
Abstract
Objective
The molecular basis of prostate cancer is highly heterogeneous. Our study aimed to perform the mutation analysis of KRAS, BRAF, PIK3CA, and immunohistochemical (IHC) evaluation of EGFR, HER2, p16, and PTEN to demonstrate new areas for targeted therapies.
Methods
A total of 24 prostatectomy samples diagnosed with adenocarcinoma were analyzed by microarray hybridization. Also, these samples were IHC stained for EGFR, HER2, P16, and PTEN. The cases were divided into two groups based on low and high Gleason scores. All findings were compared with the clinicopathological parameters of the patients.
Results
While KRAS mutation was in 3/24 (12.5%) of our cases, BRAF and PIK3CA mutations were not detected. There was no significant difference between the groups in terms of KRAS mutation frequency. HER2 was immunohistochemically negative in all samples. There was no correlation between EGFR, P16 immunopositivity, and clinicopathological features.
Conclusion
KRAS mutation frequency is similar to those in Asian populations. BRAF and PIK3CA mutation frequencies have been reported in the literature in the range of 0–15% and 0–10.4%, respectively, consistent with our study findings. HER2 immunoexpression is a controversial issue in the literature. EGFR and p16 expressions may not correlate with the stage.
Keywords:
Introduction
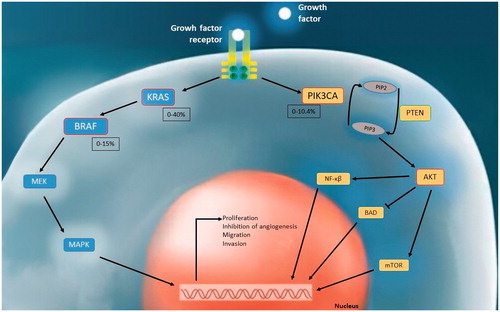
Prostate cancer (PCa) is the second most common malignancy in males except for non-melanoma skin cancer in the world. It is the fifth cause of cancer deaths worldwide [Citation1]. Although localized PCa patients benefit from androgen ablation therapy, the disease may turn into an androgen-independent state and, the risk of metastasis-recurrence increases in advanced stages. Therefore, a better understanding of the molecular basis of PCa is required to create new treatment areas. Oncogenic mutations affecting signal transduction pathways in the cell are directly related to carcinogenesis. KRAS/MAPK and PI3K/AKT pathways are the most important mechanisms in intracellular signal transduction. Molecules and genes in these pathways at different phases have been shown as targets in cancer pathogenesis. In this context, mutations of the BRAF, KRAS, and PIK3CA genes were identified with varying frequencies in many cancer types [Citation2–10]. BRAF and PI3K inhibitors have taken place in treatment schemes at various stages; studies for KRAS still continue at changing clinical and laboratory stages [Citation11–13]. In PCa, no targeted gene therapy has yet been approved [Citation14]. KRAS and BRAF mutation frequencies in PCa were reported in up to 40% and 15% in the literature, respectively [Citation15–63]. The PI3K/AKT signaling pathway is frequently affected in PCa. Deregulations in this pathway may base on different causes such as PIK3CA mutations, PIK3CA amplifications, or deletions of PTEN, an inhibitor of the pathway. PIK3CA mutations have been reported in up to 10.4% of PCa in various studies [Citation29,Citation31,Citation33,Citation38,Citation42–52,Citation57,Citation64–76].
EGFR (ErbB1) is crucial for stimulating both the RAS/MAPK and PI3K/AKT signaling pathway. While EGFR is defined especially in castration-resistant PCa, HER2 (ErbB2) has yielded conflicting results [Citation77–79]. Although there have been targeted therapy studies for ErbB receptors in PCa, it also brings problems like target therapy resistance [Citation80–81]. In patients with targeted therapy resistance to ErbB receptors, signaling pathway actors in the later stages of signalling, such as RAS/MAPK, may be actual targets in the treatment approach. Besides, RAS/MAPK and PI3K/AKT pathways are known to interact with the CDK/Cyclin D complex inhibited by p16 [Citation82]. Therefore, identifying the expressions of EGFR, HER2, p16, and PTEN together with the mutations in these pathways will guide in understanding PCa carcinogenesis.
In this study, we analyzed the genetic alterations of KRAS, BRAF, and PI3KCA mutations. We compared the genetic findings, immunoexpression profiles with clinical features to determine diagnostically relevant alterations and compared these findings with those of other studies.
