational order. On the other hand, in the columnar phases, the discs are stacked one on top of another to form columns (). Over the past thirty years, the field of DLCs has grown rapidly due to the realisation of their potential in various device applications, and to date more than 3000 compounds of this category are known.
Figure 1. Schematic representation of (a) a discotic nematic (b) a columnar phase.
Our synthetic research work on DLCs is mainly related to: (a) molecular engineering of discotic nematic liquid crystals; (b) discovery of new materials and methodologies and (c) discotic–nanoparticles hybrid systems. These will be presented briefly in the following sections.
4. Molecular engineering of discotic nematic liquid crystals
Liquid crystals are primarily known for their display applications. The twisted nematic (TN) and supertwisted nematic (STN) display devices are dominating commercial displays since their invention. The LC layer in these devices is composed of a mixture of calamitic LCs. A number of calamitic nematic LCs having room temperature mesophase stability have been synthesised and used in practical displays. The major disadvantage with these types of devices is the narrow and non-uniform viewing cone. Several methods have been developed to improve the viewing angle characteristics of these devices. We have disclosed a novel approach to overcome this problem by utilising discotic nematic LCs instead of calamitic nematic LCs (Citation 18 ). The LCD prepared using hexalkynylbenzene-based discotic nematic LC shows wide and symmetrical viewing angles and no reversal of the contrast ratio in any direction.
Only a few disc-shaped molecules are known to display a nematic phase. Moreover, the ND phase formed by these materials is high temperature with a narrow thermal range. For any LC device application, the temperature range of the mesophase and its stability well below and above room temperature are amongst the most important criteria. Therefore, we have been interested in (a) design and synthesis of organic molecules which can form discotic nematic mesophase and (b) synthetic modifications in known discotic nematic phase-forming materials to stabilise the nematic phase well below and above room temperature.
4.1 Molecular design of discotic nematic LCs
A majority of DLCs (∼95%) form columnar mesophases, probably due to intense π–π interactions of polyaromatic cores. In order to obtain nematic phases, sufficient steric hindrance around the core has to be introduced so that the rigid molecules may stay in a more or less parallel position having only orientational order but losing their long-range translational order. We anticipated that linking two discotic molecules via a short rigid spacer might result in some steric hindrance due to overlapping or interdigitating aliphatic side chains and a weak distortion of the overall planarity of the molecule. This may reduce strong π–π interactions and, therefore, is likely to exhibit discotic nematic phases.
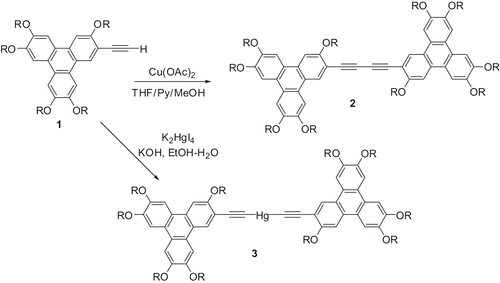
The dimers 2, consisting of two triphenylene (TP) units, were designed to verify this idea (Citation 19 ). Synthesis of these molecules is shown in . The free acetylene 1 was obtained from monohydroxytriphenylene via a multi-step synthesis (Section 5.6.5.1). As predicted, all the dimers form discotic nematic phase over a wide temperature range. Replacing one of the alkoxy chains by thioalkoxy 77 (Section 5.6.3) does not change the nature of the mesophase but displays a slightly lower isotropic transition temperature (Citation 20 ). Discotic dimers obtained by linking two discotic units via a mercury atom 3 () exhibit a metastable ND phase (Citation 21 ). However, when the TP units were replaced by simple mono-, di-or trialkoxybenzene units, the dimers do not show any nematic phase. They exhibit smectic and columnar phases, observed in normal planar molecules (Citation 22 ).
Scheme 1. Molecular design of discotic nematic liquid crystals.
4.2 Synthesis of room-temperature discotic nematic LCs
The hexa- and pentaalkynylbenzene derivatives are the molecules of choice to prepare room-temperature discotic nematic LCs because a number of compounds of this class are already known to show stable ND phases having moderate melting temperature. Therefore, some minor, but careful, structural modifications may lead to materials having room-temperature discotic nematic phases.
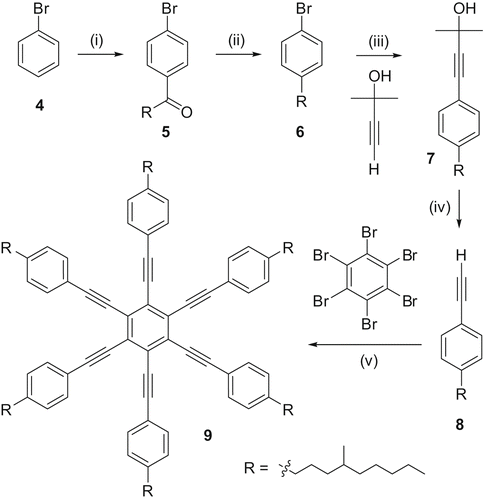
From the reported thermal data of alkyl- and alkoxy-substituted hexalkynylbenzene derivatives, it was clear that when the peripheral alkyl chains are attached to the phenyl ring in the hexaalkynylbenzene via an oxygen atom, the melting and clearing temperatures are higher, when compared with the alkyl chains directly being attached to the ring. Further, it is also known that the use of branched chains in LCs often reduces melting and clearing temperatures. The mesophase range is generally widened but the type of the mesophase formed is not affected by the introduction of branching in many cases. The decrease in the transition temperature could be due to the disorder caused by branched chains and stereoheterogeneity. This methodology has been successfully applied to reduce melting and clearing temperatures of several DLCs. Therefore, it may be anticipated that the replacement of normal alkyl chains by branched alkyl chains connected directly to the phenyl ring in a hexalkynylbenzene system would reduce the melting point of alkynylbenzene derivatives, and thus stabilise the mesophase at room temperature. Compound 9 () was designed on this basis and it was indeed found to exhibit a nematic phase at ambient temperature. The compound exhibits the nematic phase between −12°C and 68°C (Citation 23 ).
Scheme 2. Design and synthesis of a room-temperature ND phase: (i) RCOCl/AlCl3; (ii) NH2NH2/KOH; (iii) PdII, triphenylphosphine (TPP), CuI, Et3N; (iv) KOH/toluene; (v) PdII, TPP, CuI, Et3N.
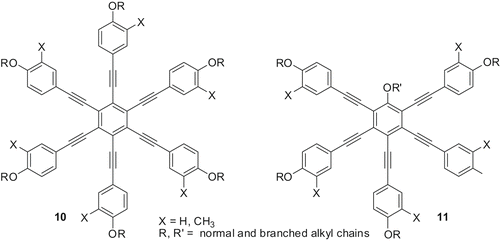
Several other new hexa- and pentaalkynylbenzene-based molecules () with a combination of normal and branched chains were prepared to obtain room-temperature discotic nematics and to understand structure–property relationships (Citation 24 , Citation 25 ). The pentaalkynylbenzene derivatives (structure 11, X = H, R = 3,7-dimethyloctane (−35 ND 40 I) and X = CH3, R = 3,7-dimethyloctane (–35 ND 73 I) displayed a stable ND phase at room temperature.
Figure 2. Molecular design for room-temperature discotic nematics.
5. New materials and methodologies
5.1 Decacyclene-based DLCs
Decacyclene (DC) is a symmetric, large polycyclic aromatic hydrocarbon present in the carbonaceous mesophase. The common DLC has been characterised as a flat molecule comprised of a rigid polycyclic aromatic core surrounded by four to nine aliphatic side chains. It was a common belief that ‘three elongated chains would insufficiently fill the space around the core and, therefore, could not allow the existence of columnar mesophases’ (Citation 26 ). We anticipated that increasing the size of the polycyclic aromatic core and keeping the minimal constraint around the core to avoid crystallisation, one could achieve liquid crystallinity regardless of the number, nature and size of the side chains. These mesophases could model the carbonaceous mesophase more closely than other known discotic systems.
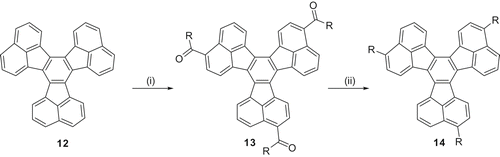
Accordingly, trialkanoyl-DC based discotic mesogens 13 were designed and synthesised by a direct Friedel–Crafts acylation of commercially available parent hydrocarbon 12 () (Citation 27 ). The reaction proceeds with very high regioselectivity and only the C3 symmetric 1,7,13-trialkanoyl-DC could be isolated. Two liquid crystalline derivatives; triheptanoyloxy- 13a and trioctanoyloxy-DC 13b have been prepared. Both display columnar mesomorphism; 13a (Cr 92.8 Colt 115 Colr 262 I) and 13b (Cr 98 Colt 108.5 Col 240.5 I). The deoxygenated hydrocarbons 14a and 14b, obtained by reduction of the keto groups, were found to be non-mesomorphic. Therefore, the dipolar interaction of carbonyl group seems to be responsible for mesophase induction. However, more materials are required to establish a clear structure–property relationship.
Scheme 3. Synthesis of decacyclene derivatives: (i) RCOCl/AlCl3/ClCH2CH2Cl/reflux, 38%; (ii) NH2NH2.H2O/KOH/reflux, 50%.
The liquid crystalline triheptanoyloxy-DC 13a has recently been used as an electron transport material in a photovoltaic device (Citation 28 ). Evidently, decacyclene-based DLCs have huge potential. More LC derivatives of this core, particularly low-temperature or room-temperature DLCs, are required to investigate the intriguing properties of these materials and to find out their applications in industry.
5.2 Tricycloquinazoline-based DLCs
The nitrogen heterocyclic hydrocarbon tricycloquinazoline (TCQ) is very attractive in materials science for a variety of reasons. It possesses a trigonal symmetry, it displays extraordinary chemical and thermal stability, its derivatives are coloured, it exhibits a low ionisation potential and interesting spectroscopic and electronic properties, it forms charge-transfer complexes with DNA and it acts as an electron acceptor.
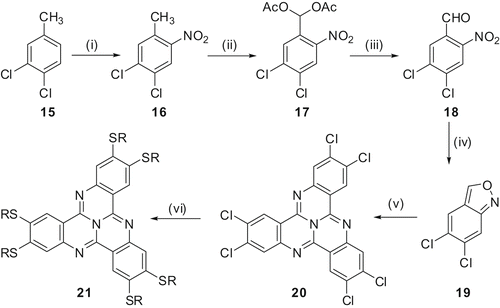
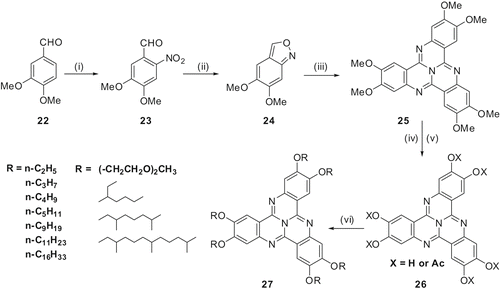
We have reported a wide spectrum of homologous 2,3,7,8,12,13-hexaalkoxy- and hexathioalkoxy-TCQ derivatives with alkyl side chain length varying from 1 to 18 carbon atoms (Citation29–31). We have also used branched alkoxy chains to prepare its room temperature LC derivatives (Citation 32 ). The synthesis of these materials is shown in and .
