Abstract
Two theoretically feasible strategies to designing the deoxyribonucleic acid (DNA) nanoconstructions are outlined. The main data on the physico-chemical properties of liquid-crystalline dispersions formed by double-stranded nucleic acid (DNA and ribonucleic acid) molecules as a result of their phase exclusion from water-salt solutions are presented and the specific features of these molecules in quasinematic layers of the liquid-crystalline particles are allocated. The basic data obtained in this field have been used as the background for the creation of the DNA nanoconstructions carrying various ‘guest’ molecules (biologically active compounds or chemical substances). The unique properties of these nanoconstructions determine the possibility of manipulation with these objects, as well as the areas of their practical application.
1. Introduction
The words nanotechnology, nanoparticles and nanomaterials are already known to a wide circle of readers. Indeed, the manipulations at the level of individual atoms provide for creating novel ‘structured’ materials and devices displaying unique prespecified properties. Such terms as nanobiotechnology and nanomedicine have been recently added to those mentioned above, thereby demonstrating that new directions of nanotechnology are being formed that utilise molecules of biological origin as building blocks for constructing nanostructures.
The nanodesign involving double-stranded (ds) nucleic acid (NA) molecules, i.e. a directed elaboration of complex spatial structures with regulated properties (nanostructures, nanoconstructions (NaCs) and nanobiomaterials) using ds NA molecules or their complexes as building blocks (Citation 1 , Citation 2 ), is in the focus of research attention in many countries. The opportunity to apply NAs for designing NaCs with regulated parameters is based on the use of several properties characteristic of only these molecules.
The current strategies of design of NA-based NaCs are divided into two groups:
-
constructing NaCs through successive modifications of the initial single-stranded (ss) NA molecules (Citation 3 ); and
-
constructing NaCs using linear ds NAs (or their complexes) fixed in the spatial structure of particles of liquid-crystalline dispersions (LCDs) (Citation 4 ).
One can briefly comment on the first group of technologies for designing NaCs. The approach to NaC design involving ss NA molecules that can be arbitrarily named ‘successive or step-by-step design’ was pioneered by Seeman in 1982 (Citation 3 ). Different versions of this approach have been described; they are based on the use of the chemically and (or) biochemically modified ss NA molecules (or synthetic oligonucleotides). A correct selection of nucleotide sequences in the initial ss NA and the application of the corresponding cruciform structures of ss NA allowed Seeman and his followers to produce various nanostructures with stiffening ribs formed by corresponding ds NA molecules (Citation 3 , Citation5–17).
The creation of nanostructures according to this technology is connected with considerable expenses for the obtaining of ss NA fragments with specified sequences of nitrogen bases, the use of a large enzyme set (restriction endonucleases and ligases) for splitting and linking the ss NA fragments at the desired sites, the isolation of specific structures from reaction mixture, the comprehensive analysis of their properties and the use of state-of-the-art control methods (such as atomic force microscopy (AFM)) at all the stages of nanodesign.
Despite the first successes in a step-by-step nanodesign based on ss NA molecules as building blocks, the most important goal of nanodesign – designing spatial structures with controllable properties carrying molecules of other compounds (‘guests’) – has not been achieved in the framework of this strategy and the question of the practical application of the nanostructures of the ss NAs still remains open to a considerable degree.
The goal of the presented review is the illustration of the second group of strategies for NaC design. This technology of NaC creation was proposed in 1996 at the Institute of Molecular Biology of the Russian Academy of Sciences (RAS) and developed in collaboration with researchers from the institutions of the other countries. It is principally different from the above-described technology and based on the concept of the self-ordering of ds deoxyribonucleic acid (DNA) molecules (or ds DNA complexes) by phase exclusion. This means that our strategy utilises not ss NA molecules but rather the preformed spatial structures spontaneously arising as a result of the phase exclusion of ds DNA molecules from water-salt solutions.
2. Formation of the deoxyribonucleic acid liquid-crystalline dispersions in polymer-containing water-salt solutions and their properties
At first, it is necessary to stress that the properties of the liquid-crystalline (LC) phases of NAs have been extensively studied in a number of laboratories from various countries since 1961 (Citation18–22). It was shown that the rigid ds DNA molecules in concentrated solutions form a variety of lyotropic LC phases (cholesteric, precholesteric, nematic, etc., see (Citation23–27)). According to an X-ray study, the ds DNA molecules are ordered in the phases at distances of 3.0–5.0 nm, i.e. they acquire the properties of a crystal, and according to the results of the analysis of their textures and the CD-spectroscopy, the DNA molecules in the neighbouring quasinematic layers are mobile, i.e. they retain the properties of a liquid (see, for instance, (Citation28–30)). The structures of these LC mesophases depend on the properties of the solvent, on the concentration and on secondary structure of the ds DNA (Citation 31 ).
Secondly, the results obtained from the study of bulk samples of ds DNA mesophases open a gate for the thorough studies of properties of particles of DNA LCDs. (The main, from my point of view, experimentalists in this interesting scientific area are presented in our book ‘Liquid-crystalline Dispersions and NaCs of DNA’, published in Russia in 2008 (Citation 32 )).
Finally, the properties of the LCDs are of interest for several reasons. From a theoretical point of view, the physico-chemical properties of LCDs can differ considerably from those of bulk phases. The main differences arise because of the ‘size effect’. This results from the contributions to the free energy of the particles that arise from the surface tension of the small droplets. There can also be packing defects within these particles caused by the curvative of the surface, and the structure within a particle may not be the same as that in the bulk LC phases. The study of LCDs of ds DNA is interesting from the biological point of view, since the structural and physico-chemical properties of particles reflect some properties of these biopolymers in biological objects. Finally, from the practical point of view, LCDs are of interest, because of their potential as multifunctional sensing elements (Citation33–35) for biosensor devices intended for detection of chemical or biologically active compounds (BACs; drugs, antibiotics, etc.) interacting with ds DNA molecules.
Because this review is not a history of the investigation of the DNA liquid crystals, we will outline only such physico-chemical properties of the ds DNA LC phases and dispersions that are necessary as a background for explanation of the ideology of transition of the ‘liquid’ structure of the ds DNA LCD to its ‘rigid’ state.
Indeed, it was shown that the mixing of ds DNA and water-salt solutions of some polymers leads to the phase exclusion of DNA molecules (DNA condensation) and the formation of dispersions. Such condensation of a single high-molecular-mass (>20 × 106 Da) ds DNA molecule is accompanied by formation of a toroid-like DNA particle. This intramolecular process is known since Lerman's experiments (1971) as ‘ψ-condensation’ (psi is the acronym for polymer-salt-induced) (Citation36–39).
In contrast, the phase exclusion (or condensation) of rigid low-molecular-mass ds DNA molecules from poly(ethylene)glycol (PEG) solutions is an intermolecular process. This process causes the ordering of the neighbouring ds DNA molecules and it is accompanied by the formation of the LC phases or LCDs of these molecules (Citation 27 , Citation 40 ). Note that the particles of the low-molecular-mass ds DNA dispersions are ‘microscopic droplets of concentrated DNA solution’, which cannot be ‘taken in hand’ or ‘directly seen’. A ‘liquid’ mode of packing of ds DNA molecules in the particles of LCDs prevents their immobilisation on the surface of a membrane filter.
The low-molecular-mass ds DNA condensation (), based on the technology of intensive mixing (stirring) the PEG-containing water-salt solutions with water-salt solutions containing a very low (5–10 μg ml−1) concentration of ds DNA (molecular mass < 1 × 106 Da) used in (Citation 32 ) is observed when the PEG concentration in the solution used for phase exclusion exceeds a certain critical value (Citation 41 ). A violation of the critical conditions for the formation of LCD particles and, in particular, a decrease in PEG concentration below the critical value, leads to the disintegration of LCD particles and the transition of DNA molecules to their initial isotropic state.
Figure 1. The formation of a particle of ds NA CLCD. Polymer molecules are shown by filled red circles (1) and the DNA CLCD particle is encircled by an oval (2); π means osmotic pressure of the solution (red arrows); ordering DNA molecules in one quasinematic layer is illustrated (2). ds DNA molecules (1) in subsequent layers (2) are located at distance d, which depends on the osmotic pressure of the solution. The formation of CLCD particles is characterised by spatially twisted packing of neighbouring layers of DNA (or RNA) molecules and is accompanied by the appearance of an abnormal negative band in the circular dichroism (CD) spectrum in the case of DNA and abnormal positive band in the case of RNA, located in the absorption region of nitrogen bases (3).
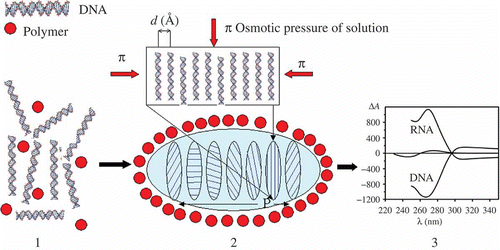
Theoretically, estimations by various methods (sedimentation analysis, ultraviolet (UV)-irradiation scattering and dynamic light scattering) demonstrate that, according to these methods, the mean diameter of LCD particles is close to 400–500 nm and that one particle, having the molecular mass of about 600–800 × 103 Da, contains approximately 104 DNA molecules (Citation 31 , Citation 32 ). However, the above valuations are approximate, as they are made by calculations where the formed ds DNA particles were considered as spherical objects. Consequently, the question as to how to visualise the LCD particles of ds DNAs and directly assess their size was still open. Until a certain stage of research, the question on how DNA molecules are packed into LCD particles was also unanswered.