Methods
Tissue samples
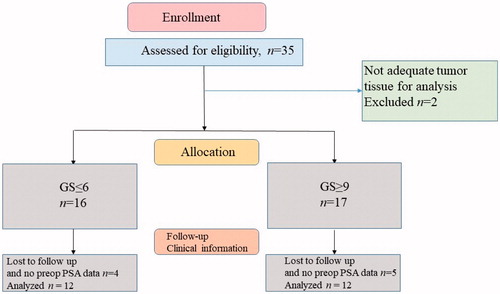
The study was performed based on the tenets of the Helsinki Declaration and according to approval by the local Ethics Committee of the Duzce University, School of Medicine (protocol number: 2018/93). It was also supported financially by the SRP unit (Scientific Research Projects: 2018.04.02.796) of Duzce University. Thirty-five radical prostatectomy samples diagnosed with PCa in our department between 2012–2018 were evaluated. The samples that did not contain sufficient tumoral tissue for genetic evaluation and the cases without clinical information were excluded from the study (). A total of 24 cases were subdivided into two groups as Gleason score = 6 (Grade Group 1, GS-L) and Gleason score ≥9 (Grade Group 5, GS-H). Age, preoperative PSA level, Gleason score, stage, surgical margin positivity, extraprostatic extension, lymphovascular invasion, perineural invasion, family history, and biochemical recurrence status of selected cases were compiled. Formalin-fixed paraffin-embedded (FFPE) blocks that included the most adequate tumoral tissue were selected for the PCR and IHC method. FFPE blocks that had large tumor tissue and no necrosis or bleeding were preferred.
Figure 1. Selection of cases.
Detection of mutations
Tumoral tissue of 5–10 µm from the selected FFPE blocks was manually removed from the surrounding intact tissue elements by macrodissection. Following the QIAamp DNA FFPE Tissue Kit (Qiagen, Germany) protocol, deparaffinization and DNA extraction were performed. After extraction, DNA values were measured with DNA-50 value in Colibri Nanodrop device. The PCR process was performed on the VERITI device, following the Multiplex PCR protocol of the KRAS/BRAF/PIK3CA Array (RANDOX) kit. Microarray Hybridization, imaging, and result acquisition procedures of PCR products were performed in Evidence Investigator (RANDOX) device by following the Microarray Hybridization, imaging, and result acquisition protocols of EV3799A/B KRAS/BRAF/PIK3CA Array kit. In this study, 20 different point mutations on the KRAS (codon 12, codon 13, codon 61, codon 146), BRAF (V600E), and PIK3CA (codon 542, codon 545, codon 1047) genes were analyzed at the same time.
Immunohistochemical analysis
Five-micrometer sections were taken on positively charged slides. The sections were deparaffinized in a 75-degree oven for 1 h. After deparaffinization, tissues stained with EGFR (Genemed Biotechnologies, clone 31G7), Cerb-B2 (Biocare Medical, clone EP1045Y), p16 (Biocare Medical, clone G175-405), and PTEN (Thermo, clone 17.A) antibodies by Ventana Benchmark XT brand automatic IHC stainer.
IHC evaluation
IHC staining intensity was scored between 0 and 3+ for P16 and EGFR. It was scored as 0 (no staining), 1+ (weak), 2+ (medium) and 3+ (strong). Also the degree of staining was scored between 0 and 3 according to the percentage of stained cells 0, <30%, 30%–60%, > 60% for P16; 0, <10%, 10%–50%, > 50% for EGFR, as previously reported [Citation83–84]. The cases were scored between 0 and 9 by obtaining a multiplicative staining score. A range of 7 to 9 was considered strong staining, a range of 4 to 6 was moderate staining, a range of 1 to 3 was considered weak staining.
Statistical analysis
Appropriate descriptive statistics (mean, standard deviation, minimum, maximum, percentage) of all data in the study were calculated. Relationships between categorical variables were examined using the Pearson chi-square, Fisher Freeman Halton (post hoc Bonferroni test), and Fisher exact tests. To examine the relationships between variables, contingency (rc) coefficient, point biserial (rp), and Kendall tau b (rk) correlation coefficients were calculated according to the types of variables. SPSS 22.0 (Statistical Package for the Social Sciences) program and special syntaxes were used for statistical evaluations. A p-value of < .05 was considered statistically significant.
Results
Twelve of the 24 individuals had a Gleason score of ≤ 6 (GS-L), while 12 had a Gleason score of ≥ 9 (GS-H). The mean age of the patients was 65.4 ± 7.9 years (Min-Max: 43–79), and the preoperative mean PSA value was 6.4 ± 3.1 (Min-Max: 0.48–13.32).
A statistically significant difference was found between GS-L and GS-H groups in terms of pT (p = .005). Stages and TNM status are presented in . There was no significant difference between the lymph node metastasis status and the rates of distant metastasis according to the groups (p = .999 for each). According to the groups, there was no significant difference between the rates of vascular invasion (p = .590). The rate of perineural invasion (33.3%) in individuals in the GS-L group was significantly lower than the rate of perineural invasion in individuals in the GS-H group (83.3%) (p = .036). While no periprostatic extension was observed in GS-L individuals, there was a periprostatic extension in 58.3% of GS-H individuals (p = .005). There was a significant difference between the rates of seminal vesicle invasion according to the groups (p = .014). No significant difference was found between the rates of surgical margin positivity, biochemical recurrence, and family history according to the groups (p = .214, p = .590, p = .193, respectively).
Table 1. Stage and TNM status of patients according to the groups.