Scheme 4. Synthesis of 2,3,7,8,12,13-hexathioalkoxy-TCQ derivatives. (i) AcOH, H2SO4, HNO3, 0–5°C, 90%. (ii) Ac2O, H2S04, CrO3, 0–5°C, 3 h, 20–60%. (iii) EtOH, H2O, HCl, reflux 1 h, 58%. (iv) AcOH, Sn, room temperature 20 h, 15–55%. (v) Sulpholane, AcOH, NH4OAc, 150°C, 7–24 h, 10–40%. (vi) n-Alkylthiol, potassium-tert-butoxide, NMP, 100°C, 10 min, 50%.
Scheme 5. Synthesis of 2,3,7,8,12,13-hexaalkoxy-TCQ derivatives: (i) HNO3, 90%; (ii) AcOH, Sn, room temperature 50%; (iii) sulpholane, AcOH, NH4OAc, reflux, 25%; (iv) HCl, Py, 230°C, 50%; (v) HCl, Py, 230°C, Py, Ac2O, 90%; (vi) KOH, DMSO, RBr, 20–80%.
TCQ derivatives having peripheral chains of 3–18 carbon atoms display the Colh phase. In general, the melting transitions and clearing temperatures decrease gradually with increasing length of peripheral chains. The use of branched alkoxy chains (e.g., 3,7-dimethyloctyloxy and 3,7,11-trimethyldodecyloxy) reduces the clearing points significantly (<200°C). The 2,3,7,8,12,13-hexakis (3,7,11-trimethyldodecyloxy)-TCQ is of particular importance as it does not crystallise down to −50°C, and thus forms a stable Colh phase at room temperature and goes to isotropic phase at 143°C. The intercolumnar distance is dependent on the peripheral chain length, whereas the interdisc distance within the discotic molecules was estimated to be approximately 0.34 nm in normal alkyl chains substituted derivatives. The small angle X-ray scattering of the 2,3,7,8,12,13- hexakis (3,7,11-trimethyldodecyloxy)-TCQ display a centre-to-centre distance of 0.329 nm, one of the lowest core–core separations known in DLCs and, therefore, making the molecule very attractive for one-dimensional charge migration.
5.3 Dibenzo[fg,op]naphthacene-based DLCs
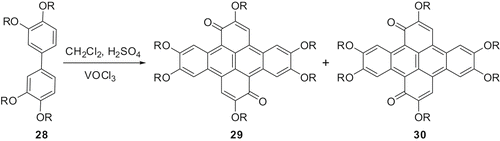
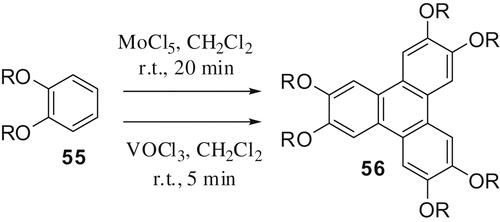
DLCs based on 1,2,5,6,8,9,12,13-octaalkoxy-dibenzo‐[fg,op]naphthacene (DBN) (also named as dibenzopyrene) have recently received good attention. These materials can be easily prepared by oxidative dimerisation of tetraalkoxybiphenyls. The reagent VOCl3, which we have realised for the efficient synthesis of triphenylene discotics, can be used to prepare DBN diketones () (Citation 33 ) which can be converted to octaalkoxy-DBN discotics.
Scheme 6. Oxidative dimerisation of tetraalkoxybiphenyl using VOCl3.
As the larger core has more delocalised π-electrons than TP, a greater degree of π–π interaction and hence higher charge carrier mobility was expected in these derivatives. However, the charge carrier mobility in the octaalkoxy-DBN derivatives was found to be one order of magnitude lower than in the columnar phase of hexaalkoxy-TP. This could be due to less ordered columnar packing caused by the steric hindrance of the ‘bay region’ alkoxy chains, overall resulting in a lower charge carrier mobility.
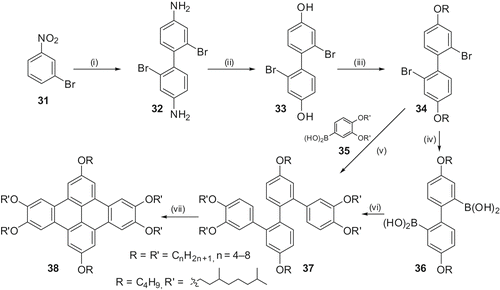
We anticipated that the removal of these alkoxy chains from the 1 and 8 positions will give a better core–core interaction and, therefore, high charge carrier mobility. Accordingly, various 2,5,6,9,12,13- hexaalkoxy-DBN derivatives () were designed. While the synthesis of octaalkoxy-DBN is very easy and well known, no synthetic route was available for hexaalkoxy-DBN derivatives. We developed a novel, versatile and regiospecific synthesis of variable degree substituted dibenzo[fg,op]-naphthacene derivatives 38 (Citation 34 , Citation 35 ). The methodology involves the preparation of a quaterphenyl 37 using a palladium-catalysed cross-coupling of arylboronic acids followed by chemical or photochemical cyclisation. All of the DBN derivatives exhibit a single mesophase, which has been identified by X-ray diffraction and optical microscopy as a hexagonal columnar phase. X-ray results confirm that hexasubstituted DBN are more ordered than octasubstituted derivatives.
Scheme 7. Synthetic route to hexaalkoxy-DBN derivatives. (i) Zn / NaOH, HCl. (ii) NaNO2 / HCl. (iii) DMSO / KOH / RBr. (iv) n-BuLi / B(OMe)3 / HCl. (v) Pd(PPh3)4 / Na2CO3. (vi) hυ / I2 or FeCl3.
5.4 Dibenzo[g,p]chrysene core
Dibenzo[g,p]chrysene (DBC) derivatives are interesting because of their notable fluorescent properties (Citation 36 ). Highly efficient blue-green emissive organic light-emitting diodes have been fabricated using bis(diphenylamino)chrysene derivatives. Non-liquid crystalline DBC chromophores have been incorporated into co-polymers for explosive detection (Citation 36 ).
The DBC architecture is expected to display mesomorphism on peripherally long alkyl chains-substitution. Accordingly, molecules 42 were designed and the strategy followed to prepare these materials is outlined in (Citation 37 ). A palladium-catalysed annulation reaction of diphenylacetylenes 40 with 2-iodobiaryls 39, followed by oxidative coupling using FeCl3 yields octaalkoxy-DBC 42. Neither the octapentyloxy- and the octaoctyloxy-DBC derivatives display mesomorphism in the virgin state but their charge-transfer (CT) complexes with trinitrofluorenone (TNF) exhibit columnar phases. Enantiotropic DLCs can be prepared by substituting the peripheral positions by dialkoxybenzoate groups (Citation 38 ).
Scheme 8. Synthesis of octaalkoxy-dibenzo[g,p]chrysene derivatives: (i) Pd(OAc)2, 40%; (ii) FeCl3, 20%.
![Scheme 8. Synthesis of octaalkoxy-dibenzo[g,p]chrysene derivatives: (i) Pd(OAc)2, 40%; (ii) FeCl3, 20%.](/cms/asset/e5ef7ad0-6f6c-454e-b68e-bc99345d88ee/tlcy_a_392219_o_sch0008g.gif)
5.5 Rufigallol-based DLCs
Rufigallol-based DLCs are interesting materials as these molecules have an elongated core with a twofold symmetry axis. They are coloured and exhibit an important polymorphism; the core is electron-deficient in nature, they are thermally stable and their chemistry is fairly easy. The first discotic liquid crystalline hexaesters of rufigallol were prepared in 1980 and since then about 100 different discotic liquid crystalline derivatives of this molecule have been prepared and studied (Citation 39 ).
5.5.1 Microwave-assisted easy synthesis of rufigallol and its derivatives
The synthesis of rufigallol was reported as early as in 1836 by Robiquet in poor yield by the action of sulphuric acid on gallic acid. Grimshaw and Haworth reported its purification in 1956 (Citation 39 ). Since then little further work has appeared and no new efficient methods to prepare rufigallol were reported. We have observed (Citation 40 ) that the self-condensation of gallic acid in the presence of sulphuric acid can be achieved in high yield in about one minute using microwave (MW) heating ().
Scheme 9. Synthesis of novel liquid crystalline rufigallol derivatives: (i) H2SO4, MW, 90 s, 80%; (ii) RBr or R'Br, DMSO, NaOH, 90°C 20 h; (iii) RBr or R'Br, Cs2CO3, NMP, MW, 3–10 min.
It should be noted that MW-assisted high-speed chemical synthesis has attracted a considerable amount of attention in the past decade. Almost all types of organic reactions have been performed using the efficiency of MW-flash heating. This is not only due to the fact that reactions proceed significantly faster and more selective than under conventional thermal conditions but also because of the operational simplicity, high yield of products, and cleaner reactions with easier work-up. Surprisingly, this technology has not been much exploited for the synthesis of LC materials. We used the potential of MW dielectric heating to quickly prepare a variety of LC samples (Citation40–47). We realised that some molecules which are difficult to prepare under classical conditions, can be easily synthesised using microwaves.
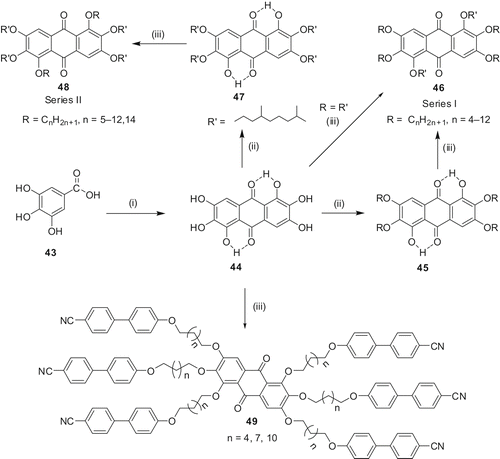
The rufigallol was converted to several novel DLC derivatives by substitution with straight and branched-alkyl chains. We were particularly interested in preparing room-temperature DLCs. In order to prepare novel room-temperature rufigallol discotics, we initially replaced all six peripheral n-alkyl chains with 3,7-dimethyloctyl chains. The reaction of rufigallol with 1-bromo-3,7-dimethyloctane in the presence of caesium carbonate under MW heating produced 46 (R = R' = 3,7-dimethyloctyl) within three minutes. However, it was found to be a viscous oil. The synthesis of the unsymmetrical hexaethers was achieved by a two-step alkylation process. The unequal reactivity of the six phenolic groups, two of which are less reactive by virtue of being intramolecularly hydrogen bonded to the adjacent quinone carbonyls, was exploited. Etherification of rufigallol 44 under mild conditions produced 1,5-dihydroxy-2,3,6,7-tetraalkoxy-9,10-anthraquinone (45 and 47) without alkylating the hydrogen-bonded C-1 and C-5 positions. These tetraalkoxy derivatives were further alkylated with the help of MW dielectric heating as shown in under mild basic conditions to give the unsymmetrical hexaethers in very good yield within 3–4 minutes. It should be noted that under classical reaction conditions no product could be obtained in such a short reaction time. In this way, we prepared 20 different novel rufigallol derivatives. Out of those, 13 displayed stable columnar mesophases at room temperature. Compound 47 is particularly interesting as it has only four branched alkoxy chains and exhibited a very broad mesophase, stable at room temperature (Colh132 I). The two free hydroxyl groups can be used to prepare main chain polymeric LCs.