The LCD particles formed by the phase exclusion of linear low-molecular-mass ds DNAs display several specific features. Firstly, the polymer (PEG) is not included in the content of the formed particles (), and the LCD particles of ds DNAs retain both the chemical reactivity of the DNA structural elements (nitrogen bases and so on) and a high (in the range of 160–600 mg ml−1) DNA local concentration with the ordered arrangement of the DNA molecules in the neighbouring layers. Secondly, as follows from the results of the X-ray studies of the LC phases formed as result of low-speed sedimentation of the ds DNA LCD particles, the interhelical distances (d inter) between adjacent DNA molecules in LCD particles can be regulated in the range of 2.5–5.0 nm by specifying the PEG concentration in the solution used for DNA condensation. For instance, at a concentration of PEG in the final solution equal to 170 mg ml−1, the value of d inter is close to 3.4 nm (Citation 31 , Citation 32 ). Thirdly, DNA molecules, due to their ‘double’ inherent anisotropy (related with helical structure and the presence of optically active carbon atoms in sugar residues), tend to form spatially twisted layered or a so-called ‘cholesteric’ structure of the LCDs (CLCDs).
It is necessary to stress that, in favour of the layered structure of particles of CLCD shown in , speak the results of reconstructions of spatial structure of the chromosomes of Dinoflagellate, based on the investigation of their cross-sections performed by Livolant (Citation42–44), Rill et al. (Citation 45 ), Bouligand and Norris (Citation 46 ) and Sartori et al. (Citation 47 ) and the results of comparison of the textures and the CD spectra of the DNA LC objects performed by Tinoko et al. (Citation 48 ) and Livolant and Maestre (Citation 49 ), as well as other scientific teams (Citation 50 ).
The formed cholesteric is ‘coloured’, because DNA molecules contain ‘chromophores’ (nitrogen bases), which absorb UV irradiation. The twisted structure of the DNA cholesteric means that the average orientation of the ‘director’ (n→ ) of the layer, which reflects the mean direction of electronic transitions of all nitrogen DNA bases in each quasinematic layer, is changed in the subsequent quasinematic layers on a fixed angle. Various approaches have been used for the description of optical properties of the particles of CLCDs of many compounds, including NAs, since 1951 (Citation 48 , Citation51–56).
However, a realistic theory capable not only of describing but also predicting the many optical peculiarities of the particles of ds DNA CLCD formed in PEG-containing solutions namely was developed by Belyakov et al. (Citation 57 ). This theory takes into account the layered structure of the packing ds DNA molecules and the above-shown parameters of the ds DNA CLCD particles. The theory predicts the appearance of an intense (abnormal) band in the CD spectrum located in the region of absorption of the nitrogen bases (UV region) of the ds DNA. The abnormal CD bands can be expected, according to this theory, in the absorption regions of any coloured compounds not only intercalating between ds DNA base pairs, but also anisotropically fixed in the space between the neighbouring DNA molecules that form CLCD particles. The theory imposes no limitations on the number of coloured molecules that can be introduced into the DNA structure. In the scheme () of ds DNA CLCD formation, with the stage of mixing of DNA and PEG solutions, the packing mode of DNA molecules in dispersion particles is determined ‘at the moment’ of their recognition during the juxtaposition of DNA molecules with the negative charges on phosphate groups neutralised by counterions (Citation 58 ). In this process, the anisotropic character of the rigid linear ds DNA molecules specifies cholesteric packing of the neighbouring DNA molecules in particles formed. The ds DNA molecules (B-family) with a right-handed helical twist of the secondary structure usually form particles with a left-handed helical packing of the quasinematic layers (Citation 50 , Citation 59 , Citation 60 ). This is demonstrated by the appearance of an abnormal negative CD band in the absorption region of DNA nitrogen bases. If the above-described scheme for formation of dispersion particles and the developed theory (Citation 57 ) are correct, then the parameters of the secondary structure of ds DNA molecules and the distribution pattern of positive charges on their surface can influence the direction of their helical packing in particles of CLCD. Indeed, fine variation in these parameters can lead to both a decrease in the amplitude of the abnormal CD band and a change in its sign. This statement of the theory was proven experimentally. For instance, the CLCDs were formed by the right-handed synthetic ds polynucleotides poly(dA-dT)*poly(dA-dT) and poly(dA)*poly(dT) with identical molecule masses, but different base sequences. The CD spectra of these CLCDs are a mirror image of each other (Citation 31 , Citation 32 ). This result shows that very small alterations in the parameters of the secondary structure of ds molecules can be sufficient to cause the change from the left-handed to the right-handed twist of the quasinematic layers of the particles of CLCDs.
The theory (Citation 57 ) predicts as well the dependence of the amplitude of the abnormal CD band on such parameters as the value of the pitch (P) of helical twist of neighbouring quasinematic layers formed by ds DNA molecules in particles, on the diameter of particles of the CLCD, etc. (Citation 61 ). This theory was applied later for description of the complex changes in abnormal optical properties of the CLCD formed by ds DNA molecules induced by the interaction with various compounds (Citation 62 ).
It is necessary to add that, according to Parcegian et al. (Citation 63 ), Podgornik et al. (Citation 64 , Citation 65 ) and Odijk (Citation 66 ), the osmotic pressure of the solvent plays a very important role in the stabilisation of the spatial structure of ds DNA LC particles formed under various conditions. Due to constant (fixed) osmotic pressure determined by PEG concentration, the spatial structure of DNA CLCD particles in a PEG-containing solution is sterically limited (‘frozen’) (Citation 61 ), although DNA molecules retain some diffusion degrees of freedom, determining a ‘liquid’ character of the packing mode of neighbouring DNA molecules in quasinematic layers.
At a high PEG concentration in the water-salt solution, the ds DNA dispersion particles are still formed, yet their abnormal CD band disappears. In this case, the packing density of ds DNA molecules corresponds to their packing in a hexagonal LC phase (Citation 42 , Citation 67 ), which lacks an abnormal optical activity. In other words, a certain limit of PEG concentration in solution causes the transition from the cholesteric mode of packing of neighbouring ds DNA molecules in particles to their hexagonal packing. This transition is accompanied by the unwinding of the helical spatial structure of particles and, consequently, by a decrease in the value of an abnormal CD band of these particles to its full disappearance.
The size of the DNA CLCD particles shown in is determined by a fine balance between the free energy of these particles and their surface free energy (Citation68–70). The competition between the free energy of a dispersion particle (tending to increase the particle size) and its surface free energy (which depends on the surface tension between the cholesteric phase and isotropic phase and is directed to a decrease in the surface between the DNA-rich phase and the ambient isotropic solution (Citation 69 , Citation 70 ) suggests the existence of a critical size of dispersion particles below which they are unstable or do not form at all. This statement complies with the results of the analysis of the formation kinetics of DNA dispersion particles and the dependence of particle formation efficiency on various factors. According to theoretical estimations, the average size of dispersion particles depends on both the DNA concentration and PEG concentration used for phase exclusion (Citation 31 , Citation 32 , Citation 71 ). Note that unlike a conventional aggregation of DNA molecules, which is independent of their lengths and is not accompanied by an appearance of abnormal optical activity, a CLCD is formed only when the length of the ds DNA molecules exceeds 15 nm (∼50 base pairs). This result demonstrates the existence of a lower limit of DNA length for the formation of an optically active dispersion. The upper limit of ds DNA molecular mass, when these molecules preserve the ability to form a CLCD, is close to 3 × 106 Da (Citation 31 ). A further increase in DNA length decreases the amplitude of the abnormal CD band displayed by dispersion, despite still being formed, as detected by the appearance of the apparent optical density in the absorption spectrum. For DNA molecular mass exceeding 10 × 106 Da, the CD spectra for dispersions are similar to the CD spectra of the initial isotropic linear ds DNA solution. The absence of an abnormal optical activity of the dispersions formed by high molecular-mass DNA demonstrates an important role of kinetic factors in the packing mode of ds DNA molecules.
In addition, one can stress that due to the ‘liquid’ character of the ds DNA molecules packing, strong modification of the DNA structure under the influence of chemical or BACs can result in distortion of the cholesteric structure of particles of the CLCD, and hence, in the disappearance of an abnormal optical activity.
Thus, not only the osmotic pressure of the PEG solution, determined by PEG concentration, but also properties of the ds DNA molecules control the packing mode of neighbouring linear rigid ds DNA molecules in dispersion particles (Citation 68 , Citation 70 ). The constant osmotic pressure of the solution determines a constant spatial structure of CLCD particles and, consequently, a fixed distance between neighbouring linear ds DNA molecules in the quasinematic layers of CLCD particles and a constant value of the amplitude of their abnormal CD band in the absorption region of DNA nitrogen bases.
shows that because of the ‘liquid’ mode of ds DNA molecules packing in quasinematic layers, low-molecular-mass chemical or BACs (antitumour drugs, peptides, chemicals, metal cations, etc.) can readily diffuse into particles of CLCD and, due to the high concentration of ds DNA molecules in the layers, can quickly interact with all reactive groups of these molecules (Citation 72 ).
Figure 2. The scheme of ordering ds DNA molecules in a quasinematic layer. π-osmotic pressure of the solution is shown by red arrows. Due to the ‘liquid’ character of ds DNA packing, various chemical substances or BACs (shown by yellow, green and blue arrows) can quickly diffuse into this layer. The sign X means that the neighbouring layers of ds DNA molecules are twisted in the structure of the CLCD particle.