KRAS mutation was detected in 3/24 (12.5%) of our PCa cases, while BRAF and PIK3CA mutations were not found in any of the cases. 2 of the KRAS mutations were at codon 12, and 1 of them was at codon 61 localization. All cases with KRAS mutations were Gleason score 3 + 3 = 6 and their pathological stages were pT2. While lymph node involvement, distant metastasis, vascular invasion and periprostatic extension were not observed in 3 of these 3 cases, perineural invasion was detected in 1 case. There was no significant difference between the rates of KRAS mutation according to the groups. In addition, there were no correlation between KRAS mutation and age (rp = −.249 p = .240), preoperative PSA (rp = −.359 p = .085), Gleason grade (rk = −.364 p = .075), pT (rk = −.233 p = .250), stage (rk = −.381 p = .050), lymph node metastasis (N) (rc = .141 p = .999), distant metastasis (M) (rc = .113 p = .999), vascular invasion (rc = .167 p = .999), perineural invasion (rc = .188 p = .550), periprostatic extension (rc = .236 p = .530), seminal vesicle invasion (rc = .213 p = .546), surgical margin positivity (rc = .188 p = .550), biochemical recurrence (rc = .167 p =.999) and family history (rc =.292 p = .209).
Immunohistochemically, there was no staining for HER2 in either group (). While most (62.5%) of the cases showed weak staining for EGFR, 8 cases (33.4%) had a moderate or strong expression (). p16 were immunopositive for 83.3% of cases (). There was no significant difference in EGFR and P16 immunopositivity between the groups. Detailed IHC staining and mutation positivity status in the GS-L and GS-H groups are shown in . PTEN analysis was not performed for technical reasons.
Figure 2. (A) Weak staining with EGFR in neoplastic glands (EGFRx200). (B) EGFR strong staining (EGFRx200). (C) Intense and strong staining with P16 in tumoral glands, negativity in the benign gland adjacent (P16 × 200). (D) No staining in neoplastic glands with HER2 (HER2 × 400).
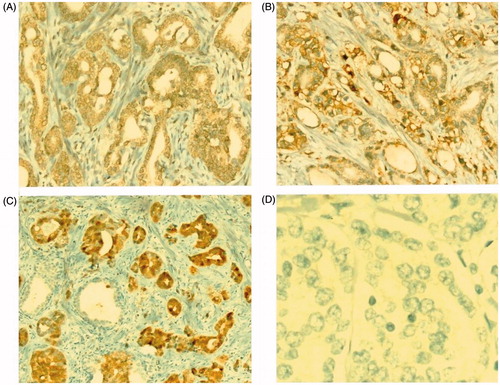
Table 2. KRAS, BRAF and PIK3CA mutation and HER2, EGFR, and p16 expression status of cases according to groups.
There were no correlation between EGFR immunopositivity and age (rk = .160 p = .349), preoperative PSA (rk = .295 p = .080), Gleason grade (rk = .328 p = .098), pT (rk = .165 p = .399), stage (rk = .206 p = .274), lymph node metastasis (N) (rk = −.216 p = .284), distant metastasis (M) (rk = .197 p = .329), vascular invasion (rk = −.201 p = .319) , perineural invasion (rk = .290 p = .151), periprostatic extension (rk = .127 p = .528), seminal vesicle invasion (rk = .189 p = .350), surgical margin positivity (rk = −.179 p = .374) , biochemical recurrence (rk = .146 p = .469), presence of family history (rk = .175 p = .395).
Also no significant relationship was observed P16 immunopositivity vs. age (rk = .062 p = .707), preoperative PSA (rk = .039 p = .810), Gleason grade (rk = .088 p = .649), pT (rk = .102 p = .591), stage (rk = .126 p = .492), lymph node metastasis (N) (rk = .330 p = .092), distant metastasis (M) (rk = .055 p = .780), vascular invasion (rk = −.024 p = .901), perineural invasion (rk = .006 p = .975), periprostatic extension (rk = .253 p = .196), seminal vesicle invasion (rk = .182 p = .353), surgical margin (rk = −.240 p = .221), biochemical recurrence (rk = .081 p = .678), presence of family history (rk = −.080 p = .691).
Discussion
The molecular basis of PCa has not been elucidated for many years. Although much progress has been made in recent years, it remains complex. The main difficulty in understanding the molecular basis of PCa is heterogeneity. In this study, we analyzed the intracellular signal transduction mechanisms of both PI3K and MAPK pathways at the same time. We evaluated the frequency of KRAS, BRAF, and PIK3CA mutation frequency and immunohistochemical expression molecules belonging to these two different pathways in PCa.
In the literature, the frequency of KRAS mutations in PCa is reported between 0% and 40% [Citation15–52]. KRAS mutations have yielded variable results in different populations depending on the ethnic origins of the populations. KRAS mutation frequencies are reported to be higher in Asian populations than in Western populations. Shen et al. [Citation30] reported the frequency of KRAS mutation as 9.1% in the study performed on 121 patients in the Chinese population. Similarly, Cho et al. [Citation28] and Salmaninejad et al. [Citation40] reported KRAS mutation frequency as 7.3% and 5.7% in Korean and Iranian patients, respectively. Similar to these studies, the frequency of KRAS mutations in the current study was detected to be 12.5%. As shown in , KRAS mutation frequency is reported to be as low as 0, 3% in the USA, and several Western societies, unlike Asian populations [Citation29,Citation31,Citation33,Citation37,Citation38]. We think these differences are mainly related to ethnic origin. However, Watanabe et al. [Citation22] found the frequency of KRAS mutations in Japanese patients as 14.2%, in Hawaiian Japanese as 5%. The rate is somewhat low in Hawaiian Japanese compared to this study and other studies in Japanese patients. This result suggests that environmental and epigenetic factors may affect the KRAS gene. Also, the significantly low KRAS mutation frequency in studies in the United States may be because of its relatively low proportion due to the excess of non-genetic factors in the etiology of PCa.