Novel liquid crystalline oligomers 49, containing six rod-like cyanobiphenyl moieties connected to the rufigallol core via flexible alkyl spacers, were prepared by reacting rufigallol with terminally bromo-substituted alkoxycyanobiphenyls (Citation 45 ). The synthesis of the target compounds was challenging as classical reactions failed to produce these oligomers. Differential scanning calorimetry (DSC) and polarised optical microscopy (POM) analysis revealed the existence of nematic phases with a shorter spacer and SmA phases with longer spacers. The compound with medium alkyl spacer shows both nematic and SmA phases at higher temperature and a re-entrant nematic phase at lower temperature.
5.5.2 DLCs derived from rufigallol using conventional synthetic methods
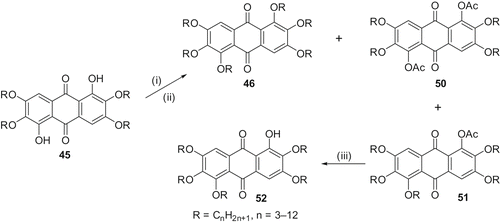
We have primarily been interested in synthesising 1-hydroxy-2,3,5,6,7-pentaalkoxyanthra-9,10-quinones (mono-functionalised rufigallol) so that rufigallol-based novel electron-deficient dimers, oligomers, polymers, mixed-tails derivatives and discotic donor-acceptor systems can be prepared. As mentioned earlier, in rufigallol, the hydroxyl groups at 1- and 5-positions are hydrogen bonded and, therefore, are less reactive. Under milder etherification conditions, the hydrogen-bonded 1- and 5-positions do not get alkylated and thus 1,5-dihydroxy-2,3,6,7-tetraalkoxy-9,10-anthraquinone (di-functionalised rufigallol) 45 () forms. In principle, partial alkylation of this material should give a mixture of unreacted (tetraalkylated), pentaalkylated and hexaalkylated products from which the desired mono-functionalised rufigallol can be isolated, but all efforts to isolate it in pure form from this mixture were met with failure. However, we realised that acetylation of the crude product furnished tetraalkoxydiacetoxy-rufigallol 50 (), pentaalkoxymonoacetoxy-rufigallol 51 and hexaalkoxy-rufigallol 46 having a significant difference in retention factor (R f ) values on a chromatographic column and thus all three products could be separated readily by column chromatography. Pure monoacetoxy-pentaalkoxy-rufigallol 51 on hydrolysis afforded the desired mono-functionalised rufigallol 52. The unreacted material can be recycled and the hexaalkylated by-product can be used for various physical studies. All the monohydroxy-pentaalkoxy-rufigallols 52 () display hexagonal columnar phases over a wide temperature range (Citation 48 ).
Scheme 10. Synthesis of mono-functionalised rufigallol and derived metallomesogens: (i) DMSO/KOH/RBr; (ii) Ac2O/Py; (iii) Aq. NaOH/EtOH.
5.5.3 Rufigallol-based discotic metallomesogens
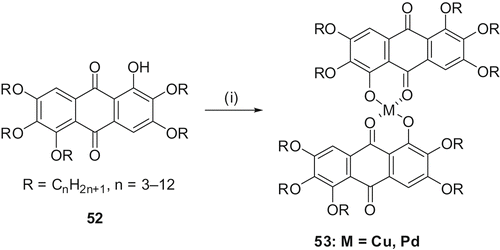
Metal complexes of β-diketonates are well known to form calamitic and discotic LCs. A comparison of the structure of β-diketone molecule with that of a 1-hydroxyanthraquinone derivative clearly demonstrates a similarity between the two systems. Therefore, similar to a β-diketone system, a large number of metallomesogens can be prepared from monofunctionalised rufigallols. Refluxing monohydroxy-pentaalkoxy-rufigallols with metal acetate in acetonitrile-pyridine easily furnished metal-bridged rufigallol dimers 53 (). Two series of complexes; one with copper (Cu) and the other with Palladium (Pd) were prepared in this way (Citation49–51). While the Pd complexes were found to be thermally unstable at higher temperature, the Cu complexes of the same ligand were stable. Lower members of both series are not liquid crystalline but columnar mesophase can be induced by doping them with an electron acceptor, TNF.
Scheme 11. Synthesis of rufigallol-bases metallomesogens: (i) metal acetate/CH3CN-Py.
5.5.4 Rufigallol-based discotic main chain polymers
It is interesting to note that the branched chain substituted tetraalkoxy compound 47 exhibits liquid crystalline behaviour at room temperature, contrary to its straight chain analogue which does not show any liquid crystalline properties. The columnar phase was stable below −30°C and cleared at 115°C. The two unreacted hydroxyl groups of this tetramer can be utilised to prepare main chain polymers simply by reacting with α, ω-dibromoalkanes (). The so-prepared polymer displayed a Colr phase at room temperature which clears at 56.3°C. The mesophase structure was confirmed by X-ray studies (Citation 52 ).
Scheme 12. Synthesis of rufigallol-based main chain polymers; (i) 1,12-dibromododecane/ Cs2CO3/ o-dichlorobenzene/90°C/ 10 days.
5.6 Triphenylene core
5.6.1 Development of new reagents for the synthesis of hexaalkoxy‐triphenylenes
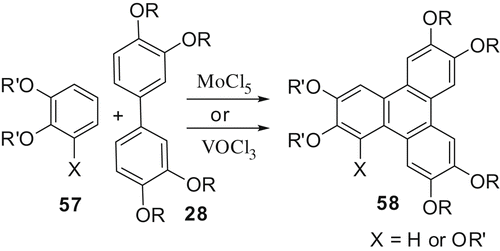
Hexasubstituted TPs are the most widely synthesised and studied discotic mesogens and a number of methods have been developed for their synthesis (Citation 10 b). We have found that molybdenum(v) chloride is a novel reagent for the oxidative trimerisation of 1,2 dialkoxybenzenes to hexaalkoxy-TPs in high yield (Citation 53 ). The reaction occurs under very mild conditions with or without an acid catalyst and in a very short time at room temperature (). The synthesis of unsymmetrical TPs was achieved by coupling a 3,3',4,4'-tetraalkoxybiphenyl and appropriate 1,2-dialkoxy- or 1,2,3-trialkoxybenzene () under similar reaction conditions.
Scheme 13. Oxidative trimerisation of 1,2-dialkoxybenzene to hexaalkoxytriphenylene using MoCl5 or VOCl3.
Scheme 14. Synthesis of unsymmetricaland heptaalkoxy‐ triphenylenes using using MoCl5 or VOCl3.
We have also discovered that vanadium oxytrichloride, a liquid oxidising agent, may be used as a novel reagent for the oxidative trimerisation of 1,2-dialkoxyloxybenzenes to hexaalkoxy-TP in high yields () (Citation 53 ). Unlike other reagents, which are solid and insoluble in organic solvents, VOCl3 can be readily handled using standard syringe techniques or by using an addition funnel. VOCl3 has a high solubility in various organic solvents, which could be a reason for the almost spontaneous trimerisation of dialkoxy benzenes into hexaalkoxy-TPs. The handling of the reagent and work-up of the reaction are very easy; there is no need to use concentrated sulphuric acid, which is an obvious advantage, and the product yields were high. Effects of reagent concentration, solvent and temperature have been studied in detail. Unsymmetrical and heptaalkoxy-TPs can also be prepared using VOCl3 ().
5.6.2 Nitration of hexaalkoxytriphenylene discotics
Electrophilic aromatic substitution in unsubstituted TP is directed by steric and electronic effects. Substitution at the β- or 2-position is favoured compared with the α- or 1-position, presumably owing to a steric hindrance effect. However, the electronic effect plays a major role in the nitration of TP and results in a mixture of 1-nitro and 2-nitro-TP. During the nitration of pentapentyloxy-TP (Section 5.6.5.1) we realised that the nitration occurred preferentially in the sterically hindered α-position. Evidently electronic effects dominate in the nitration of alkoxy-TP. This prompted us to examine the nitration of TP discotics more carefully (Citation 55 , Citation 56 ). The functionalisation of the nucleus at the α-position is important not only to induce colour, molecular dipole, enhanced liquid crystalline properties and a chemical reaction site but also to helically deform the normally planar TP core.
In hexaalkoxy-TP, only the α-positions (‘bay regions’ 1, 4, 5, 8, 9 and 12 positions) are free for further substitution. Nitration of 2,3,6,7,10,11-hexaalkoxy-TP provides the α-nitrated product in very high yields. Solvents play an important role in the nitration of hexaalkoxy-TP. In a mixture of ether-acetic acid almost exclusively the α-nitro product is formed. Even under exhaustive conditions, in this solvent system, only a small amount of trinitro-hexaalkoxy-TP forms. Changing the solvent system from ether-acetic acid to dichloromethane-nitromethane imparts a dramatic effect and all the three rings of TP can be successively nitrated under very mild conditions. The trinitration proceeds with high regioselectivity to give exclusively one isomer having C3 symmetry, i.e. 1,5,9-trinitro-2,3,6,7,10,11-hexaalkoxy-TP. Alkoxynitro-TP are valuable precursors to several other derivatives like amino, mono- and di-alkylamino, acylamino-, azo- etc., and thus a number of new triphenylene derivatives can be prepared ().
Scheme 15. Synthesis of nitrotriphenylenes and their derivatives: (i) HNO3/CH3NO2; (ii) NH2NH2/Pd or NiCl2/NaBH4; (iii) Ac2O/Py.
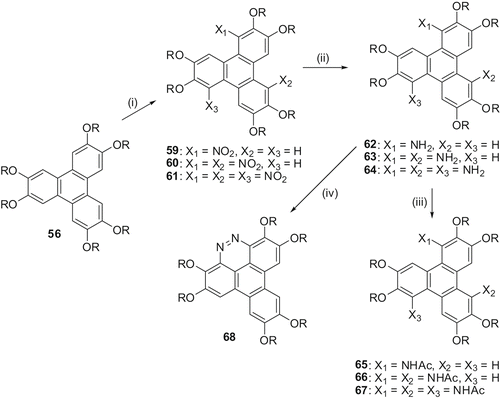
Nitro-TPs 59, 60 and 61 were reduced to the corresponding amino derivatives 62, 63 and 64 with hydrazine and palladium or with nickel chloride and sodium borohydride (). The amino derivative 62 was readily converted to α, α-diazo compound 68 by diazotisation of the amine with nitrous acid followed by cyclisation. Acylation of amines with acetic anhydride in pyridine converted them in to their N-acyl derivatives 65, 66 and 67. Many of these derivatives display columnar mesophases.