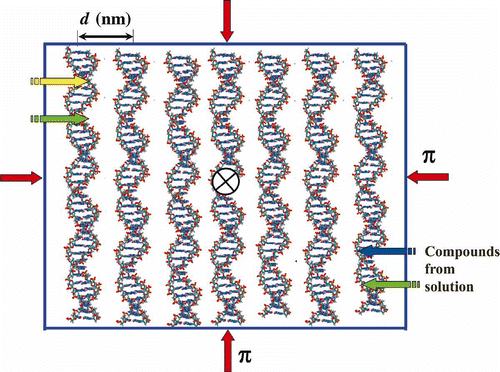
Note that a low extent of intercalation of BACs between pairs of DNA nitrogen bases does not interfere with the packing mode of neighbouring ds DNA molecules in quasinematic layers, leaving the overall spatial structure of ds DNA CLCD particles intact.
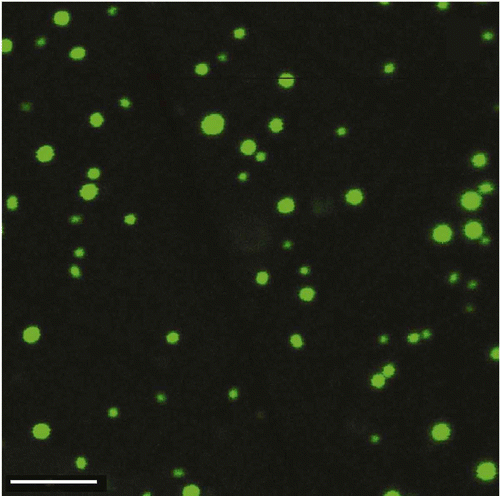
The development of new microscopic techniques, in combination with the use of new intercalating fluorescent compounds of the ‘SYBR Green’ family, opens a gate for obtaining additional information on the peculiarities of the ds DNA CLCD particles existing in the PEG-containing solutions. These compounds are capable not only of intercalating between nitrogen bases of ds DNA, but also of retaining their high fluorescence, being incorporated into the DNA CLCD particles, formed in PEG-containing solutions. As an example, shows the ‘fluorescence images’ of particles of the ds DNA CLCD obtained by the confocal microscope. The study of such pictures obtained under various conditions shows that these particles exist as independent objects, the process of formation of perfect ds DNA CLCD takes time, and the mean size of the ds DNA CLCD particles depends on the initial concentration of the ds DNA molecules used for the phase exclusion.
Figure 3. The ‘fluorescence images’ of particles of the DNA CLCD treated with the fluorescent intercalator – SYBR Green. The images were obtained by the confocal microscope Leica TCS SP5; bar – 4 μm; С SYBR Green = 4.9 × 10−6 М; С DNA = 50 μg ml−1; 10 min; С PEG = 170 mg ml−1; 0.3 M NaCl; 0.002 М Na-phosphate solution; pH 6.8.
The above data, obtained by experimentalists from the various countries and different laboratories, demonstrate that the study of the physico-chemical properties of the ds DNA CLCD particles gave sufficiently detailed information about the conditions required for the formation of ds DNA CLCDs and on the properties of CLCD particles, as well as the factors providing for the control of the properties of ds DNA CLCD particles. This information formed the background for the further elaboration of the methods, allowing the liquid ds DNA CLCD particles to be transformed into ‘rigid’ ds DNA NaCs.
3. Approaches to designing rigid nanoconstructions using double-stranded deoxyribonucleic acid molecules fixed within quasinematic layers of cholesteric liquid-crystalline dispersion particles
3.1 First approach: formation of nanobridges between ds DNA molecules
The structure of the quasinematic layer formed by ds DNA molecules in particles of LCDs () at a certain osmotic pressure of PEG solution explains the essence of our technology of designing ds DNA-based NaCs containing a high concentration of BACs (‘guest’ molecules).
When analysing , it is necessary to take into account several facts that were important for the first attempt to solve the problem of accumulating ‘guest’ molecules.
-
The chemical reactivity of the ds DNA molecules fixed within quasinematic layers of CLCD particles does not change.
-
The local concentration of ds DNA molecules in CLCD particles is very high and amounts to several hundred milligrams per millilitre.
-
At a fixed osmotic pressure of the solution, the mutual orientation of neighbouring ds DNA molecules in the quasinematic layer is ‘frozen’, although DNA molecules retain some diffusion degrees of freedom, which determines a ‘liquid’ packing pattern of neighbouring ds DNA molecules.
-
It is possible to regulate the distance between neighbouring DNA molecules in the quasinematic layer from 2.5 to 5.0 nm only by changing the osmotic pressure of the PEG solution or, at a fixed PEG concentration and molecular mass, by changing the temperature of the solution.
-
There is a ‘free space’ between the ds DNA molecules ordered in the quasinematic layer.
-
A considerable distance between ds DNA molecules, the liquid mode of their packing and their high concentration in the quasinematic layer provide the conditions for a quick ‘one-dimensional’ diffusion of molecules of ‘guest’ molecules between both the ds DNA molecules in one layer and the ds DNA molecules in neighbouring layers of CLCD particles.
-
The interaction of BACs with nitrogen bases of ds DNA molecules (under certain conditions) does not interfere with the packing pattern of neighbouring DNA molecules in the layer.
-
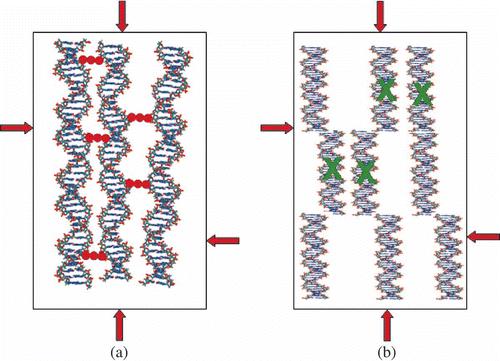
From the theoretical standpoint, there are no limitations for the incorporation of ‘guest’ molecules in the free space between ds DNA molecules in the quasinematic layer; hence, it is possible to achieve a high concentration of ‘guests’ by incorporating these molecules into the content of cross-links formed between neighbouring ds DNA molecules ((a)).
Figure 4. Two possibilities for spatial fixation of ds DNA molecules in a quasinematic layer. π-osmotic pressure of the solution is shown by red arrows. (a) Neighbouring ds DNA molecules can be chemically linked by ‘nanobridges’ composed of ‘guest’ molecules (red dots) without altering the spatial location of the DNA molecules. (b) Modification of the secondary structure of ds DNA molecules induced by interaction with agent ‘X’ (shown in green) is accompanied by modification of the ds DNA secondary structure and by the increase in interaction of fragments of neighbouring molecules and their spatial fixation. In this case the irregular location of the DNA fragments appears. In both cases, the fixation of the ds DNA molecules or DNA fragments results in formation of ‘rigid’ structures, capable of existing in the absence of the solvent osmotic pressure.
-
To form cross-links, it is necessary that the ds DNA surface has the sites of their beginning and ending (both a metal ion bound to a nitrogen base and the molecule of a ligand additionally introduced into the system can serve as such sites).
-
Taking into account the sterical location of the reactive groups (in particular, the nitrogen atom of N-7 of purines) in the groove on the surface of the ds DNA molecules, it is evident that two neighbouring ds DNA molecules can be linked only when the spatial orientations of these groups are coordinated (i.e. the neighbouring ds DNA molecules must be sterically ‘phased’ to realise the linking of the same reactive groups that belong to neighbouring DNA molecules). This means that the formation of the cross-links requires meeting a number of conditions.
-
The theory predicts that when constructing cross-links containing chromophores and these cross-links are specifically oriented in respect to the long axes of ds DNA molecules in quasinematic layers, we should expect the appearance of an additional abnormal CD band in the absorption region of these chromophores or its amplification.
All of these points show that the fundamental problem is reduced to the formation of artificial cross-links with adjustable properties between neighbouring ds DNA molecules fixed (due to constant osmotic pressure of the used PEG-containing solution) at an interhelical distance of about 3.5 nm in the structure of CLCD particles without alteration of the spatial organisation of CLCD particles. Taking into account this distance, one can call the cross-links ‘nanobridges’ between ds DNA molecules.
The stated problem was first formulated by Yevdokimov et al. in 1996 (Citation 73 ) and solved only recently as a result of collaboration of scientific teams from Russia, Italy and Germany (Citation74–79).
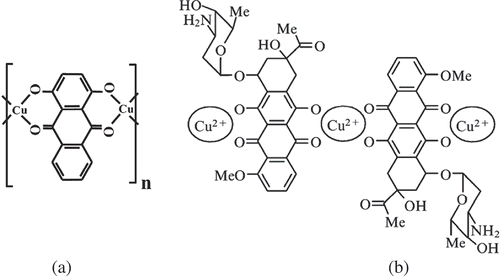
Obviously, the formation of nanobridges between neighbouring ds DNA molecules requires the presence of terminal sites on the surface of ds DNA molecules. If such sites are located at neighbouring molecules, a nanobridge will cross-link the neighbouring ds DNAs. Theoretically, such a terminal site can be metal ions specifically fixed in a groove (or grooves) on the DNA surface that is capable both of forming complexes with the DNA nitrogen bases and attaching other ligands that are additionally introduced into the system. In addition, molecules of planar compounds also tend to form external complexes with DNA molecules. In view of these circumstances, antibiotics of the anthraquinone or anthracycline group are of interest (). These compounds, owing to their chemical structure, can form planar chelate complexes with cations of divalent metals, in particular, with Cu2+ ions. In particular, anthracycline chelate complexes ((b)) can contain up to 10 repeating units; i.e. under certain conditions, anthracyclines can form extended planar structures with Cu2+ ions (Citation 75 ). Antibiotics of this group (in particular, daunomycin (DAU)) can intercalate between pairs of bases of NAs and synthetic polynucleotides only of the B-family. Upon intercalation, the reactive DAU groups (keto-oxygen and peri-OH-groups, (b)) are inaccessible for chemical reactions. However, it is known that DAU and its analogues can form so-called ‘external’ complexes with the NAs both of the B- and A-families of ds polynucleotides (Citation 74 ). As a result of the formation of an external complex, reactive DAU oxygen groups become available for chemical reactions, in particular, for chelate formation. Therefore, DAU molecules can also play the role of terminal sites for nanobridges.