Table 3. KRAS, BRAF and PIK3CA mutations in human PCa tissues in the literature.
Two studies have analyzed KRAS mutations in PCa in Turkey. Vural et al. [Citation41] reported a rate of 8.8%, similar to the current study. Besides, gene regions in which KRAS mutations occurred (exon 2, codon 12/13; exon 3, codon 61) were similar to our study. Silan et al. [Citation36] reported KRAS mutation as 40% in the study that included PCa patients diagnosed in the middle Anatolian region. This is the highest rate ever reported in the literature. The relatively high number of Gleason score ≥ 7 cases in that study or the regional/environmental changes within the country may have caused the difference. Also, an automatic reverse hybridization method was used in that study, while the microarray hybridization method was used in ours. Another remarkable finding in the study of Silan et al. is the combined codon 12–13 mutations in 9 cases (30%). Another combined mutation was reported by Konishi et al. in the literature [Citation24]. In that study, a combined 13–61 mutation was observed in 1 case (11.1%). In our study, no combined mutation was found.
Ras expression has been observed to increase the ability of malign transformation and resistance to castration in PCa cell lines (LNCaP). Bakin et al. showed that increased RAS expression and sustained activation of MAP kinase decreased androgen requirement for growth, PSA expression, and tumorigenesis [Citation85–87]. It was also suggested that the KRAS mutation in PCa increases the susceptibility to bone metastasis through CD24a expression [Citation88]. In parallel with these studies, Anwar et al. [Citation20] found that the frequency of KRAS mutations in patients with stage III and IV was two times higher than in patients with stage I and II. Moreover, all cases with KRAS mutation were poorly differentiated PCa in the study of Suzuki et al. [Citation23]. On the other hand, there was no significant relationship between KRAS mutation status vs. stage, high Gleason score, or metastatic disease in the current study as in the studies of Shen et al. [Citation30] and Cho et al. [Citation28]. Additionally, survival analysis could not be performed because all our patients except one were alive.
BRAF is another target of the MAPK signaling pathway in carcinogenesis. BRAF mutation rates in PCa have yielded different results in various studies. Although results reaching 15% were reported in several studies in Asian populations, in many other studies in Western countries and Asia, no mutations were detected consistent with our study () [Citation28–34,Citation38,Citation40,Citation42,Citation44–46,Citation48,Citation53–63].
According to The Cancer Genome Atlas (TCGA) data, the frequency of BRAF mutation in PCa is specified as 1.4% [Citation89]. These mutations are several activating mutations other than the canonical V600E. Cho et al. [Citation28] reported the BRAF mutation rate as 10%. Likewise, most of the mutations detected in that study were V600A and V600M variants, which are rare and were not evaluated by us. Considering only V600E mutation positivity in that study, it shows a low rate (1%) similar to ours. In a recent study, Suh et al. [Citation61] reported the BRAF mutation rate in locally advanced PCa as 15%, which is the highest frequency in the literature. We think that the high BRAF mutation rate in that study resulted from the new generation sequencing (NGS) method. As a matter of fact, 2/3 of the detected BRAF mutations are K601E changes. We examined the V600E mutation in exon 15, which is a hotspot class I mutation of BRAF. Therefore, other mutations in exon 15 and mutations in exon 11 were not included in the study. V600E mutation is a hotspot mutation whose oncogenic potential is well known. BRAF mutations other than V600E are categorized as class II (kinase-active non V600E), class III (RAS dependent), and variants of unknown significance (VUS). Class II BRAF mutations show moderate/high kinase activity, while class III mutations lead to weak or no kinase activity [Citation90]. With the NGS method, many gene regions, including VUS, can be analyzed simultaneously. However, NGS is not cost-effective for many institutions, and it is a disadvantage in that it provides results that do not reflect the pathogenic potential exactly due to random genetic variants.
In the report of Jafarian et al., which is one of the studies only V600E mutation was examined, mutation frequency was found to be 4% [Citation60]. The mean age of patients in that study was higher than in the current study (71.83; 65.4, respectively), and all patients with mutations were between the ages of 70 and 90 years. Also, Gleason score> 6 cases consist of more than 80% of all cases in that study, while 50% in ours. On the other hand, Agell et al. reported the BRAF mutation frequency as 0%, in which a Gleason score> 6 cases predominant serial, in line with our study [Citation33].