5.6.3 Triphenylene discotics with three different types of peripheral substituent
The nature of peripheral substituents around the core has a great influence on the mesophase structure and its stability. For example, hexaesters of TP form Colr and Colh mesophases, TP hexabenzoates form ND and Col phases, hexaphenylacetylene derivatives of TP exhibit ND phases, symmetrically substituted hexaether derivatives of TP having 5 to 13 carbon atoms in the aliphatic chains form single Colh mesophase while hexabutyloxy-TP display a more ordered Colp phase and hexahexylthio-TP exhibit a highly ordered helical phase, in addition to a Colh phase. Numerous rod-like molecules with a variety of peripheral substitutions are known to exhibit various types of mesophases. However, probably because of synthesis problems, such a rich variety of peripheral substitutions are uncommon in DLCs. Almost all of the TP derivatives reported in the literature have either a single type of peripheral substitution pattern, or two types of peripheral substitution. Efforts have not been made to prepare DLCs having more than two types of peripheral substituent. We took on this challenging problem and prepared a variety of novel TP derivatives having three different types of peripheral substitution () (Citation 20 ).
Scheme 16. Reagents and conditions: (i) iodine/iodic acid, CH2Cl2-AcOH, reflux, 45%; (ii) C6H5B(OH)2, [(C6H5 )3P]4Pd, Na2CO3, THF, reflux, 41%; (iii) CH2Cl2, VOCl3, room temperature, 80%; (iv) Br2, CH2Cl2, 60%; (v) C5H11SK, NMP, [(C6H5)3 P]4Pd, 73%; (vi) Br2, CH2Cl2, 28%; (vii) CuCN, NMP, reflux, 35%; (viii) 2-methyl-3-butyn-2-ol, CuI, PdCl2 (PPh3)2, Et3N, 60–70°C, 40%; (ix) KOH, toluene, reflux, 98%; (x) Cu(Ac)2, THF–C5H5N–CH3OH, reflux, 18%.
![Scheme 16. Reagents and conditions: (i) iodine/iodic acid, CH2Cl2-AcOH, reflux, 45%; (ii) C6H5B(OH)2, [(C6H5 )3P]4Pd, Na2CO3, THF, reflux, 41%; (iii) CH2Cl2, VOCl3, room temperature, 80%; (iv) Br2, CH2Cl2, 60%; (v) C5H11SK, NMP, [(C6H5)3 P]4Pd, 73%; (vi) Br2, CH2Cl2, 28%; (vii) CuCN, NMP, reflux, 35%; (viii) 2-methyl-3-butyn-2-ol, CuI, PdCl2 (PPh3)2, Et3N, 60–70°C, 40%; (ix) KOH, toluene, reflux, 98%; (x) Cu(Ac)2, THF–C5H5N–CH3OH, reflux, 18%.](/cms/asset/79cc76b6-74fc-41e6-870c-29658b7cf11d/tlcy_a_392219_o_sch0016g.gif)
Monobromination of 2, 3,6,7-tetrajus(pentyloxy)-TP 70, prepared by Syzuki coupling of 2-iodo-3',4,4', 5-tetrakis(pentyloxy)biphenyl 39 and phenylboronic acid followed by cyclisation, yields 10-bromo-2,3,6,7-tetrakis(pentyloxy)-TP 71. Nucleophilic aromatic displacement of the bromine with the potassium salt of pentanethiol, followed by bromination, yields 2-bromo- 6,7,10,11-tetrakis(pentyloxy)-3-(pentylsulphanyl)-TP 73 having a bromo, thioalkyl and alkoxy-substitutedperiphery of the TP nucleus. The reaction of 73 with copper(I) cyanide gives the cyano-TP derivative 74, while palladium-copper catalysed alkynylation of 73 results in the synthesis of the substituted alkyne derivative 75. The deprotected alkyne 76 was converted to dimer 77 where two molecules of a monothioalkyl-tetra-alkoxy-TP are connected via a rigid π-conjugated diacetylene bridge. Compounds 73–76 form hexagonal columnar phases while the dimer 77 shows a discotic nematic phase.
5.6.4 Hydroxy-functionalised triphenylenes
Hydroxy-functionalised TPs are required to prepare a variety of DLCs such as unsymmetrical DLCs, mixed-tail derivatives, discotic dimers, oligomers and polymers. The physical properties of these non-conventional LCs are significantly different from those of conventional LCs. However, because of synthetic problems in obtaining functionalised TPs, the potential utility of these intriguing materials has not been fully explored. We have developed several methods to prepare various hydroxyl-alkoxy-TPs (Citation 41 , Citation57–59). One of these methods is given below (Citation 58 ).
5.6.4.1 B-Bromocatecholborane for selective ether cleavage of hexaalkoxytriphenylenes
B-Bromocatecholborane is a monofunctionalised bulky Lewis acid. We anticipated that it may show a better regioselectivity than commonly used boron tribromide and cleave selectively one, two and three alkoxy chains of hexaalkoxy-TP. The reagent can be readily prepared by reacting catechol with boron tribromide. The colourless solid is stable for months, but can be stored more easily as CH2Cl2 solutions. However, we observed that the freshly distilled product gives better results, and very different reaction conditions are required for the commercial reagent.
When hexapentyloxy-TP (H5TP) 56 (R = C5H11) was treated with 1–1.2 equivalents of the reagent in dichloromethane at room temperature for 24–48 hours, it furnished mainly the monohydroxy-H5TP 78 with a minor amount of unreacted H5TP (). Increase in reaction time and temperature did not change the yield of monohydroxy-H5TP significantly. An increase in the concentration of the reagent started cleaving of the other alkoxy chains and variable amounts of mono-, di-, and trifunctionalised-H5TP were obtained. Reaction of H5TP with 2–2.4 equivalents of Cat-B-Br at room temperature to prepare difunctionalised-H5TP always yields a mixture of mono- 78, di- 79 + 80, and trifunctionalised-H5TP 81 and 82. However, the formation of trihydroxytriphenylenes can be checked by performing the reaction at 0°C and the dihydroxy derivatives can be isolated in reasonably good quantity.
Scheme 17. Synthesis of mono-, di- and trihydroxy-TPs using B-bromocatecholborane: (i) Cat-B-Br, 1.2 eq.; (ii) Cat-B-Br, 2.5 eq.; (iii) Cat-B-Br, 3.6 eq.
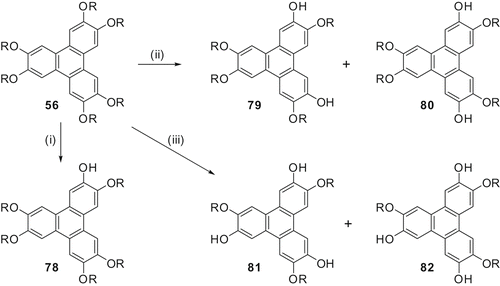
Out of the four possible isomers of dihydroxy-H5TP, we found that only 2,6- and 2,7-isomers, 79 and 80, respectively, were the major products. These two isomers can be separated by careful and repeated chromatography over basic alumina in about 6:4 ratio. The main advantage of this monofunctionalised, bulky Lewis acid was found in the preparation of trifunctional TPs. Treatment of H5TP with 3.6 equivalents of Cat-B-Br in dichloromethane at room temperature for 36 hours gives exclusively two products, the symmetrical 2,6,10-trihydroxy-3,7,11-tris(pentyloxy)-TP 81 (61%) and non-symmetrical 2,7,10-trihydroxy-3,6,11-tris(pentyloxy)-TP 82 (38%).
5.6.4.2 Green chemistry approach to the synthesis of hydroxy-functionalised triphenylenes
Several methods are available for the synthesis of mono-functionalised TPs. Most of the reagents used to prepare these materials are expensive, hazardous and difficult to handle. We were interested in finding an inexpensive and less hazardous method to produce these materials quickly. We found that ionic reagents can be used to prepare mono-functionalised TPs in moderate yield (Citation 41 ). This methodology avoids all type of toxic, volatile and hazardous reagents. Developing green chemistry methodologies is one of the main themes of modern synthetic chemistry. In this context, the use of ionic liquids and microwaves are powerful tools.
We applied several pyridinium and imidazolium salts to examine the possibility of selective ether dealkylation in hexaalkoxy-TP under classical as well as MW heating conditions (). When different hexaalkoxy-TPs [R = −C4H9 (H4TP), −C5H11 (H5TP), −C6H13 (H6TP)] were treated with various ionic reagents (I–V), mono-functionalised-TP formed in moderate yields. For example, when hexabutyloxy-TP was heated with pyridine hydrochloride (no solvent) for 48 hours, about 35–40% of monohydroxy-pentabutyloxy-TP is formed. Other reagents gave slightly lower yields. We investigated the potential of these reagents (I–V) to prepare mono-functionalised TP derivatives under MW irradiation. While reagents I, III and IV did not give any mono-functionalised TP under MW heating up to 10 minutes, reagents II and V produced the desired product. Thus, when a mixture of hexabutyloxy-TP and pyridine hydrobromide (3 equivalents) was irradiated at 360 W for 3 minutes, monohydroxy-pentabutyloxy-TP is formed in about 36% yield. Enhancing the MW power to 600 W or 800 W did not increase the yield significantly. Compared with thermal heating, the yield of monohydroxy-TP was low under MW heating conditions but the reaction can be finished in a very short time.
Scheme 18. Synthesis of monohydroxy-TP using ionic reagents.
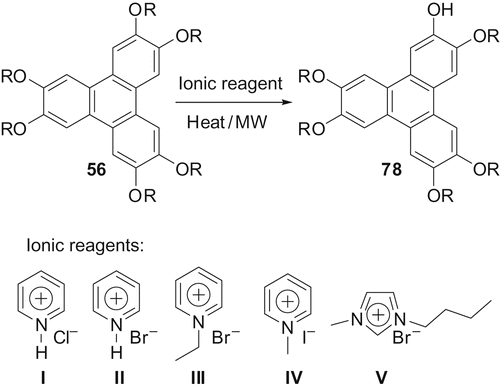
Although this method is not the best yield-wise, it is an economic and green synthesis method for the preparation of monohydroxy-hexaalkoxy-TP derivatives. In all cases, no side reaction occurred and the unreacted starting material can be isolated easily, and can be recycled.
5.6.5 Discotics derived from hydroxy-functionalised triphenylenes
The hydroxy-functionalised TPs are very valuable precursors for the synthesis of dimers, oligomers, polymers, networks, mixed tails, lower and higher degree substituted derivatives. Though a number of different alkoxy-hydroxy-TP are possible, seven different types of alkoxy-hydroxy-TP derivatives; monohydroxy, 2,3-dihydroxy, 2,6-dihydroxy, 2,7-dihydroxy, 2,11-dihydroxy, 2,6,10-trihydroxy and 2,6,11-trihydroxy, have been prepared using selective or non-selective chemical methods (Citation 10 b). These derivatives have been used to prepare a number of triphenylene discotics. Here, we present some of the derivatives prepared by us using these precursors.