Figure 5. Examples of chelate complexes formed between anthraquinone (a) or anthracycline (b) molecules and Cu2+ ions possessing planar structures.
Thus, the technology used by us is based on the combination of several known experimental results and some theoretical possibilities, specifically: spatial ordering of neighbouring, closely spaced ds DNA molecules in CLCD particles and the relative limitation of their diffusion mobility; the formation of planar chelate complexes consisting of alternating metal ions and anthracycline molecules under conditions of the phase exclusion of DNA molecules; and the use of copper ions and (or) anthracycline molecules as terminal sites for nanobridges.
Formation of nanobridges was realised according to the following scheme: the CLCD was formed from ds DNA molecules in a PEG-containing solution, according to our technology of mixing, the CD spectrum was checked, DAU solution was added to it up to the formation of an external complex, and then the dispersion was treated with CuCl2 solution. One should bear in mind that the abnormal band in the absorption region of DNA (λ ∼ 270 nm, ) reflects the CLCD ‘quality’: the more native the secondary DNA structure and the more ordered the cholesteric packing of DNA molecules, the stronger this band. The amplitude of this band depends linearly on the DNA concentration: the higher the DNA concentration used in the preparation of the CLCD, the stronger the band. The addition of CuCl2 to the DNA CLCD processed with DAU and having an equilibrium value of the band amplitude at λ ∼ 500 nm, specific to DAU chromophores, leads to an amplification of this band and reflects the optical properties of the chromophore of the [DAU-Cu2+] complex. Under the conditions used (the ds DNA molar mass is 8 × 105 Da and the DNA concentration is 5 μg ml−1), the maximum amplitude of the band at λ ∼ 500 nm is approximately 2500 (in ΔA units).
The prerequisites for formation of nanobridges are the fixed distance between ds DNA molecules in the content of CLCD, determined by the PEG concentration, the planar structure of the [DAU-Cu2+] complex and the sterical ‘phasing’ (displacement and rotation around the long DNA axis) of neighbouring DNA molecules, because the reactive groups are located in the grooves on the surface of the DNA molecules (Citation 78 ). These requirements clearly show that the creation of these nanobridges is a very delicate stereochemical process, which can be realised only under rather strict conditions (Citation 79 ).
The number of Cu2+ ions contained in the nanobridges under conditions used for the formation of the ds DNA CLCD was estimated directly by low-temperature magnetometry (Citation 80 ). If Cu2+ ion forms a chelate complex with four reactive oxygen atoms, this ion is in d 9 state, which exhibits a non-zero magnetic moment. The results obtained by the low-temperature magnetometry (Citation 80 ) and by the theoretical calculations (Citation 77 ) show that the nanobridge [–Cu2+-DAU-Cu2+-…-Cu2+-DAU-Cu2+-] between neighbouring ds DNA molecules contains six copper ions (II) and possesses the structure shown in (b).
Figure 6. The structure of nanobridges between two adjacent ds DNA molecules fixed in the spatial structure of a CLCD particle. (a)Top-view projection of the nanobridge between ds DNA (I) and ds DNA (II) along the long axes of these molecules. (b) Nanobridges between DNA molecules (for clarity, the nanobridges are turned on 90° in respect to their real position). Note that in order to realise the formation of nanobridges between suitable reactive groups located in the DNA grooves on the surfaces of neighbouring molecules, it is necessary to rotate one molecule by 180° about the vertical axis.
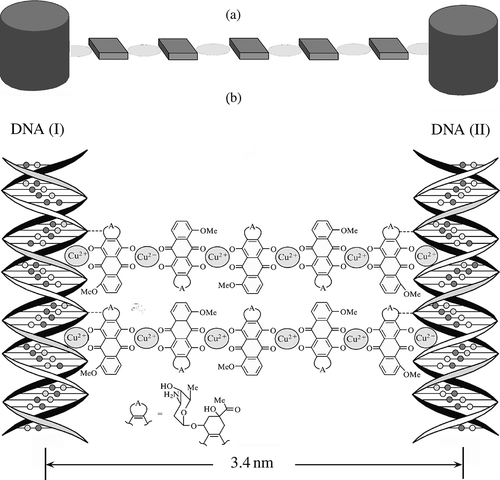
The nanobridges, composed of alternating molecules of anthracycline antibiotic and copper ions, link the DNA molecules () located both in the same and neighbouring quasinematic layers of CLCD particles.
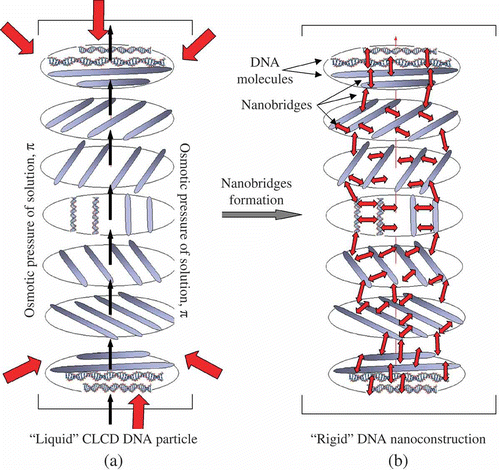
Figure 7. The transition of a ‘liquid’ structure of a DNA CLCD particle into a ‘rigid’ state. π-osmotic pressure of the solution in (a) is shown by red arrows. The ds DNA molecules in the neighbouring layers are shown as rods (a), and each layer is turned by a certain angle relative to the previous layer. The CLCD particle in which ds DNA molecules are ordered exists only under certain osmotic pressure of the solution. The nanobridges in (b) are shown by red double-headed arrows between adjacent ds DNA molecules. Note that the formation of nanobridges does not distort the total spatial structure of the CLCD particle, but this particle can exist under conditions of absence of the osmotic pressure of the solvent.
The efficiency of the formation of nanobridges depends on the concentrations of both DAU molecules and copper ions (Citation 81 ).
The formation of nanobridges between neighbouring ds DNA molecules can be expected to lead to the creation of a spatially fixed structure of the CLCD. The stability of this structure is determined by the number and properties of nanobridges rather than the properties of the initial PEG-containing solution. This means that a new structure would persist even in the absence of the high osmotic pressure of this solution. In this case it appears a possibility to investigate the properties of this structure not only by theoretical, but also by experimental, techniques. For instance, an opportunity is opened to visualise the particles of ds DNA CLCD existing after their transformation into a spatially fixed structure and directly assess the size of the formed structure.
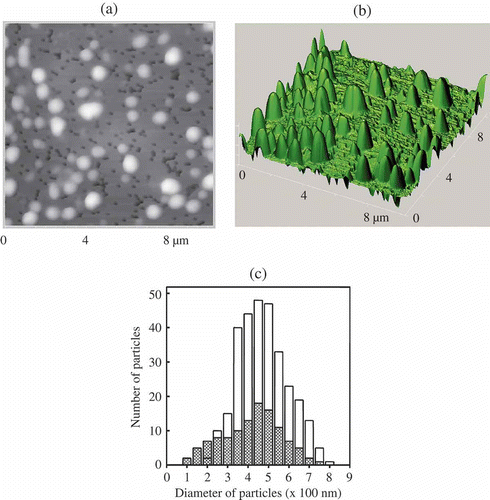
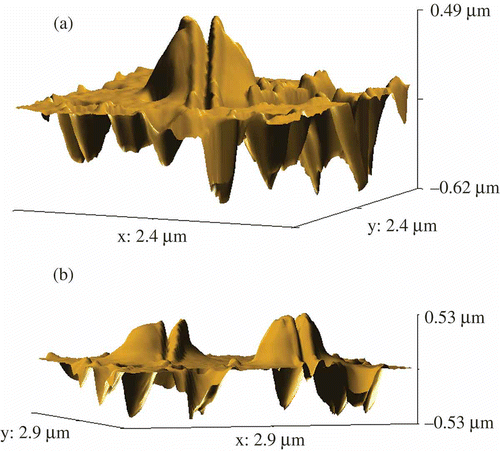
For this purpose, the formed particles were immobilised on the surface of a nuclear membrane filter (Citation 76 , Citation 78 ) and the AFM images of particles were registered ((a) and (b)). The shape of the particles is close to elongated cylinders. Estimation of the size of 400 particles demonstrates that the sizes varied from 0.1–0.2 μm to 0.8 μm with 0.4–0.5 μm as the average ((c)), which is in good agreement with the data on the particle size theoretically calculated for the solutions with a constant osmotic pressure for the initial ds DNA CLCD (Citation 31 ). This means that the initial size of the CLCD particles was not changed in the nanodesign process used. Hence, as a result of the formation of nanobridges between neighbouring ds DNA molecules fixed in the structure of CLCD particles, a new structure of CLCD particles arises, in which the diffusion mobility of neighbouring ds DNA molecules is indeed ‘frozen’. Our international team was first to obtain directly the data characterising the size and shape of artificial structures formed by low-molecular-mass ds DNA molecules.