Genetic changes have been identified at almost every step of the PI3K pathway in PCa carcinogenesis [Citation91]. Some of those are the increased PI3K synthesis by PIK3CA mutations, functional losses in PTEN protein suppressing AKT, or increased AKT synthesis by mutations in the PTEN gene [Citation44,Citation92]. Besides, the synergistic interaction with the RAS signaling pathway is also important in the effect of this pathway on carcinogenesis. RAS effect is minimal in stimulating the PI3K pathway under normal conditions. However, oncogenic RAS is an effective activator of this pathway. PI3K pathway deregulation was reported at a rate of 42% in localized PCa, and 100% in metastatic PCa. This suggests that the pathway also plays a role in castration resistance [Citation31]. PIK3CA is the gene encoding the catalytic subunit of PI3K. In the literature, the frequency of PIK3CA mutations in PCa has been reported between 0% −10.4% (). MacConaill et al. [Citation29], Taylor et al. [Citation31], and Toomey et al. [Citation45] found the PIK3CA mutation at a rate of 2.1% in 95 patients, 1.2% in 156 patients, 4.8% in 83 patients, respectively. The mutation rates are slightly higher than in the current study in these three studies, which included a relatively large number of cases. This may be due to the mass spectrometry-based method with high-throughput genotyping (OncoMap, iPLEX, and Agena, respectively) in all three studies. In addition, fresh tumor samples were analyzed in the study of MacConaill [Citation29] et al. Additionally, Barnett et al. [Citation38] detected PIK3CA mutation in 2 of 40 patients by the mass spectrometric method. While one of these mutations is the H1047R mutation that we analyzed in the current study, the other is the Q546F mutation that we did not.
Figure 3. Interaction of intracellular signal transduction pathways and mutation frequency ranges detected in KRAS, BRAF and PIK3CA in human PCa tissues in the literature.
Similar to our study, Agell et al. found no mutation in 102 patients [Citation33]. Likewise, Jaratlerdrishi achieved a PIK3CA mutation rate of 0% in 15 cases with the whole genome sequencing method [Citation43]. Boorman et al. [Citation66] had the same result as ours by automatic sequencing from fresh tumor tissues in 63 patients. On the other hand, Beltran et al. [Citation67] detected the PIK3CA mutation rate as 4% in 45 patients. The difference between this result and ours is likely to be since the number of metastatic PCa in that study is almost twice that of localized PCa. We think that the proportionally lower number of metastatic PCa in our study had a negative effect on the PIK3CA mutation profile. Indeed, Dawson et al. detected PIK3CA frequency as 4.5% by NGS in 1010 cases [Citation72]. Approximately half of the tissues consist of metastatic biopsies in that large sample analysis.
It is thought that both intertumoral and intratumoral heterogeneity plays a role in the molecular heterogeneity of PCa. This is because the tumor consists of more than one clone developing with a step-by-step branched progression through its evolutionary process [Citation93–94]. We believe that intratumoral heterogeneity may also have a role in the low results of BRAF and PIK3CA in our study.
EGFR is the main trigger of activation of the MAPK and PI3K signaling pathways. EGFR stimulation also activates the androgen receptor even in the absence of androgen hormone [Citation95]. Even more, castration leads to an increase in EGFR expression [Citation96]. Supporting this crosstalk between AR and EGFR, several studies have found a significant relationship between EGFR expression and PCa progression [Citation96–102]. In this study, no correlation was found in this direction. This result may be due to differences in immunohistochemical interpretation. EGFR immunostaining was strongly associated with advanced tumor stage, high Gleason grade, and preoperative PSA according to the study of Schlomm et al. However, as shown in the same study, EGFR expressions were not correlated with EGFR copy number changes in PCa [Citation77,Citation97,Citation103].
HER2 overexpression/gene amplification, a predictive parameter in breast cancer and is involved in treatment, has yielded variable results in PCa. We have no HER2 positive case. In the literature, studies have reported low or no HER2 expression/amplification levels in parallel with our study, as well as studies reporting high rates up to 100% [Citation77,Citation101,Citation104–108]. Kankaya et al. reported HER2 expression as 72.9%, while they found HER2 gene amplification as 4.05% in the same study. A meta-analysis of 5976 PCa cases reported an association between HER2 overexpression with mortality and recurrence [Citation109]. However, it has been observed that HER2 target therapies alone have a limited antitumor effect on PCa [Citation81,Citation110]. Combination therapies or immunotherapy options can be promising for a specific subpopulation.
p16, a major member of the cell cycle control mechanism, interacts with various mitogenic signaling pathways through cyclin D. It has a role in inhibiting the MAPK signaling pathway. p16 immunohistochemical expression has been reported between 23% and 100% in PCa in the literature [Citation111–115]. Evaluation only nuclear staining positively may be the reason for the low rate of 23% in the study of Vlachostergios PJ et al. The rate was %86 in our study. Many other studies that reported similar to ours have considered both cytoplasmic and nuclear positivity [Citation111–113]. Cut-off value variabilities, immunohistochemical technics, or fixation differences are the other points that might cause different results. A recent analysis based on TCGA data showed a reverse association with p16 expression and Gleason score [Citation116]. The reason for not reaching such a result in our study may be the small sample size. Another point we were surprised that there was no association between KRAS mutation positivity and p16 expression levels.
PI3K pathway dysregulations are very common in PCa carcinogenesis. PTEN deletions and inactivations are blamed for the majority of dysregulations of this pathway [Citation44,Citation46,Citation57]. In our study, we could not evaluate PTEN due to technical reasons.