5.6.5.1 Discotics derived from monohydroxy-pentaalkoxy-triphenylenes
The monohydroxy-TP can be directly converted to chiral esters 83 by reacting with (S)-2-chloro-4-methylpentanoic acid chloride or (2S, 3S)-2-chloro-3-methylpentanoic acid chloride. Induction of a cholesteric phase from a nematic-discotic phase was observed by doping with these chiral discotic LCs (Citation 60 ).
Two novel series of liquid crystalline non-symmetrical hexaalkoxy-TPs containing a branched alkyl chain have been prepared using MW dielectric heating () (Citation 44 ). Series-1 (84) contains 2-ethyl hexyloxy group as the branched chain whereas series-2 (85) contains 3,7-dimethyl octyloxy as the branched chain along with five normal alkoxy chains. The number of carbon atoms varies from 4 to 8 in the normal alkoxy chains. All the compounds show enantiotropic mesophase transitions with Colh structure. In series-1 the mesophase range and transition temperatures of all the compounds are lowered as compared with the parent compounds, whereas in series-2 the transition temperatures of all the compounds are lowered. The mesophase range for lower members is decreased; however, higher members show larger mesophase stability. Both melting and clearing temperatures of series-2 show a strong odd–even effect. The intercolumnar distance increases as expected for compounds of both series with increasing alkyl chain length along with some degree of interdigitation of the alkyl chains. The intercolumnar distances for the compounds of series-2 are slightly higher than those of the compounds of series-1.
Scheme 19. Triphenylenes derived from mono-functionalised-TP (21): (i) Py/R*COCl (83) or RBr, Cs2CO3, NMP, MW (84, 85) or R = substituted carbazole (86); (ii) CF3SO2Cl/Py; (iii) 5-chloro-1-phenyl-tetrazole/K2CO3; Pd/C, EtOH/H2; (iv) HNO3; (v) AlCl3/RCOCl; (vi) Br2/CH2Cl2; (vii) CuCN/NMP; (viii) RC≡CH/Pd/Cu/PPh3/Et3N; (ix) alcoholic KOH; (x) H2/Pd/C.
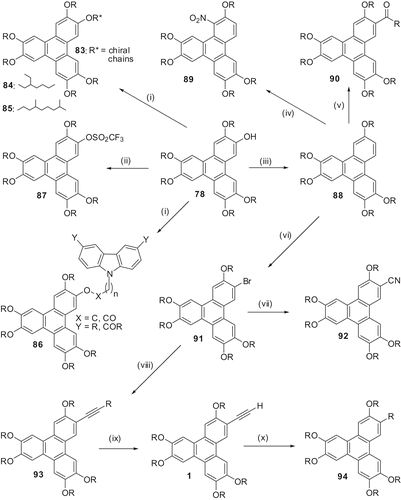
We have prepared several derivatives 86 incorporating a carbazole unit in TP to obtain added physical properties (Citation61–63). These modifications have been achieved by the alkylation or esterification of monohydroxy-TP derivatives with alkyl bromides and carboxylic acids incorporating the carbazole moiety. The pure compounds are not liquid crystalline in nature but when doped with TNF, Colh phases were induced. These mesophases exist below room temperature. The mesophase clearing temperatures are dependent on several factors including the chain length separating the carbazole moiety from the TP core, the nature of the ether or ester linkage and the amount of TNF doping. Corresponding 3,6-bisalkylated or 3,6-bisacylated carbazole derivatives do not show mesomorphism either in pure form or when doped with TNF (Citation 63 ).
We have been interested in synthesising fluorescent, low-symmetry triphenylene DLCs having a single electron-withdrawing group (and consequently a large dipole moment) connected directly to the polyaromatic core. To achieve such materials, we required a pentaalkoxy-TP 88. We first attempted its synthesis via triflate 87 reduction which can be easily prepared from hydroxyalkoxy-TP 78. Although the reduction was not successful, we discovered that triflate 87 is mesogenic over a broad temperature range with an unidentified phase occurring below the hexagonal discotic phase (Citation 64 ). Reduction of the phenolic groups of hydroxyalkoxy-TP has ultimately been accomplished by activation with a tetrazole unit followed by catalytic hydrogenation. Compounds 88 now possess reactive sites capable of derivatisation via classical aromatic substitution chemistry. Thus, the bromination of 88 with Br2 in methylene chloride provides the mesogenic bromoalkoxy-TP 91 in 98% yield (Citation 64 ). Friedel–Crafts acylation of 88 with acetyl chloride and aluminium trichloride proceeds in high yield with acylation occurring in the 2-position (90). Somewhat surprisingly, classical nitration of 88 with nitric acid in acetic acid occurs preferentially in the sterically hindered α-position (89) as unambiguously indicated by 1H NMR. Reaction of 91 with copper cyanide gives the cyanotriphenylene 92 in excellent yield. Palladium/copper-catalysed alkynylation of 91 can be accomplished in excellent-to-quantitative yields with a variety of substituted alkynes, as demonstrated with compounds 93 (R = (a) TMS, (b) (CH3)2COH, (c) C4H9). In addition, TPs 93a and 93b can be deprotected to yield free phenylacetylenes for further elaboration or potential polymerisation. Compound 93c on hydrogenation with catalytic palladium yields the alkyl/alkoxy-TP derivative 94. All of the new LCs show a significantly broader range of mesogenicity relative to the parent compound 2,3,6,7,10,11-hexakis(pentyloxy)-TP. Moreover, some of the new mesogens exhibit a more ordered mesophase relative to the hexagonal columnar phase at lower temperatures (Citation 65 ). The free acetylene compound can be easily dimerised to 2 () to generate the ND phase.
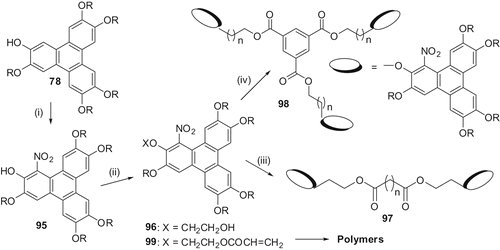
The monohydroxypentaalkoxy-TP can be nitrated at the α-position but this reaction is complicated and often only the oxidised products, the 3,6,7,10,11-pentaalkoxytriphenylene-1,2-diones, can be isolated. Under very carefully controlled conditions, we prepared 1-nitro-2-hydroxy-3,6,7,10,11-pentaalkoxy-TPs 95 (). These double-functionalised TP derivatives are extremely important precursors as the functional group such as nitro, amino, azo, etc., can be utilised to modify the electronic nature of the core and at the same time the hydroxy functional group may be converted to a polymerisable group. Thus, processable oligomers and polymers can be synthesised. The 1-nitro-2-hydroxy-3,6,7,10,11-pentaalkoxy-TP was alkylated with 2-bromoethanol and the resultant alcohol 96 was coupled with various diacids to obtain a number of functionalised dimers 97 (). These yellow-coloured materials with polar nitro group have a broad mesophase range, in many cases stable at room temperature (Citation 66 ). Similarly, condensation of this alcohol 96 with 1,3,5-benzenetricarboxylicacid chloride easily furnished a trimer 98 which displays a monotropic columnar mesophase. The product does not crystallise at room temperature over a long period of time or on cooling to very low temperature. The corresponding unfunctionalised trimer was not liquid crystalline. The dipolar interaction of nitro groups is probably responsible for the induction of mesogenicity (Citation 67 ). The acrylate 99 derived from the alcohol also exhibited a very broad room temperature columnar phase (Citation 68 ). The material was also polymerised but because of the paucity of the material, full characterisation could not be carried out.
Scheme 20. Synthesis of nitro-hydroxy double functionalised TP discotics; (i) HNO3/CH2Cl2; (ii) BrCH2CH2OH/K2CO3; (iii) ClCO(CH2)nCOCl/Py; (iv) 1,3,5-benzenetricarboxylicacid chloride/Py.
Recently, we examined the variation of the relative orientation of two discotic mesogenic moieties around the benzene ring 100–102 () (Citation 69 ). Two series of novel TP-based benzene-bridged symmetric discotic dimers were prepared and characterised. Two TP discotics were connected to a rigid benzene ring via flexible methylene spacers. In one series, the TP moiety was tethered with benzene via an ester linkage while in the second series it is via an ether linkage. Within each series, the orientation of linkage of the TP core around the benzene core was changed by substituting the benzene ring at o, m and p-positions. None of the virgin compounds display any mesomorphism; however, their CT complexes with TNF exhibit columnar mesophases.
Scheme 21. Synthesis of DLCs derived from monohydroxy-TP: (i) ceric ammonium nitrate, CH3CN; (ii) o-phenylenediamine, AcOH; (iii) Zn Ac2O; (iv) DMSO, KOH, RBr; (v) Br(CH2)nBr, Cs2CO3; (vi) Py/toluene/reflux; (vii) imidazole/THF/NaH/reflux; (viii) 1-methylimidazole, toluene, reflux; (ix) CH3I, room temperature.
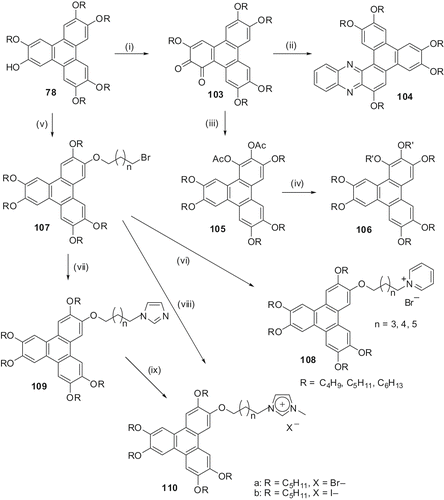
As mentioned earlier, oxidation of monohydroxy-TP furnished the 3,6,7,10,11-pentaalkoxytriphenylene-1,2-diones 103 in high yield. We utilised these diones to prepare a novel discotic phenazine ring structure 104 (). Two homologues with butyloxy- and pentyloxy-peripheral chains were prepared. Both display columnar mesophase over a wide temperature range (Citation 70 ).
Reductive acetylation of these o-quinone furnished the diacetate 105 that can be directly alkylated to various 1,2,3,6,7,10,11-heptaalkoxy-TP derivatives 106 () (Citation 71 ). These unsymmetrically substituted TP derivatives are of three varieties, (i) with seven identical peripheral alkoxy chains, (ii) two or three out of the seven alkoxy chains have different chain lengths and (iii) the mode of the attachment of two of the peripheral chains is different, i.e. via ester linkages. Unsymmetrical substitution has a large effect on thermal behaviour but the type of mesophase formed does not change. All 21 liquid crystalline compounds prepared show very broad Colh phase. Mixed ether–ester derivatives have smaller core–core separations and higher correlation length, therefore are better candidates for charge transport studies (Citation 72 ).
Reaction of monohydroxy-TP with α,ω-dibromoalkanes gives terminally brominated TP 107. Treatment of this bromide with pyridine easily furnished discotic ionic pyridinium salts 108 (Citation 73 ). This way several members with different peripheral chains and spacers were prepared and characterised. Increasing the number of carbon atoms on the peripheral chains of the TP core stabilised the columnar phase while increasing the spacer length connecting the TP unit with the pyridine moiety destabilised the mesophase (Citation 73 ).