Figure 8. The AFM images of NaCs formed by ds DNA CLCD particles cross-linked by nanobridges and immobilised onto the surface of the nuclear membrane filter (two-dimensional and 3D images, (a) and (b), respectively); the dark spots are ‘pores’ in the filter, polyethylene terephthalate (PETP); (c) size distribution of the DNA NaCs in two sets of experiments. The images were taken by Smart SPM produced by AIST-NT (Zelenorgad, Russia).
Because the nanobridges can arise both between neighbouring ds DNA of the same quasinematic layer and between ds DNA molecules of neighbouring layers, the formation of nanobridges, in principle, means that, instead of the osmotic pressure of the PEG-containing solution, another factor of the spatial structure stabilisation arises, specifically, the number and stability of nanobridges.
To determine the state of NA molecules in the ds DNA CLCD particles treated with DAU and copper ions, X-ray diffraction analysis of the phases obtained from these particles was performed (Citation 75 ). The X-ray scattering curves contain only one maximum in the range of angles 1° < 2θ < 5°, which corresponds to the average distance between molecules in the CLCD particles. The value of the measured d Bragg for ds DNA molecules fixed in the spatial structure of CLCD particles (i.e. between the ds DNA molecules cross-linked by nanobridges) is 3.01 nm (i.e. d inter is about 3.4 nm). The absence of high-order reflections in the X-ray diffraction patterns indicates the absence of regular three-dimensional (3D) order in the arrangement of molecules of the [DNA-DAU-Cu2+] complexes in CLCD particles; therefore, we can only speak about the existence of short-range order in the arrangement of molecules in these particles.
The high local DNA concentration in the formed ds DNA CLCDs corresponds to the concentration range in the phase diagram of ds DNA, where these molecules are in the cholesteric state (Citation 31 ).
Comparison of the results of X-ray diffraction analysis with the above results of low-temperature magnetometry, as well as the results of AFM studies, shows that the formation of nanobridges between ds DNA molecules does not violate the initial spatial packing of ds DNA molecules; moreover, it leads to its stabilisation.
The formed spatial structure was named rigid NaC (). The created NaC has unique properties. Firstly, unlike the initial CLCD particles, the main factor stabilising NaCs is the number and ‘strength’ of nanobridges rather than the osmotic pressure of the water-salt polymer-containing solution. Secondly, a ‘liquid’ packing pattern of neighbouring DNA molecules in CLCD particles and DNA diffusion mobility disappeared; the structure became rigid, and the particle acquired the properties of a solid material. Thirdly, characteristic of the DNA NaC is not only an abnormal optical activity, reflected as an amplified CD band in the DNA absorption region, but also an amplified band in the absorption region of antibiotic chromophores. Fourthly, the NaC is characterised by two thermal structural transitions: one of them corresponds to the melting of nanobridges and the other to the melting of DNA cholesteric. The temperature of melting (τm) of nanobridges, determined by the temperature alteration of amplitude of the CD band, grows with their concentration, remaining below the τm of the initial ds DNA cholesteric (Citation 81 ). Both the τm value and the shape of melting curves become the function of the number of nanobridges (i.e. their size) and change according to the laws very close to those found for the nanoparticles made of other compounds. Fifthly, the NaC not only retained a high local concentration of DNA molecules (reaching 400 mg ml−1), but also acquired a high concentration of the antitumour antibiotic, DAU. Last but not least, the abnormal optical activity provides for controlling the changes in the secondary structure of initial ds DNA molecules, as well as an appearance of the molecules forming nanobridges in the NaC structure, and the integrity of nanobridges.
In contrast to the initial ‘liquid’ particles of the CLCD, the rigid ds DNA NaCs open a gate for easy nanosized manipulations with these structures (this is a procedure of technical importance). As an example, demonstrates the splitting mode of the individual structures by means of the cantilever of SmartSPM – the scanning probe microscope. The various spatial views of the split rigid ds DNA NaCs are shown in .
Figure 9. The AFM images of initial DNA NaCs (a) and these NaCs split with the help of a microscopic cantilever (b). For conditions see .
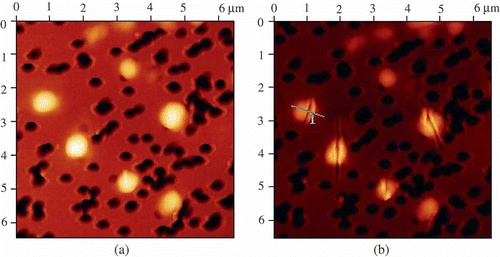
Figure 10. The spatial images of the split rigid DNA NaCs. For conditions see .
Thus, using the first approach ((a)), we have developed technology for producing rigid ds DNA NaCs capable of carrying a high concentration of ‘guests’, i.e. a novel type of biomaterial with controllable properties has been designed. It is quite clear that destroying the integrity of nanobridges between ds DNA molecules () under the action of various chemical or biologically relevant compounds will result in disintegration of the spatial structure of DNA NaCs. This process is accompanied by the decrease in the value of the easily detected CD band located in the region of absorption of the structural elements of nanobridges. This means that ds DNA NaCs by themselves, or being immobilised on the surface optically transparent and optically isotropic nuclear membrane filter, can be used as a microscopic size multifunctional sensing unit (chip) for biological or chemical needs (Citation 82 , Citation 83 ).
In conclusion, one can add that this technology has not yet been fully completed and can be impro-ved by selecting other components for nanobridges or using other DNA molecules, including DNA complexes with synthetic or natural polymers.
3.2 Second approach: the induction of interaction between the ds DNA fragments fixed within quasinematic layers of CLCD particles
Coming back to the structure of the quasinematic layer of ds DNA molecules within the CLCD particle () permits one to pay attention to a number of additional specific features of ds DNA molecules in this layer, which suggested the development of the second approach to the problem of ‘guest’ molecule accumulation.
-
Some chemical substances and BACs, when interacting with nitrogen base pairs in ds DNA molecules in quasinematic layers, can cause local (nanoscale) distortions of the regular ordering of nitrogen bases, leading to inhomogeneity in the ds DNA secondary structure.
-
The steric limitations resulting from both dense packing of ds DNA molecules in quasinematic layers and fixed PEG concentration in solution prevent sterically the separation (denaturation) of the strands of the neighbouring ds DNA molecules. This means that the distortions of the secondary structure caused by the action of various compounds on ds DNA must be ‘transformed’ by this or that way into the changes in the energy of interaction between neighbouring ds DNA molecules (fragments) in quasinematic layers.
-
Positively charged counterions can efficiently neutralise the negative charges of ds DNA molecules in quasinematic layers. Taking into account the polyphosphate nature of DNA molecules, two parameters become important – the efficiency of interaction between counterions and the DNA phosphate groups, carrying negative charges, and the solubility of the (DNA–counterion) complex.
-
At fixed osmotic pressure and ionic strength of a PEG-containing solution, the transition of ds DNA molecules in quasinematic layers into an insoluble state must change the mode of interaction between these molecules. This means that the ds DNA CLCD particles can acquire a rigid structure instead of a ‘liquid’ structure.
-
The theory predicts that the change in the efficiency of interaction between ds DNA molecules in quasinematic layers must be accompanied by changes in the parameters of the spatial helical structure of CLCD particles (in particular, the cholesteric helix pitch, P, value) and, consequently, by change in the abnormal optical activity of CLCD particles.
This allowed us to formulate the fundamental problem as follows: at the action of chemical compounds (‘guests’) on the ds DNA molecules ordered in quasinematic layers of CLCD particles formed in PEG-containing solution under conditions used by us, to decrease the solubility of these molecules so that the interaction between neighbouring ds DNA molecules (or fragments of neighbouring DNA molecules, (b)) would lead to the transition of ds DNA CLCD particles into a rigid state. In this case the created structure would be able to exist in the absence of the osmotic pressure of PEG-containing solution and contain ‘guest’ molecules introduced into the system ((b)).
The solution of this problem will mean that a high concentration of ‘guest’ molecules can be accumulated within a new structure.
It is evident that there are many compounds (counterions) that are able to neutralise the negative charges of phosphate groups in the ds DNA molecules, causing a decrease in their solubility. In particular, these substances are various polycations and multivalence cations. However, polycation molecules can not only neutralise the negative charges of DNA phosphate groups, but also form irregular polycation cross-links between neighbouring ds DNA molecules. In this case, polycations can play the role of a dielectric medium influencing the mode and efficiency of interaction between neighbouring ds DNA molecules (Citation 62 ). In the case of polycations, the combination of these factors leads to very complex dependences not only of the pattern of interaction between ds DNA molecules on the chemical nature and structure of polycations, but also of the value of abnormal optical activity of the produced particles of (DNA–polycation) complexes on these parameters.
Multiple charged cations, such as Al3+ or Fe3+, can neutralise the charges of DNA phosphate groups (Citation84–86); however, they exist in the solution as a set of hydrated forms, causing uncontrollable changes in the orientation of ds DNA base pairs. In addition, their complexes with phosphate groups have high solubility and are poorly formed in solutions of a high ionic strength, used for the phase exclusion of ds DNA molecules.