Unlike PCa, almost no KRAS or BRAF mutations were detected in benign prostate hyperplasia (BPH) in several studies in the literature [Citation20,Citation21,Citation25,Citation35,Citation60]. In fact, in a recent NGS study, no evidence of driver genomic alterations was found in BPH cases, inconsistent with a neoplastic process. Even they observed multiple signatures related to the inactivation of KRAS signaling [Citation117].
Best of our knowledge, this is the first report that analyses KRAS, BRAF, and PIK3CA mutations simultaneously in PCa from Turkey. The small number of cases and the relatively narrow gene mutation panel for BRAF and PIK3CA are among the limitations of our study. In addition, the proportionally low number of metastatic PCa in our study is another limitation.
Our study provides new data to the literature as a basis for pharmacological target therapy research. We analyzed the various targets of two main signal transduction pathways, MAPK and PI3K, in the cell. In the patient group with KRAS mutation, targeted KRAS inhibitor agents that are in the research stage may be a ray of hope in treatment. Although the BRAF V600E and PIK3CA mutations were determined to be 0% in our study, consistent with many studies in the literature, there are also studies that determined different rates than ours. To more clearly determine the significance of these pathways in PCa, studies are needed in large series with predominantly advanced cases. The use of BRAF and PI3K inhibitors, which are approved in the treatment of different types of cancer and with satisfactory results may potentially prevent organ loss and increase the quality of life of patients with PCa.
Acknowledgement
The authors wish to thank Cem Horozoglu, phD from Molecular Medicine Dept., Aziz Sancar Research Institute, Istanbul University; for his critical reading of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
- Lima J, Teixeira Y, Pimenta C, et al. Fecal genetic mutations and human DNA in colorectal cancer and polyps patients. Asian Pac J Cancer Prev. 2019;20(10):2929–2934.
- Solassol J, Vendrell J, Märkl B, et al. Multi-center evaluation of the fully automated PCR-based Idylla™ KRAS mutation assay for rapid KRAS mutation status determination on formalin-fixed paraffin-embedded tissue of human colorectal cancer. PLoS One. 2016;11(9):e0163444.
- Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–4689.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954.
- Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314–341.
- Gamsizkan M, Yilmaz I, Simsek HA, et al. Mutation analysis of metastatic melanomas in the central nervous system: results of a panel of 5 genes in 48 cases. Clin Neuropathol. 2016;35(4):178–185.
- Yilmaz I, Gamsizkan M, Sari SO, et al. Molecular alterations in malignant blue nevi and related blue lesions. Virchows Arch. 2015;467(6):723–732.
- Şahpaz A, Önal B, Yeşilyurt A, et al. BRAF(V600E) mutation, RET/PTC1 and PAX8-PPAR gamma rearrangements in follicular epithelium derived thyroid lesions - institutional experience and literature review. Balkan Med J. 2015;32(2):156–166.
- Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer. 2006;94(4):455–459.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239–1246.
- Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223.
- Yang J, Nie J, Ma X, et al. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. 2019;18(1):26.
- National Compherensive Cancer Network (NCCN) [Internet]. Clinical practice guidelines in oncology, prostate cancer, version 2. 2020 [cited 2020 May 21] Available from: https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf
- Carter BS, Epstein JI, Isaacs WB. ras gene mutations in human prostate cancer. Cancer Res. 1990;50(21):6830–6832.
- Capella G, Cronauer-Mitra S, Pienado MA, et al. Frequency and spectrum of mutations at codons 12 and 13 of the c-K-ras gene in human tumors. Environ Health Perspect. 1991;93:125–131.
- Gumerlock PH, Poonamallee UR, Meyers FJ, et al. Activated ras alleles in human carcinoma of the prostate are rare. Cancer Res. 1991;51(6):1632–1637.
- Konishi N, Enomoto T, Buzard G, et al. K-ras activation and ras p21 expression in latent prostatic carcinoma in Japanese men. Cancer. 1992;69(9):2293–2299.
- Moul JW, Friedrichs PA, Lance RS, et al. Infrequent RAS oncogene mutations in human prostate cancer. Prostate. 1992;20(4):327–338.
- Anwar K, Nakakuki K, Shiraishi T, et al. Presence of ras oncogene mutations and human papillomavirus DNA in human prostate carcinomas. Cancer Res. 1992;52(21):5991–5996.
- Pergolizzi RG, Kreis W, Rottach C, et al. Mutational status of codons 12 and 13 of the N- and K-RAS genes in tissue and cell lines derived from primary and metastatic prostate carcinomas. Cancer Invest. 1993;11(1):25–32.
- Watanabe M, Shiraishi T, Yatani R, et al. International comparison on ras gene mutations in latent prostate carcinoma. Int J Cancer. 1994;58(2):174–178.
- Suzuki H, Aida S, Akimoto S, et al. State of adenomatous polyposis coli gene and ras oncogenes in Japanese prostate cancer. Jpn J Cancer Res. 1994;85(8):847–852.
- Konishi N, Hiasa Y, Matsuda H, et al. Intratumor cellular heterogeneity and alterations in ras oncogene and p53 tumor suppressor gene in human prostate carcinoma. Am J Pathol. 1995;147(4):1112–1122.