Similarly, two novel TP-tethered imidazolium salts 110 were synthesised either by the quaternisation of 1-methylimidazole with an ω-bromo-substituted TP or by the quaternisation of a TP-substituted imidazole with methyl iodide () (Citation 74 ). These TP-based imidazolium salts with bromide or iodide as counterion show columnar mesophase properties over a wide temperature range. The iodide salt exhibited a slightly lower isotropic transition temperature than the bromide salt. This could be due to the bulky nature of the counterion. Interestingly, the non-ionic imidazole-substituted TP derivative 109 was found to be non-mesogenic. Therefore, the mesomorphism in ionic salts is induced due to ionic self-assembly.
It may be noted that ionic liquids are currently attracting considerable attention as ‘green’ solvents for various chemical reactions. Moreover, it is well known that ionic molecules form amphotropic LCs. They have great potential as ordered reaction media that can impart selectivity in reactions by ordering reactants. The formation of supramolecular assemblies containing ionic liquids may find applications as heat carriers in solar–thermal energy generators and as electrolytes for batteries and capacitors.
The coupling of ω-brominated products 107 with imidazole-substituted triphenylene 109 under MW irradiation furnished the discotic dimers 111 () in about 1 minute (Citation 41 ). Similarly, we have prepared several calamitic-calamitic and calamitic-discotic dimers (). POM and X-ray diffraction experiments showed smectic and columnar phases of the calamitic-calamitic and discotic-discotic hybrids, respectively. The calamitic-discotic hybrid was found to be non-liquid crystalline, probably due to non-compatibility of the two different types of cores (Citation 41 )
Figure 3. Structure of a triphenylene-based discotic-discotic ionic dimer.
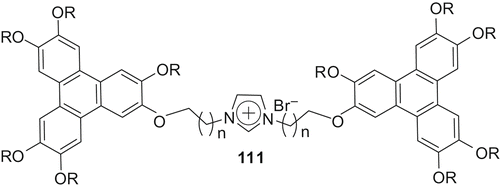
Figure 4. Imidazole-based calamitic–calamitic, calamitic–discotic and discotic–discotic ionic dimmers.
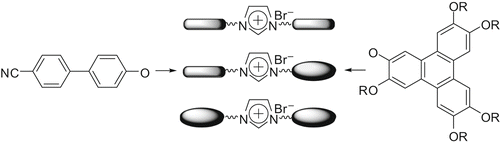
1-Vinylimidazole can be easily coupled with bromo-terminated-TP to produce compound 112. The photopolymerisation of TP-substituted 1-vinylimidazole 112 () yielded a novel ionic discotic liquid crystalline polymer 113 (Citation 75 ). Both the monomer and polymer displayed the Colr phase over a wide temperature range. However, the columnar order in the polymer was found to be small compared with that of the monomer. These materials are not only important for new possibilities of polymeric molten salts in materials science, but also contribute to the development of novel anisotropic soft materials for directional ion conductivity and charge transport at the nanoscale.
Scheme 22. Synthesis of imidazole-based ionic polymer with pendent TP discotic.
A number of discotic donor–acceptor systems can be derived by coupling a monohydroxy-TP with an appropriate functionalised acceptor molecule. These kinds of materials are expected to behave as intrinsic, non-composite p/n-type semiconductors. These molecular double-cables may stack one on top of the other in the columns, which could eventually provide side-by-side percolation pathways for electrons and holes in solar cells. Thus, we designed various discotic donor–acceptor dimers and oligomers as shown in .
Figure 5. Novel discotic donor–acceptor systems.
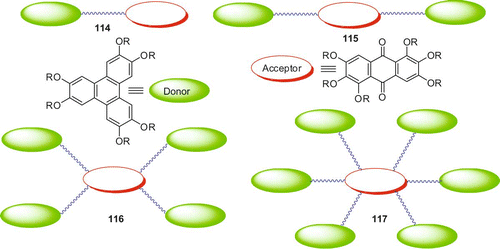
The dimer 114 was prepared in two steps (). Alkylation of monohydroxy-pentahexyloxy-TP with excess of 1,12-dibromododecane furnished the triphenylenedodecylbromide 107 carrying one terminal bromine substituent. The desired dimer 114 was prepared by reacting 107 with monohydroxyanthraquinone under classical alkylation reaction conditions. The dimer displayed a Col mesophase over a wide temperature range. It exhibited one weak broad peak, probably a glass transition, in the DSC at about 40°C and the mesophase to isotropic phase transition at 96.9°C. Upon cooling, the isotropic to columnar phase transition appeared at 94.3°C. The DSC did not show any crystallisation or glass transition peak down to room temperature. On subsequent heating, the DSC showed only a mesophase to isotropic transition and the weak transition at 40°C was not discernible (Citation 76 ).
Scheme 23. Synthesis of discotic donor-acceptor oligomers: (i) K2CO3/DMF/monofunctionalised rufigallol; (ii) Cs2CO3/NMP/difunctionalised rufigallol/MW; (iii) DMSO/NaOH/ rufigallol/90°C; (iv) Cs2CO3/NMP/rufigallol/MW.
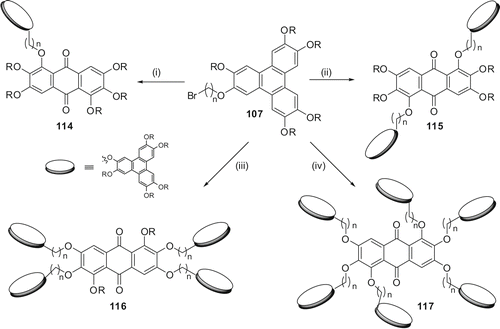
Similarly, triphenylene-anthraquinone-based symmetric discotic liquid crystalline trimers 115 can be prepared. Thus, two series of D-A-D triads with different spacer length and peripheral chains were prepared (Citation 47 ). These triads were synthesised using MW dielectric heating. The etherification of H-bonded hydroxyl groups of tetraalkoxy anthraquinone with bulky ω-bromo-substituted TP failed to produce the desired triads under classical reaction conditions. Mesophase behaviour of the symmetrical trimers was studied by POM and DSC. They exhibited columnar mesophase over a wide range of temperature. Hexagonal columnar structure of the mesophase of these donor-acceptor-donor triads was established by X-ray diffraction studies. Longer spacer length, smaller peripheral alkyl chain length and branching in peripheral alkyl chain of anthraquinone favour mesogenic behaviour in these symmetrical trimers.
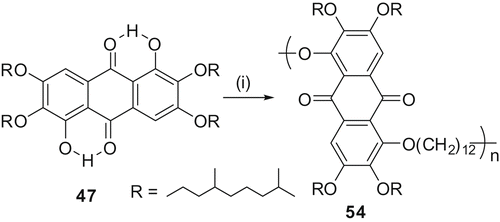
Alkylation of rufigallol with ω-bromo-substituted TP under mild etherification conditions furnished novel pentamer 116 leaving the less reactive intramolecular hydrogen-bonded hydroxyl groups at the 1- and 5-positions unreacted (Citation 77 ). The dihydroxyl functionalised pentamer 116 was acetylated to its corresponding diacetate (116diAc) with acetic anhydride and sulphuric acid under classical conditions. Differential scanning colorimetry measurements of both the pentamers revealed only one first order transition for cooling and heating runs, corresponding to the mesophase to isotropic transition as observed by POM. The pentamer 116 showed the transition from columnar to isotropic at 143.6°C. On cooling it exhibited an isotropic to columnar transition at 139°C. There was no other detectable transition down to −40°C. The pentamer 116diAc upon heating showed a transition at 112.5°C while on cooling the transition is at 106°C. In subsequent heating and cooling cycles the transitions were highly reproducible. No actual glass transition could be detected in the range between−40°C and the clearing points for both the pentamers. The mesophase structure was established by preliminary X-ray diffraction studies.
On the other hand alkylation of rufigallol with ω-bromo-substituted TP under MW irradiation produced a novel star-shaped oligomer 117 (Citation 78 ). The compound exhibited a very broad colh phase in between −40°C and 123°C. Full characterisation of the material is in progress. We are now interested in synthesising various discotic donor-acceptor polymers. These dimer, oligomers and polymers certainly hold immense potential for device applications.
5.6.5.2 Discotics derived from dihydroxy-tetralkoxy-triphenylenes
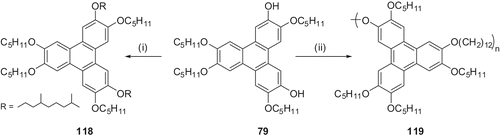
Difunctionalised discotics are primarily useful for the preparation of main-chain polymers and mixed-tail discotics. We utilised the 2,6-dihydroxy-TP 79 to prepare a novel room-temperature liquid crystalline triphenylene monomer 118 and polymer 119 ( ). Upon replacement of two pentyloxy chains by 3,7-dimethyloctyloxy branched chains, compound 118 exhibits liquid crystalline behaviour at room temperature. This compound shows a columnar hexagonal to isotropic transition at 69°C while heating and an isotropic to columnar hexagonal transition at 67°C while cooling. No other transitions are observed while heating and cooling in between −30°C and the clearing temperature (Citation 52 ). Alkylation of 2,6-dihydroxy-TP 79 with 1,12-dibromododecane furnished the polymer 119. This highly viscous compound does not show any peak during heating and cooling in the DSC thermogram, but it exhibits a birefringence texture in polarising microscopy when cooled from the isotropic state. When heated from room temperature this polymer passes slowly to the isotropic state at 89°C and upon cooling the birefringence texture appears slowly. The mesomorphic nature of the polymer was confirmed by X-ray diffraction studies.
Scheme 24. Room-temperature discotic monomer and polymer derived from a dihydroxy-TP: (i) 3,7-dimethyloctylbromide, Cs2CO3, MEK; (ii) 1,12-dibromododecane, Cs2CO3, NMP.
Oxidation of 2,3-dihydroxy-6,7,10,11-tetrapentyloxy-TP 120 yields 6,7,10,11-tetrapentyloxy-triphenylene-2,3-dione 121 (). Reaction of this o-quinone with equimolar amount of 1,2-phenylenediamine yields 2,3,6,7-tetrapentyloxy-phennanthro[b]pheazine 122 which exhibited a metastable monotropic columnar mesophase (Citation 79 ).
Scheme 25. Synthesis of phenanthro[b]phenazine derivaties; (i) CAN, CH3CN, 90%; (ii) 1,2-diaminobenzene, C6H6-AcOH, 80%.
![Scheme 25. Synthesis of phenanthro[b]phenazine derivaties; (i) CAN, CH3CN, 90%; (ii) 1,2-diaminobenzene, C6H6-AcOH, 80%.](/cms/asset/941d4d40-8531-4af6-84e5-dac8b86cf9eb/tlcy_a_392219_o_sch0025g.gif)
The synthesis of mesogenic phennanthro[a]pheazine and phennanthro[b]pheazine ring structures opens a route to prepare a variety of new materials using various alkyl and alkoxy-substituted 1,2-phenylenediamines. These unsymmetrical, coloured, polar heteroaromatic supramolecular architectures could be potential candidates for various devices such as photovoltaic solar cells, light-emitting diodes, etc. It would also be interesting to investigate the medicinal properties of these novel phenazines.