The cations of rare earth elements are interesting from the standpoint of making the ds DNA molecules in quasinematic layers poorly soluble. Firstly, these cations neutralise the negative charges of DNA phosphate groups under a wide range of conditions, and the complexes of rare earth elements with phosphates are practically insoluble (Citation87–90). Secondly, when interacting with base pairs of linear ds DNA molecules, rare earth cations cause local (nanoscale) changes in the secondary structure of these molecules, which are analogous to the known B→Z transition (Citation 91 , Citation 92 ). Last but not least, some of these cations, in particular, gadolinium, have found a wide practical application (Citation 93 , Citation 94 ). Consequently, we used salts of rare earth elements when elaborating the technology based on the second approach to accumulate ‘guest’ molecules.
We have realised the transition, schematically shown in , by treating the ds DNA CLCD particles formed at high ionic strength of PEG-containing solution with, for instance, gadolinium salts (Citation95–97). It was shown that, in this case, a ‘liquid’ structure of ds DNA CLCD particles transformed into a ‘rigid’ spatial structure.
Figure 11. The transition from ‘liquid’ to ‘rigid’ structure of the particle of the CLCD induced by gadolinium cations. The osmotic pressure of the solution in (a) is shown by red arrows. One can see (structure in (b)), that here the interaction of ds DNA molecules with Gd-cations results in the practical absence of the regular spatial ordering of neighbouring ds DNA molecules in quasinematic layers. A low solubility of [DNA-Gd] complexes is accompanied by a decrease in the solubility of the whole structure. This ‘rigid’ structure can exist in the absence of the osmotic pressure of a solvent.
![Figure 11. The transition from ‘liquid’ to ‘rigid’ structure of the particle of the CLCD induced by gadolinium cations. The osmotic pressure of the solution in (a) is shown by red arrows. One can see (structure in (b)), that here the interaction of ds DNA molecules with Gd-cations results in the practical absence of the regular spatial ordering of neighbouring ds DNA molecules in quasinematic layers. A low solubility of [DNA-Gd] complexes is accompanied by a decrease in the solubility of the whole structure. This ‘rigid’ structure can exist in the absence of the osmotic pressure of a solvent.](/cms/asset/5414f0e7-ff61-491c-b0ee-edb490f3ae08/tlcy_a_549300_o_f0011g.jpg)
According to the magnetometric and neutron activation analyses, the particles of a new structure contain a high concentration of gadolinium ions (Citation 95 ). These results demonstrate that these particles contain approximately one gadolinium atom per one phosphate group of the ds DNA molecule in the quasinematic layer (under saturation conditions). The gadolinium ions neutralising the negative charges of the phosphate groups of the ds DNAs make particles of the CLCD insoluble. Indeed, in the presence of a large excess of gadolinium ions, these ions, displacing sodium ions, are bonded to the phosphate groups of the ds DNAs. It is worth noting that, when Gd3+ ions are bonded to polyphosphates, poorly soluble Gd-polyphosphate is formed (solubility constant is equal to about 10−12 M) (Citation87–90, Citation 98 ).
Since ds DNA molecules have a ‘polyphosphate’ origin, these molecules in the presence of saturating gadolinium concentrations become poorly soluble in PEG-salt-aqueous solutions. Moreover, gadolinium ions, neutralising the charges of the phosphate groups of the ds DNAs create the residual (non-compensated) positive surface charge on particles of the CLCD and aggregation of these particles becomes impossible. In addition, an appearance of the surface charge on the CLCD particles can result in the stabilisation of the spatial structure of these particles after their treatment with GdCl3. This means that, as a result of interaction of gadolinium ions with the ds DNA in the content of the CLCD particles, these particles can lose their ability to coalescence. Particles of the CLCD lose their solubility and can sediment in the solution. The sedimentation is accompanied by a decrease in the amplitude of the abnormal band in the CD spectrum. However, the intense shaking of the solution containing the resulting sediment leads to the complete recovery of the initial abnormal optical activity, i.e. the aggregation of neighbouring particles of the CLCD of the DNA-Gd complex does not occur. The tenfold dilution of the CLCD formed in the PEG-containing solution (CPEG = 170 mg ml−1) of the DNA-Gd complex does not lead to change in the abnormal optical properties of the dispersion (with allowance for the decrease in the concentration of particles of the CLCD upon such dilution).
In addition, the inhomogeneous chemical nature of the nitrogen bases in ds DNA molecules leads to the fact that the interaction between gadolinium cations and ds DNA molecules is accompanied by nanoscale conformational changes (similar to the changes that appeared at the B→Z transition) (Citation 91 , Citation 92 ) only in the fragments of these molecules. Under these conditions, a homogeneity of the secondary structure of the ds DNA molecules in the quasinematic layers is impaired, which, in turn, is manifested as the decrease (to its complete disappearance) in the intensity of the peak in the small-angle X-ray scattering curves as the ds DNA CLCD particles are treated by gadolinium salts (Citation 96 , Citation 97 ). Because ds DNA molecules cannot ‘leave’ the physical volume of CLCD particles, due to the fixed osmotic pressure of the PEG-containing solution, the loss of solubility of individual neighbouring ds DNA molecules, combined with an increase in the interaction between their fragments with different conformations (Citation 91 , Citation 92 ), can initiate the transition of the overall structure of CLCD particles from a ‘liquid’ to a ‘rigid’ state (Citation 97 ).
Thus, particles of the CLCD of the ds DNAs whose phosphate groups are neutralised by gadolinium ions become poorly soluble and can exist in the absence of the high osmotic pressure of the PEG-containing solution. This mean that the presence of PEG is not required in order to stabilise the structure of the particles of the CLCD and the osmotic pressure of the water-salt solution is sufficient for supporting the cholesteric packing of molecules of the DNA–gadolinium complexes in particles of the CLCD. Such particles can be immobilised on a membrane filter, and their shape can be visualised after washing with water to prevent NaCl crystallisation and/or PEG film formation.
The combination of these specific features opens the possibility for easy immobilisation of the formed rigid particles onto the surface of a nuclear membrane filter and their visualisation by AFM. shows the AFM images of CLCD particles from ds (DNA-Gd3+) complexes immobilised onto the surface of a nuclear membrane filter. It is evident that the CLCD particles of the (DNA-Gd3+) complex exist as independent individual objects, which are easy to visualise and manipulate. The existence of independent particles confirms the presence of the uncompensated surface charge on the CLCD particles, which prevents their aggregation. The visualisation of individual particles demonstrate that, in full agreement with the scheme shown in (b), a ‘liquid’ character of the ds DNA molecules packing in the ds DNA CLCD particles disappeared after they were treated by GdCl3 solution, and the particles acquire ‘rigid’ spatial structure. The average size (0.5 μm) of CLCD particles from the ds (DNA-Gd3+) complex () practically coincides with the theoretically estimated size of the CLCD particles from the initial ds DNA molecules (Citation 31 , Citation 32 ). This result demonstrates that the mean packing density of DNA molecules in the CLCD particles from the ds (DNA-Gd3+) complex is sufficiently close to the packing density of the ds DNA molecules in the CLCD particles formed from the initial molecules. The latter fact is quite important, because it means that the mean DNA packing density in the LC particles is not changed by Gd cation incorporation, i.e. the ‘chromophore’(nitrogen base) density in the quasinematic layers of CLCD particles of the ds (DNA-Gd3+) complex remains high enough to sustain their abnormal optical activity.
Figure 12. AFM images of the particles of CLCD formed by (DNA-Gd3+) complexes and immobilised onto the surface of the nuclear membrane filter. CDNA= 1.07 μg ml−1; CNaCl = 0.03 M; CPEG =17 mg ml−1; CGdCl3 = 0.23 mM; the dark spots are ‘pores’ in the nuclear membrane filter; two sites on the PETP filter are shown.

Figure 13. Size distribution of the ds DNA NaCs formed by (DNA-Gd3+) complexes (1) and the pores (2) in the membrane filter. For conditions see .
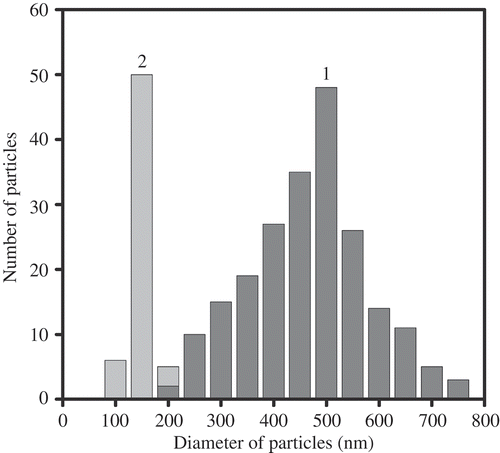
Taking into account the previous points, the alteration in the interaction mode of the ds DNA molecules (or ds DNA fragments) in quasinematic layers is accompanied by the decrease in the helical pitch, P, of the spatial structure of ds DNA CLCD particles (Citation 95 , Citation 96 ) and determines a drastic increase in the amplitude of an abnormal negative CD band in the absorption region of DNA nitrogen bases, when the CLCD particles are transformed into their ‘rigid’ state.
The hypothetical structure of such a ‘rigid’ particle is shown in . The above-shown specific features of the ds DNA CLCD particles treated by the salts of rare earth elements mean that, by applying the second approach ((b)), we succeeded in creating the ds DNA NaCs that are capable of carrying a high concentration of ‘guest’ molecules (in the considered case, gadolinium ions). Consequently, the ds DNA NaCs saturated with high concentrations of rare earth elements have been formed; this opens the possibilities for their practical application. Indeed, the preliminary experiments demonstrated high efficiency of the NaCs of the (DNA-Gd3+) complex for destroying living cells under action of the thermal neutron flux (Citation 95 , Citation 97 ).