- Moyret-Lalle C, Marçais C, Jacquemier J, et al. ras, p53 and HPV status in benign and malignant prostate tumors. Int J Cancer. 1995;64(2):124–129.
- Konishi N, Hiasa Y, Tsuzuki T, et al. Comparison of ras activation in prostate carcinoma in Japanese and American men. Prostate. 1997;30(1):53–57.
- Shiraishi T, Muneyuki T, Fukutome K, et al. Mutations of ras genes are relatively frequent in Japanese prostat5e cancers: pointing to genetic differences between populations. Anticancer Res. 1998;18(4B):2789–2792.
- Cho NY, Choi M, Kim BH, et al. BRAF and KRAS mutations in prostatic adenocarcinoma. Int J Cancer. 2006;119(8):1858–1862.
- MacConaill LE, Campbell CD, Kehoe SM, et al. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One. 2009;4(11):e7887.
- Shen Y, Lu Y, Yin X, et al. KRAS and BRAF mutations in prostate carcinomas of Chinese patients. Cancer Genet Cytogenet. 2010;198(1):35–39.
- Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; Jul 1318(1):11–22.
- Choi S, Chun JM. 741 clinical significance of braf and kras mutations in prostate cancer. Eur Urol Suppl. 2010;9(2):240.
- Agell L, Hernandez S, Salido M, et al. PI3K signaling pathway is activated by PIK3CA mRNA overexpression and copy gain in prostate tumors, but PIK3CA, BRAF, KRAS and AKT1 mutations are infrequent events. Mod Pathol. 2011;24(3):443–452.
- Ren G, Liu X, Mao X, et al. Identification of frequent BRAF copy number gain and alterations of RAF genes in Chinese prostate cancer. Genes Chromosom Cancer. 2012;51(11):1014–1023.
- Sha J, Liang G, Pan J, et al. Application of pyrosequencing technique for improved detection of K-Ras mutation in formalin-fixed and paraffin-embedded prostate carcinoma tissues in Chinese patients. Clin Chim Acta. 2012;413(19–20):1532–1535.
- Silan F, Gultekin Y, Atik S, et al. Combined point mutations in codon 12 and 13 of KRAS oncogene in prostate carcinomas. Mol Biol Rep. 2012;39(2):1595–1599.
- Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243.
- Barnett CM, Heinrich MC, Lim J, et al. Genetic profiling to determine risk of relapse-free survival in high-risk localized prostate cancer. Clin Cancer Res. 2014;20(5):1306–1312.
- Fu M, Zhang W, Shan L, et al. Mutation status of somatic EGFR and KRAS genes in Chinese patients with prostate cancer (PCa). Virchows Arch. 2014;464(5):575–581.
- Salmaninejad A, Ghadami S, Dizaji MZ, et al. Molecular characterization of KRAS, BRAF, and EGFR genes in cases with prostatic adenocarcinoma; reporting bioinformatics description and recurrent mutations. Clin Lab. 2015;61(7):749–759.
- Vural H, Kurar E, Avcı E, et al. KRAS mutations: a possible biomarker for advanced prostate cancer. Cancer Sci Res. 2018;1(4):1–4.
- Martinez-Gonzalez LJ, Pascual Geler M, Robles Fernandez I, et al. Improving the genetic signature of prostate cancer, the somatic mutations. Urol Oncol. 2018;36(6):312.e17–312.e23.
- Jaratlerdsiri W, Chan EKF, Gong T, et al. Whole-genome sequencing reveals elevated tumor mutational burden and initiating driver mutations in african men with treatment-naïve, high-risk prostate cancer . Cancer Res. 2018;78(24):6736–6746.
- Ikeda S, Elkin SK, Tomson BN, et al. Next-generation sequencing of prostate cancer: genomic and pathway alterations, potential actionability patterns, and relative rate of use of clinical-grade testing. Cancer Biol Ther. 2019;20(2):219–226.
- Toomey S, Carr A, Mezynski MJ, et al. Identification and clinical impact of potentially actionable somatic oncogenic mutations in solid tumor samples. J Transl Med. 2020;18(1):99.
- Abeshouse A, Ahn J, Akbani R, et al. The molecular taxonomy of primary prostate cancer. Cell. 2015;163(4):1011–1025.
- Hovelson DH, McDaniel AS, Cani AK, et al. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia. 2015;17(4):385–399.
- Grasso CS, Cani AK, Hovelson DH, et al. Integrative molecular profiling of routine clinical prostate cancer specimens. Ann Oncol. 2015;26(6):1110–1118.
- Barata PC, Mendiratta P, Heald B, et al. Targeted next-generation sequencing in men with metastatic prostate cancer: a pilot study. Target Oncol. 2018;13(4):495–500.
- Mitri ZI, Parmar S, Johnson B, et al. Implementing a comprehensive translational oncology platform: from molecular testing to actionability. J Transl Med. 2018;16(1):358.
- Abida W, Cyrta J, Heller G, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci USA. 2019;116(23):11428–11436.
- Crumbaker M, Chan EKF, Gong T, et al. The impact of whole genome data on therapeutic decision-making in metastatic prostate cancer: a retrospective analysis. Cancers (Basel). 2020;12(5):1178.