5.6.5.3 Discotics derived from trihydroxy-trialkoxy-triphenylenes
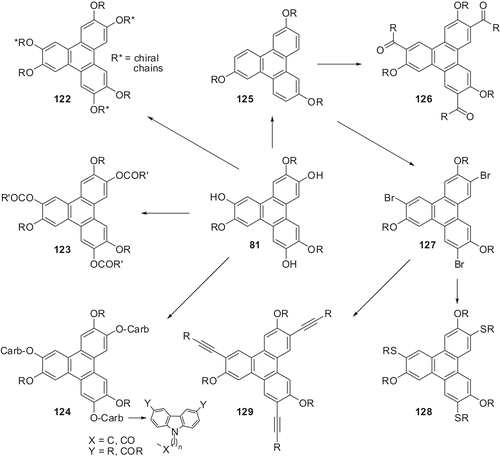
Both symmetric 81 and unsymmetric 82 trihydroxy-trialkoxy-TPs can be easily prepared from hexaalkoxy-TP. Following the chemistry developed for the synthesis of TP derivatives derived from monohydroxy-pentalkoxy-TPs (), we have prepared a number of new materials from symmetric as well as unsymmetric trihydroxy-trialkoxy-TPs (Citation 60 , Citation 62 , Citation 74 , Citation 80 ). Some of these derivatives obtained from symmetrical trifunctionalised-TP are shown in .
Scheme 26. Discotics derived from trifunctionalised-TP. Reagents and conditions are as given in .
6. Discotic-functionalised nanomaterials
In general, the properties of materials change drastically when dimensions are scaled down to the nanometer length scale. For instance, the conductivity of bulk gold is very high (4.3 × 107 Ω−1 m−1) while gold nanoparticles (GNPs) of 1–2 nm size are only semiconducting (1.4 × 10−3 Ω−1 m−1). The incorporation of nanoparticles in the supramolecular order of DLCs would provide materials that possess the properties of nanoparticles as well as the processing, handling and self-assembling properties of LCs, and therefore are likely to lead to novel materials for many device applications. With this view, we have initiated a research programme to insert nanomaterials, such as GNPs and carbon nanotubes (CNTs) into the supramolecular order of DLCs (Citation 52 , Citation81–85).
6.1 GNP–DLC Composites
GNPs present fascinating aspects such as their self-assembly, size-related electronic, magnetic and optical properties, and their applications in catalysis and in the bottom-up approach of nanotechnology (Citation 86 ). Monolayer-protected GNPs can be handled as simple organic materials and a variety of chemical reactions can be performed on functionalised GNPs. We prepared a variety of monolayer-protected GNPs as shown in . These are: (a) alkanethiol-capped GNPs; (b) alkanethiol and discotic mixed monolayer-protected GNPs; (c) GNPs fully covered with TP-based DLCs; and (d) alkoxycyanobiphenyl-covered GNPs. These monolayer-protected GNPs were prepared by reducing gold salt in the presence of an appropriate thiol. A ligand exchange reaction was used to prepare mixed monolayer-covered GNPs. All of the virgin monolayer-protected GNPs were found to be non-mesogenic. Binary mixtures of GNPs with DLCs were prepared by mixing the two components in dichloromethane followed by removal of the solvent and drying in vacuum. Several compositions by mixing alkanethiol-covered GNPs (a) and TP-covered GNPs (c) in HHTT and H6TP were prepared and characterised for their mesomorphic properties (Citation81–84).
Figure 6. Schematic representation of monolayer-protected GNPs.
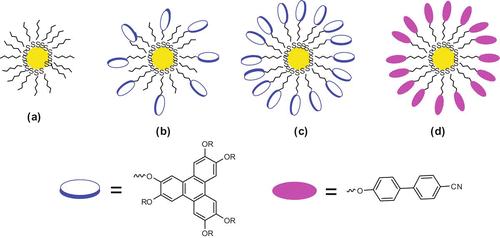
The thermophysical properties of these nanocomposites, studied by POM, DSC and small angle X-ray diffraction, confirm their insertion into the columnar matrix. The presence of the GNPs in the TP-based DLCs does not disturb their mesomorphic behaviour other than altering the transition temperatures. Increasing the amount of GNPs increases the tendency of phase segregation and decreases the clearing temperature. DC conductivity measurements show an enhancement of electrical conductivity by more than a factor of 106 upon doping of DLCs with the TP-capped nanoparticles under ambient conditions (Citation 83 ). The dispersion of discotic-capped nanoparticles in a liquid crystalline matrix can provide a route for synthesising similar composites of varying properties that may find applications in many device developments.
Reduction of gold salts in the presence of novel terminally thiol-functionalised cyanobiphenyls (Citation 87 , Citation 88 ) yields alkoxycyanobiphenyl-covered GNPs ((d)). These thiol-functionalised cyanobiphenyls form stable self-assembled monolayers (SAMs) on a gold surface which have been studied using cyclic voltammetry and electrochemical impedance spectroscopy (Citation 89 ).
6.2 Single-wall carbon nanotubes in discotic liquid crystals
Carbon nanotubes, the one-dimensional carbon allotropes, are well-ordered all-carbon hollow cylinders of graphite with a high aspect ratio. The combination of superlative mechanical, thermal and electronic properties displayed by CNTs make them ideal for a wide range of applications, such as conductive and high-strength composites, catalyst supports in heterogeneous catalysis, energy-storage and energy-conversion devices, field emitters, transistors, sensors, gas storage media, tips for scanning probe microscopy and molecular wires (Citation 90 ).
There has been growing interest in the field of dispersion of CNTs in thermotropic LCs (Citation 91 ). The insertion (dispersion) of CNTs in the supramolecular order of discotic liquid crystalline monomers and polymers, particularly having stable columnar phase at room temperature, may lead to novel materials with interesting properties useful for device applications. With this in mind, we have initiated a research programme to disperse functionalised CNTs into the matrix of liquid crystalline discotic monomers and polymers. TP-functionalised single-wall carbon nanotubes (SWNTs) and commercially available octadecylamine (ODA)-functionalised SWNTs were used to disperse in hexaalkoxy-TP discotics as well as in rufigallol and TP-based room-temperature monomeric and polymeric DLCs (Citation 52 , Citation 85 ).
Discotic-functionalised SWNTs were prepared by mixing SWNT-COCl with hydroxyl-terminated TP and heating at 80°C for 48 hours under anhydrous reaction conditions (). These functionalised SWNTs were highly soluble in common organic solvents such as dichloromethane, chloroform, THF, etc. The formation of discotic-functionalised SWNTs was confirmed by IR, 1H NMR, 13C NMR, TGA and STM studies.
Scheme 27. Schematic representation of discotic-decorated SWNTs.
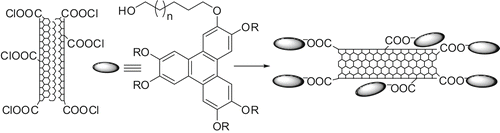
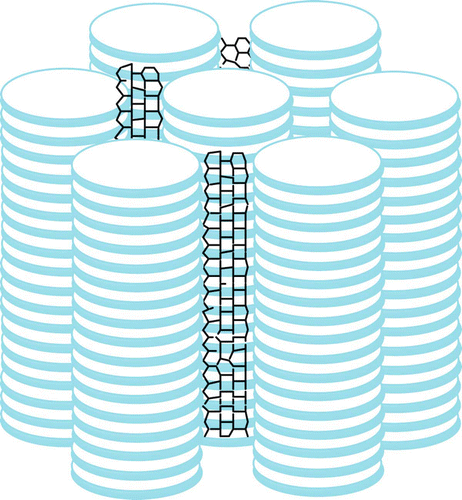
The insertion of either discotic-functionalised SWNTs or commercially available ODA-functionalised SWNTs does not affect the mesophase structure of the pure compounds except in reducing the clearing temperatures. With an increase in the amount of CNTs, the clearing temperature decreases in all the composites. Commercial ODA-functionalised SWNTs can be dispersed in DLCs only by small amounts (∼1%) while a large amount (10%) of discotic-functionalised SWNTs can be dispersed in a columnar matrix. X-ray studies indicate that SWNTs align in the hexagonal columnar phase along the director (). The room-temperature liquid crystalline nanocomposites with broad mesophase ranges and different electronic properties may be important for many device applications.
Figure 7. Dispersion of SWNTs in a columnar matrix.
7. Summary and outlook
The LC industry is a multi-billion dollar industry. However, only a few liquid crystalline compounds are used in practical displays and other devices. New materials are required not only to understand structure–property relationships, but also to find applications in devices. DLCs have recently emerged as a new class of organic semiconductor and may find application in many organic-electronic devices. Liquid crystals are important not only in display and semiconducting industries but also in the pharmaceutical industries, and therefore their future is bright.
In this review article we have briefly summarised our research work mainly on the chemistry of DLCs. We wish to continue our discotic synthesis to realise many more novel discogens which may find applications in industry.
References
- Lehn , J. -M. 1995 . Supramolecular Chemistry , Weinheim : VCH .
- Ringsdorf , H. , Schlarb , B. and Venzmer , J. 1988 . Angew. Chem. Int. Ed. , 27 : 113 – 158 .
- Whitesides , G. M. and Grzybowski , B. 2002 . Science , 295 : 2418 – 2421 .
- de Gennes , P. G. and Prost , J. 1994 . The Physics of Liquid Crystals , Oxford : Clarendon Press .
- Kato , T. 2002 . Science , 295 : 2414 – 2417 .
- Elemans , J. A.A.W. , Rowan , A. E. and Nolte , R. J.M. 2003 . J. Mater. Chem. , 13 : 2661 – 2670 .
- Percec , V. , Rudick , J. G. , Peterca , M. and Heiney , P. A. 2008 . J. Am. Chem. Soc. , 130 : 7503 – 7508 .
- Bahadur , B. , ed. 1990 . Liquid Crystals: Application and Uses , Vol. 1, 2 and 3 , Singapore : World Scientific .
- Chandrasekhar , S. , Sadashiva , B. K. and Suresh , K. A. 1977 . Pramana. , 9 : 471 – 480 .
- For recent reviews on DLCs, see, (a) Kumar , S. 2006 . Chem. Soc. Rev. , 35 : 83 – 109 . (b) Kumar, S. Liq. Cryst. 2005, 32, 1089–1113; (c) Kumar, S. Liq. Cryst. 2004, 31, 1037–1059; (d) Kumar, S. Curr. Sci. 2002, 82, 256–257
- Chandrasekhar , S. and Kumar , S. 1997 . Science Spectra. , 8 : 66 – 70 .
- Laschat , S. , Baro , A. , Steinke , N. , Giesselmann , F. , Hagele , C. , Scalia , G. , Judele , R. , Kapatsina , E. , Sauer , S. , Schreivogel , A. and Tosoni , M. 2007 . Angew. Chem. Int. Ed. , 46 : 4832 – 4887 .
- Sergeyev , S. , Pisula , W. and Geerts , Y. H. 2007 . Chem. Soc. Rev. , 36 : 1902 – 1929 .
- Wu , J. , Pisula , W. and Mullen , K. 2007 . Chem. Rev. , 107 : 718 – 747 .