Thus, the results presented in this review demonstrate how the fundamental data obtained when studying the physico-chemical properties of the ds DNA CLCDs and the specific properties of these molecules in quasinematic layers have been used for the design of rigid ds DNA NaCs carrying ‘guest’ molecules and displaying unique properties.
Note that the above-described approaches to the nanodesign based on the use of the ds DNA CLCD particles are far from being completed, as many questions yet remain unanswered and the approaches to nanodesign themselves can be modified taking into account specific application problems. In particular, other chemical or BACs or their complexes can be used for the formation of nanobridges. It cannot also be excluded that the surfaces of the ds DNA molecules can be chemically modified to bring a new reactivity on them, thereby opening the way for designing a whole spectrum of biomaterials.
4. Conclusions
Independent of the technologies used for the design, the formed NaCs involving ds NA (DNA and ribonucleic acid (RNA)) molecules can find application in various fields of science and technology.
-
The NaCs formed by ds DNA CLCD particles, where the ds DNA concentration exceeding 200 mg ml−1, can be used as carriers of genetic material or various BACs and chemical substances introduced into these structures (application fields: medicine and biotechnology).
-
The NaCs formed by the CLCD particles of (DNA-polycation) complexes are sensing units of optical biosensors proving for the detection of many BACs in various media (application fields: medicine, biotechnology and ecology).
-
The NaCs with controllable physico-chemical properties incorporated into polymeric films (hydrogels) can be used in technology (in particular, as molecular sieves, optical filters, and so on) (application fields: optics and electronics).
-
The results presented in this review clearly demonstrate that, in the space available between ds molecules ordered in the CLCD particles, one can fix the chemical or BACs. In addition, one can suppose that nanoparticles with diameters of about 2–4 nm (for instance, Ag or Au-nanoparticles) can be fixed in this space as well. In this last case it is not excluded that a possibility for obtaining nanoclusters (nanorods) from these particles with tailored physico-chemical properties (for instance, with regulated surface plasmon resonance) will appear (application fields: optics and electronics).
Acknowledgements
I am greatly indebted to my collaborators and colleagues from the Institute of Molecular Biology of the RAS, from the Institute of Crystallography of the RAS, from the Institute of Spectroscopy of the RAS and the other academy institutions, as well as to our colleagues from Padova University (Italy) and Muenster University (Germany), who took part at various stages of the investigations of NaCs, for their stimulating discussions and critical remarks.
The work was supported financially (in part) by the grants from the Russian Foundation for Basic Research (project no 08-04-12010) and the State Contract no.02.512.11.2217.
References
- Niemeyer , C.M. and Mirkin , C.A. , eds. 2004 . Nanotechnology. Concept, Application and Perspectives , Wiley-VCH : Weinheim .
- Yevdokimov , Yu.M. and Sychev , V.V. 2008 . Russian Chem. Rev. , 77 : 194 – 206 .
- Seeman , N.C. 1982 . J. Theor. Biol. , 99 : 237 – 247 .
- Yevdokimov , Yu.M. , Salyanov , V.I. and Skuridin , S.G. 2009 . Mol. Biol. , 43 : 284 – 300 .
- Seeman , N.C. 2004 . Sci. Am. , 6 : 64 – 69 .
- Shin , J. and Bergstrom , D.E. 1997 . Angew. Chem., Int. Ed. , 36 : 111 – 113 .
- Winfree , E. , Liu , F. , Wenzer , L.A. and Seeman , N.C. 1998 . Nature (London, UK) , 394 : 539 – 544 .
- Yang , X. , Wenzer , L.A. , Qi , J. , Li , X. and Seeman , N.C. 1998 . J. Am. Chem. Soc. , 120 : 9779 – 9786 .
- Scheffel , M. , Dorenbeck , A. , Jordan , S. , Wustefeld , M. and von Kiedrowski , G. 1999 . Angew. Chem., Int. Ed. , 38 : 3312 – 3315 .
- La Bean , T.H. , Yan , H. , Kopatsch , J. , Liu , F. , Winfree , E. , Reif , J.H. and Seeman , N.C. 2000 . J. Am. Chem. Soc. , 122 : 1848 – 1860 .
- Yan , H. , LaBean , T.H. , Feng , L.P. and Reif , J.H. 2003 . Proc. Natl. Acad. Sci. USA , 100 : 8103 – 8108 .
- Shin , W.M. , Quispe , J.D. and Joyce , G.F. 2004 . Nature (London, UK) , 427 : 618 – 621 .
- Gothelf , K.V. , Thomsen , A. , Nielsen , M. , Ci , E. and Brown , R. 2004 . J. Am. Chem. Soc. , 126 : 1044 – 1046 .
- He , Y. , Chen , Y. , Liu , H. , Ribbe , A. and Mao , C. 2005 . J. Am. Chem. Soc. , 127 : 12202 – 12203 .
- Rothemund , P.W. 2006 . Nature (London, UK) , 440 : 297 – 302 .
- Seeman , N.C. 2007 . Mol. Biotechnol. , 37 : 246 – 257 .
- He , Y. , Ye , T. , Su , M. , Zhang , C. , Ribbe , A. , Jiang , W. and Mao , C. 2008 . Nature (London, UK) , 452 : 198 – 201 .
- Robinson , K. 1961 . Tetrahedron , 13 : 219 – 234 .
- Iizuka , I. 1977 . Polymer J. , 9 : 173 – 180 .
- Rill , R.L. , Hilliard , P.R. and Levy , G.C. 1983 . J. Biol. Chem. , 258 : 250 – 256 .
- Brandes , R. and Kearns , D.R. 1986 . Biochemistry , 25 : 5890 – 5895 .
- Potaman , V.N. , Alexeev , D.G. , Skuratovskii , I.Ya. , Rabinovich , A.Z. and Shlyakhtenko , L.S. 1981 . Nucleic Acids Res , 9 : 55 – 64 .
- Livolant , F. and Bouligand , Y. 1986 . J. Phys. , 47 : 1813 – 1827 .
- Livolant , F. 1987 . J. Phys. , 48 : 1051 – 1066 .
- Strzelecka , T.E. and Rill , R.L. 1987 . J. Am. Chem. Soc. , 109 : 4513 – 4518 .
- Yevdokimov , Yu.M. , Skuridin , S.G. and Salyanov , V.I. 1988 . Liq. Cryst. , 3 : 1443 – 1459 .
- Rill , R.L. , Strzelecka , T.E. , Davidson , M.W. and van Winkle , D.H. 1991 . Phys. A , 176 : 87 – 116 .
- Durand , D. , Doucet , J. and Livolant , F. 1992 . J. Phys. , 2 : 1769 – 1783 .
- Leforestier , A. and Livolant , F. 1993 . Biophys. J. , 65 : 56 – 72 .
- Livolant , F. and Leforestier , A. 1996 . Prog. Polym. Sci. , 21 : 1115 – 1164 .
- Yevdokimov , Yu.M. , Skuridin , S.G. and Lortkipanidze , G.B. 1992 . Liq. Cryst. , 12 : 1 – 16 .
- Yevdokimov , Yu.M. , Salyanov , V.I. , Semenov , C.V. and Skuridin , S.G. 2008 . Zhidkokristallicheskie dispersii i nanokonstruktsii DNK (Liquid Crystalline Dispersions and DNA-Based Nanoconstructions) , Edited by: Yevdokimov , Yu.M. 296 Moscow : Radiotekhnika .
- Yevdokimov , Yu.M. , Salyanov , V.I. and Semenov , S.V. 1996 . Biosens. Bioelectron. , 11 : 889 – 901 .
- Skuridin , S.G. , Yevdokimov , Yu.M. and Efimov , V.S. 1996 . Biosens. Bioelectron. , 11 : 903 – 911 .
- Skuridin , S.G. and Yevdokimov , Yu.M . 2004 . Biophysics , 49 : 445 – 462 .
- Lerman , L.S. 1971 . Proc. Natl. Acad. Sci. USA. , 68 : 1886 – 1890 .
- Yevdokimov , Yu.M. , Platonov , A.L. , Tikhonenko , A.S. and Varshavsky , Ya.M . 1972 . FEBS Lett. , 23 : 180 – 184 .
- Akimenko , N.M. , Dijakova , E.B. , Yevdokimov , Yu.M. , Frisman , E.V. and Varshavsky , Ya.M . 1973 . FEBS Lett. , 38 : 61 – 63 .
- Maniatis , T. , Venable , J.H. and Lerman , L.S. 1974 . J. Mol. Biol. , 84 : 37 – 64 .
- Kassapidou , K. , Jesse , W. , van Dijk , J.F. and van der Maarel , J.R. 1998 . Biopolymers , 46 : 31 – 37 .
- Salyanov , V.I. , Pogrebnyak , V.G. , Skuridin , S.G. , Lortkipanidze , G.B. , Chidzhavadze , Z.G. , Toryanik , A.I. and Yevdokimov , Yu.M . 1978 . Mol. Biol. , 12 : 367 – 375 .
- Livolant , F. 1984 . Eur. J. Cell Biol. , 33 : 300 – 311 .
- Livolant , F. 1991 . J. Mol. Biol. , 218 : 165 – 181 .
- Livolant , F. 1991 . Phys. A , 176 : 117 – 137 .
- Rill , R.L. , Livolant , F. , Aldrich , H.C. and Davidson , M.W. 1989 . Chromosoma , 98 : 280 – 286 .
- Bouligand , Y. and Norris , V. 2001 . Biochimie , 83 : 187 – 192 .
- Sartori , B.N. , Senn , A. , Leforestier , A. , Livolant , F. and Dubochet , J. 2001 . J. Struct. Biol. , 34 : 76 – 81 .
- Tinoko , I. , Bustamante , C. and Maestre , M.F. 1980 . Annu. Rev. Biophys. Bioeng. , 9 : 107 – 146 .
- Livolant , F. and Maestre , M.F. 1988 . Biochemistry , 27 : 3056 – 3068 .
- Gottarelli , G. and Spada , G.P. 1994 . Circular Dichroism: Principles and Applications , Edited by: Nakanishi , K. , Berova , N. and Woody , R.W. 105 – 119 . New York : VCH .
- De Vries , H. 1951 . Acta Cryst. , 4 : 219 – 226 .
- Saeva , F.D. , Sharpe , P.E. and Olin , G.R. 1973 . J. Am. Chem. Soc. , 95 : 7656 – 7659 .
- Maestre , M.F. and Reich , C. 1980 . Biochemistry , 19 : 5214 – 5223 .
- Keller , D. and Bustamante , C. 1986 . J. Chem. Phys. , 84 : 2961 – 2971 .
- Keller , D. and Bustamante , C. 1986 . J. Chem. Phys. , 84 : 2972 – 2980 .
- Kim , M.-H. , Ulibarri , L. , Keller , D. and Bustamante , C. 1986 . J. Chem. Phys. , 84 : 2981 – 2989 .
- Belyakov , V.A. , Orlov , V.P. , Semenov , S.V. , Skuridin , S.G. and Yevdokimov , Yu.M . 1996 . Liq. Cryst. , 20 : 777 – 784 .
- Yevdokimov , Yu.M. , Golo , V.L. , Salyanov , V.I. , Lortkipanidze , G.B. and Kats , E.I. 2000 . Biophysics , 45 : 998 – 1006 .
- Spada , G.P. , Brigidi , P. and Gottarelli , G. 1988 . J. Chem. Soc., Chem. Commun. , 14 : 953 – 954 .
- Piraccini , S. , Gottarelli , G. , Mariani , P. and Spada , G.P. 2001 . Chirality , 6 : 3249 – 3253 .
- Yevdokimov , Yu.M. , Skuridin , S.G. , Semenov , S.V. , Salyanov , V.I. and Lortkipanidze , G.B. 1998 . Biophysics , 43 : 215 – 229 .
- Yevdokimov , Yu.M. and Salyanov , V.I. 2003 . Liq. Cryst. , 30 : 1057 – 1074 .
- Parsegian , V.A. , Rand , R.P. , Fuller , N.I. and Rau , D.C. 1986 . Methods Enzymol. , 127 : 400 – 416 .
- Podgornik , R. , Strey , H.H. , Rau , D.C. and Parcegian , V.A. 1996 . Biophys. Chem. , 57 : 111 – 121 .
- Podgornik , R. , Strey , H.H. , Gawrish , K. , Rau , D.C. , Rupprecht , A. and Parcegian , V.A. 1996 . Proc. Natl. Acad. Sci. USA. , 40 : 4261 – 4266 .
- Odijk , T. 1998 . Biophys. Chem. , 73 : 23 – 29 .
- Livolant , F. , Levelut , A.M. , Doucet , J. and Benoit , J.P. 1989 . Nature (London, UK) , 339 : 724 – 726 .
- Stanley , C.B. , Hong , H. and Strey , H.H. 2005 . Biophys. J. , 89 : 2552 – 2557 .
- Leonard , M. , Hong , H. , Easwar , N. and Strey , H.H. 2001 . Polymer , 42 : 5823 – 5827 .
- Goldar , A. , Thomson , H. and Seddon , J.M. 2008 . J. Phys.: Condens. Matter , 20 : 1 – 9 .
- Grosberg , A.Yu. , Erukhimovich , I.Ya. and Shakhnovich , A.I. 1982 . Biopolymers , 21 : 2414 – 2432 .
- Yevdokimov , Yu.M. , Salyanov , V.I. and Palumbo , M. 1985 . Mol. Cryst. Liq. Cryst. , 131 : 285 – 297 .
- Yevdokimov , Yu.M. , Salyanov , V.I. , Gedig , E. and Spener , F. 1996 . FEBS Lett. , 393 : 269 – 273 .
- Yevdokimov , Yu.M. , Salyanov , V.I. , Spener , F. and Palumbo , M. 1996 . Int. J. Biol. Macromol. , 19 : 247 – 255 .
- Yevdokimov , Yu.M. , Salyanov , V.I. , Buligin , L.V. , Dembo , A.T. , Grdig , E. , Spener , F. and Palumbo , M. 1997 . J. Biomol. Struct. Dyn. , 15 : 97 – 105 .
- Yevdokimov , Yu.M. , Salyanov , V.I. , Mchedlishvili , B.V. , Bykov , V.A. , Belayev , A.V. , Saunin , S.A. , Spener , F. and Palumbo , M. 2000 . 19 : 1355 – 1365 .
- Nechipurenko , Yu.D. , Ryabokon , V.F. , Semenov , S.V. and Yevdokimov , Yu.M . 2003 . Biophysics , 48 : 594 – 601 .
- Yevdokimov , Yu.M. , Skuridin , S.G. , Nechipurenko , Yu.D. , Zakharov , M.A. , Salyanov , V.I. , Kurnosov , A.A. , Kuznetsov , V.D. and Nikiforov , V.N. 2005 . Int. J. Biol. Macromol. , 36 : 103 – 115 .
- Yevdokimov , Yu. M. and Sitchev , V.V. 2007 . Open Nanosci. Rev. , 1 : 1 – 13 .
- Nikiforov , V.N. , Kuznetsov , V.D. , Nechipurenko , Yu.D. , Salyanov , V.I. and Yevdokimov , Yu.M . 2005 . JETP Lett. , 81 : 264 – 266 .
- Zakharov , M.A. , Sokolovkaya , L.G. , Nechipurenko , Yu.D. , Lortkipanidze , G.B. and Yevdokimov , Yu.M . 2005 . Biophysics , 50 : 721 – 727 .
- Yevdokimov , Yu.M . 2000 . Biochem. Soc. Trans. , 28 : 77 – 81 .
- Yevdokimov , Yu.M. , Salyanov , V.I. and Zakharov , M.A. 2001 . Lab Chip , 1 : 35 – 41 .
- Karlik , S.J. , Eichhorn , G.L. , Lewis , P.N. and Crapper , D.R. 1980 . Biochemistry , 19 : 5991 – 5998 .
- Wedrychowski , A. , Schmidt , W.N. and Hlinica , L.S. 1986 . J. Biol. Chem. , 261 : 3370 – 3376 .
- Corain , B. , Bombi , G.G. and Zatta , P. 1988 . Neurobiol. Aging , 9 : 413 – 414 .
- Mumper , R.J. and Jay , M. 1992 . J. Phys. Chem. , 96 : 8626 – 8631 .
- Qi , Y.-H. , Zhang , Q.-Y. and Xu , L. 2002 . J. Chem. Inf. Comput. Sci. , 42 : 1471 – 1475 .
- Lessing , P.A. and Erickson , A.W. 2003 . J. Eur. Ceram. Soc. , 23 : 3049 – 3057 .
- Zhang , P. and Kimura , T. 2006 . Solvent Extr. Ion Exch. , 24 : 146 – 163 .
- Gersanovsky , D. , Colson , P. , Houssier , C. and Fredericq , E. 1985 . Biochim. Biophys. Acta , 35 : 313 – 325 .
- Ha , S.C. , Lowenhaupt , K. , Rich , A. , Kim , Y. and Kim , K.K. 2005 . Nature (London, UK) , 437 : 1183 – 1186 .
- Shih , J.-L.A. and Brugger , R.M. 1992 . Med. Phys. , 19 : 733 – 744 .
- Martin , R.F. , D'Cunha , G. , Pardee , M. and Allen , B.J. 1988 . Int. J. Radiat. Res. , 54 : 205 – 208 .
- Yevdokimov , Yu.M. , Salyanov , V.I. , Kondrashina , O.V. , Borshevsky , V.I. , Semenov , S.V. , Gasanov , A.A. , Reshetov , I.V. , Kuznetsov , V.D. , Nikiforov , V.N. , Akulinichev , S.V. , Mordovskoi , M.V. , Potashev , S.I. and Skorkin , V.M. 2005 . Int. J. Biol. Macromol. , 37 : 165 – 173 .
- Yevdokimov , Yu.M. , Salyanov , V.I. , Kondrashina , O.V. , Gasanov , A.A. , Shtykova , E.V. and Dembo , K.A. 2007 . JETP , 104 : 499 – 507 .
- Yevdokimov , Yu.M. , Salyanov , V.I. , Shtykova , E.V. , Dembo , K.A. , Volkov , V.V. , Spirin , P.V. , Slusheva , A.S. and Prassolov , V.S. 2008 . Open Nanosci. J. , 2 : 17 – 28 .
- Li , L. , Yang , J. , Wu , X. , Sun , Ch. and Zhou , G. 2003 . J. Lumin. , 101 : 141 – 146 .