- Shimizu , Y. , Oikawa , K. , Nakayama , K. and Guillon , D. 2007 . J. Mater. Chem. , 17 : 4223 – 4229 .
- Bushby , R. J. and Lozman , O. R. 2002 . Curr. Opin. Colloid Interface Sci. , 7 : 343 – 354 .
- Demus , D. , Goodby , J. , Gray , G. W. , Spiess , H. -W. and Vill , V. , eds. 1998 . Handbook of Liquid Crystals , Weinheim : Wiley-VCH . Vol. 2B
- Chandrasekhar , S. , Prasad , K. S. , Nair , G. G. , Shankar Rao , D. S. , Kumar , S. and Manickam , M. EuroDisplay '99, The 19th International Display Research Conference Late-News Papers , 9 1999
- Kumar , S. and Varshney , S. K. 2002 . Org. Lett. , 4 : 157 – 159 .
- Kumar , S. and Naidu , J. J. 2002 . Liq. Cryst. , 29 : 899 – 906 .
- Kumar , S. and Varshney , S. K. 2001 . Liq. Cryst. , 28 : 161 – 163 .
- Kumar , S. , Varshney , S. K. , Shankar Rao , D. S. and Prasad , S. K. 2001 . Mol. Cryst. Liq. Cryst. , 357 : 55 – 65 .
- Kumar , S. and Varshney , S. K. 2000 . Angew. Chem. Int. Ed. Eng. , 39/17 : 3140 – 3142 .
- Kumar , S. 2003 . Pramana. , 61 : 199 – 203 .
- Kumar , S. , Varshney , S. K. and Chauhan , D. 2003 . Mol. Cryst. Liq. Cryst. , 396 : 241 – 250 .
- Malthite , J. and Collet , A. 1987 . J. Am. Chem. Soc. , 109 : 7544 – 7545 .
- Keinan , E. , Kumar , S. , Moshenberg , R. , Ghirlando , R. and Wachtel , E. J. 1991 . Adv. Mater. , 30 : 251 – 254 .
- Kouske , H. , Keisuke , T. and Kazuhito , H. 2007 . Synthetic Metals , 157 : 290 – 296 .
- Keinan , E. , Kumar , S. , Singh , S. P. , Girlando , R. and Wachtel , E. J. 1992 . Liq. Cryst. , 11 : 157 – 173 .
- Kumar , S. , Wachtel , E. J. and Keinan , E. 1993 . J. Org. Chem. , 58 : 3821 – 3827 .
- Kumar , S. 1996 . Mol. Cryst. Liq. Cryst. , 289 : 247 – 253 .
- Kumar , S. , Shankar Rao , D. S. and Prasad , K. S. 1999 . J. Mater. Chem. , 9 : 2751 – 2754 .
- Kumar , S. and Varshney , S. K. 2001 . Synthesis , : 305 – 311 .
- Kumar , S. and Naidu , J. J. 2001 . Liq. Cryst , 28 : 1435 – 1437 .
- Kumar , S. , Naidu , J. J. and Shankar Rao , D. S. 2002 . J. Mater. Chem. , 12 : 1335 – 1341 .
- Yamaguchi , S. and Swager , T. M. 2001 . J. Am. Chem. Soc. , 123 : 12087 – 12088 .
- Kumar , S. and Varshney , S. 2002 . Mol. Cryst. Liq. Cryst. , 378 : 59 – 64 .
- Chaudhuri , R. , Hsu , M. -Y. , Li , C. -W. , Wang , C. -I. , Chen , C. -J. , Lai , C. K. , Li-Y , Chen , Liu , S. -H. , Wu , C. -C. and Liu , R. -S. 2008 . Org. Lett. , 10 : 3053 – 3056 .
- Kumar , S. 2008 . Phase Trans. , 81 : 113 – 128 .
- Kumar , S. and Bisoyi , H. K. 2006 . Phase Trans. , 79 : 285 – 292 .
- Pal , S. K. and Kumar , S. 2006 . Tet. Lett. , 47 : 8993 – 8997 .
- Bisoyi , H. K. and Kumar , S. 2007 . Tet. Lett. , 48 : 4399 – 4402 .
- Kumar , S. , Bisoyi , H. K. and Pal , S. K. 2008 . Mol. Cryst. Liq. Cryst. , 480 : 287 – 294 .
- Bisoyi , H. K. and Kumar , S. 2008 . J. Phys. Org. Chem. , 21 : 47 – 52 .
- Pal , S. K. , Kumar , S. and Seth , J. 2008 . Liq. Cryst. , 35 : 521 – 525 .
- Bisoyi , H. K. and Kumar , S. 2008 . New J. Chem. , 32 : 1974 – 1980 .
- Gupta , S. K. , Raghunathan , V. A. and Kumar , S. 2008 . New J. Chem. ,
- Kumar , S. , Naidu , J. J. and Varshney , S. K. 2003 . Liq. Cryst. , 30 : 319 – 323 .
- Kumar , S. and Naidu , J. J. 2002 . Mol. Cryst. Liq. Cryst. , 378 : 123 – 128 .
- Kumar , S. and Naidu , J. J. 2002 . Liq. Cryst. , 29 : 1369 – 1371 .
- Naidu , J. J. and Kumar , S. 2003 . Mol. Cryst. Liq. Cryst. , 397 : 17[317] – 24[324] .
- Bisoyi , H. K. and Kumar , S. 2008 . J. Mater. Chem. , 18 : 3032 – 3039 .
- Kumar , S. and Manickam , M. 1997 . Chem. Commun. , : 615 – 1616 .
- Kumar , S. and Varshney , K. S. 1999 . Liq. Cryst. , 26 : 1841 – 1843 .
- Kumar , S. and Manickam , M. 1998 . Mol. Cryst. Liq. Cryst. , 309 : 291 – 295 .
- Kumar , S. , Manickam , M. , Balagurusamy , V. S.K. and Schonherr , H. 1999 . Liq. Cryst. , 26 : 1455 – 1466 .
- Kumar , S. , Schuhmacher , P. , Henderson , P. , Rego , J. A. and Ringsdorf , H. 1996 . Mol. Cryst. Liq. Cryst. , 288 : 211 – 222 .
- Kumar , S. and Manickam , M. 1998 . Synthesis , : 1119 – 1122 .
- Kumar , S. and Lakshmi , B. 2005 . Tet. Lett. , 46 : 2603 – 2605 .
- Yelamaggad , C. V. , Prasad , V. , Manickam , M. and Kumar , S. 1998 . Mol. Cryst. Liq. Cryst. , 325 : 33 – 41 .
- Manickam , M. , Kumar , S. , Preece , J. A. and Spencer , N. 2000 . Liq. Cryst. , 27 : 703 – 706 .
- Manickam , M. , Belloni , M. , Kumar , S. , Varshney , S. K. , Shankar Rao , D. S. , Ashton , P. R. , Preece , J. A. and Spencer , N. 2001 . J. Mater. Chem. , 11 : 2790 – 2800 .
- Manickam , M. , Cooke , G. , Kumar , S. , Ashton , P. R. , Preece , J. A. and Spencer , N. 2003 . Mol. Cryst. Liq. Cryst , 397 : 99[399] – 116[416] .
- Henderson , P. , Kumar , S. , Rego , J. A. , Ringsdorf , H. and Schuhmacher , P. 1995 . J. Chem. Soc. Chem. Commun. , : 1059 – 1060 .
- Rego , J. A. , Kumar , S and Ringsdorf , H. 1996 . Chem. Mater. , 8 : 1402 – 1409 .
- Kumar , S. , Manickam , M. and Schonherr , H. 1999 . Liq. Cryst. , 26 : 1567 – 1571 .
- Kumar , S. and Manickam , M. 1999 . Liq. Cryst. , 26 : 939 – 941 .
- Schonherr , H. , Manickam , M. and Kumar , S. 2002 . Langmuir. , 18 : 7082 – 7085 .
- Gupta , S. K. , Raghunathan , V. A. and Kumar , S. (To be submitted)
- Kumar , S. and Manickam , M. 1999 . Liq. Cryst. , 26 : 1097 – 1099 .
- Kumar , S. and Manickam , M. 1998 . J. Chem. Soc. Chem. Commun. , : 1427 – 1428 .
- Kumar , S. , Manickam , M. , Varshney , S. K. , Shankar Rao , D. S. and Prasad , S. K. 2000 . J. Mater. Chem. , 10 : 2483 – 2489 .
- Kumar , S. and Pal , S. K. 2005 . Tet. Lett. , 46 : 4127 – 4130 .
- Kumar , S. and Pal , S. K. 2005 . Tet. Lett. , 46 : 2607 – 2610 .
- Pal , S. K. and Kumar , S. 2008 . Liq. Cryst. , 35 : 381 – 384 .
- Kumar , S. , Naidu , J. J. and Varshney , S. K. 2004 . Mol. Cryst. Liq. Cryst. , 411 : 355[1397] – 362[1404] .
- Bisoyi , H. K. and Kumar , S. 2008 . Tet. Lett. , 49 : 3628 – 3631 .
- Bisoyi , H. K. and Kumar , S. to be communicated
- Kumar , S. and Manickam , M. 2000 . Mol. Cryst. Liq. Cryst. , 338 : 175 – 179 .
- Schonherr , H. , Kremer , F. J.B. , Kumar , S. , Rego , J. A. , Wolf , H. , Ringsdorf , H. , Jaschke , M. , Butt , H. -J. and Bamberg , E. 1996 . J. Am. Chem. Soc. , 118 : 13051 – 13057 .
- Kumar , S. and Lakshminarayanan , V. 2004 . Chem. Commun. , : 1600 – 1601 .
- Kumar , S. , Pal , S. K. and Lakshminarayanan , V. 2005 . Mol. Cryst. Liq. Cryst. , 434 : 251[579] – 258[586] .
- Kumar , S. , Pal , S. K. , Suresh , K. P. and Lakshminarayanan , V. 2007 . Soft Matter , 2 : 896 – 900 .
- Kumar , S. 2007 . Synth. React. Inorg. Met.-Org. Nano-Metal Chem. , 37 : 327 – 331 .
- Kumar , S. and Bisoyi , H. K. 2007 . Angew. Chem. Int. Ed. , 46 : 1501 – 1503 .
- Hutchings , G.J , Brust , M. and Schmidbaur , H. 2008 . 37 : 1759 – 1765 .
- Kumar , S. and Pal , S. K. 2005 . Liq. Cryst. , 32 : 659 – 661 .
- Pal , S. K. , Raghunathan , V. A. and Kumar , S. 2007 . Liq Cryst. , 34 : 135 – 141 .
- Ganesh , V. , Pal , S. K. , Kumar , S. and Lakshminarayanan , V. 2006 . J. Colloid. Int. Sci. , 296 : 195 – 203 .
- Dresselhaus , M. , Dresselhaus , G. and Avouris , P. , eds. 2002 . Acc. Chem. Res 997 – 1113 . 35
- Dierking , I. , Scalia , G. , Morales , P. and LeClere , D. 2004 . Adv. Mater. , 16 : 865 – 868 . Dierking, I.; Scalia, G.; Morales, P. J. Appl. Phys. 2005, 97, 044309