- Cohen Y, Xing M, Mambo E, et al. BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst. 2003;95(8):625–627.
- Burger M, Denzinger S, Hammerschmied C, et al. Mitogen-activated protein kinase signaling is activated in prostate tumors but not mediated by B-RAF mutations. Eur Urol. 2006;50(5):1102–1110.
- Liu T, Willmore-Payne C, Layfield LJ, et al. Lack of BRAF activating mutations in prostate adenocarcinoma: a study of 93 cases. Appl Immunohistochem Mol Morphol. 2009;17(2):121–125.
- Köllermann J, Albrecht H, Schlomm T, et al. Activating BRAF gene mutations are uncommon in hormone refractory prostate cancer in Caucasian patients. Oncol Lett. 2010;1(4):729–732.
- Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228.
- Beltran H, Prandi D, Mosquera JM, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298–305.
- Cohn AL, Day BM, Abhyankar S, et al. BRAFV600 mutations in solid tumors, other than metastatic melanoma and papillary thyroid cancer, or multiple myeloma: a screening study. Onco Targets Ther. 2017;10:965–971.
- Jafarian AH, Mirshekar Nasirabadi K, Etemad S, et al. Molecular status of BRAF mutation in prostate adenocarcinoma: the analysis of 100 cases in north-east of IRAN. Iran J Pathol. 2018;13(4):415–421.
- Suh J, Jeong CW, Choi S, et al. Targeted next-generation sequencing for locally advanced prostate cancer in the Korean population. Investig Clin Urol. 2020;61(2):127–135.
- Kasajima R, Yamaguchi R, Shimizu E, et al. Variant analysis of prostate cancer in Japanese patients and a new attempt to predict related biological pathways. Oncol Rep. 2020;43(3):943–952.
- Zhao SG, Chen WS, Li H, et al. The DNA methylation landscape of advanced prostate cancer. Nat Genet. 2020;52(8):778–789.
- Müller CI, Miller CW, Hofmann WK, et al. Rare mutations of the PIK3CA gene in malignancies of the hematopoietic system as well as endometrium, ovary, prostate and osteosarcomas, and discovery of a PIK3CA pseudogene. Leuk Res. 2007;31(1):27–32.
- Sun X, Huang J, Homma T, et al. Genetic alterations in the PI3K pathway in prostate cancer. Anticancer Res. 2009;29(5):1739–1743.
- Boormans JL, Korsten H, Ziel-van der Made AC, et al. E17K substitution in AKT1 in prostate cancer. Br J Cancer. 2010;102(10):1491–1494.
- Beltran H, Yelensky R, Frampton GM, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63(5):920–926.
- Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–689.
- Millis SZ, Ikeda S, Reddy S, et al. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19 784 diverse solid tumors. JAMA Oncol. 2016;2(12):1565–1573.
- Kumar A, Coleman I, Morrissey C, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22(4):369–378.
- Huang FW, Mosquera JM, Garofalo A, et al. Exome sequencing of African-American prostate cancer reveals loss-of-function ERF mutations. Cancer Discov. 2017;7(9):973–983.
- Dawson NA, Zibelman M, Lindsay T, et al. An emerging landscape for canonical and actionable molecular alterations in primary and metastatic prostate cancer. Mol Cancer Ther. 2020;19(6):1373–1382.
- Lo Iacono M, Buttigliero C, Monica V, et al. Retrospective study testing next generation sequencing of selected cancer-associated genes in resected prostate cancer. Oncotarget. 2016;7(12):14394–14404.
- Seed G, Yuan W, Mateo J, et al. Gene copy number estimation from targeted next-generation sequencing of prostate cancer biopsies: analytic validation and clinical qualification. Clin Cancer Res. 2017;23(20):6070–6077.
- Hussain M, Daignault-Newton S, Twardowski PW, et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: results from NCI 9012. J Clin Oncol. 2018;36(10):991–999.
- Rescigno P, Rediti M, Dolling D, et al. PI3K/AKT pathway deleterious mutations in lethal prostate cancer. Ann Oncol. 2018;29(8):viii293.
- Baek KH, Hong ME, Jung YY, et al. Correlation of AR, EGFR, and HER2 expression levels in prostate cancer: immunohistochemical analysis and chromogenic in situ hybridization. Cancer Res Treat. 2012;44(1):50–56.
- Day KC, Lorenzatti Hiles G, Kozminsky M, et al. HER2 and EGFR overexpression support metastatic progression of prostate cancer to bone. Cancer Res. 2017;77(1):74–85.
- Sanchez KM, Sweeney CJ, Mass R, et al. Evaluation of HER-2/neu expression in prostatic adenocarcinoma: a requested for a standardized, organ specific methodology. Cancer. 2002;95(8):1650–1655.
- Liao Y, Guo Z, Xia X, et al. Inhibition of EGFR signaling with Spautin-1 represents a novel therapeutics for prostate cancer. J Exp Clin Cancer Res. 2019;38(1):157.
- Fu W, Madan E, Yee M, et al. Progress of molecular targeted therapies for prostate cancers. Biochim Biophys Acta. 2012;1825(2):140–152.
- Choi Y, Anders L. Signaling through cyclin D-dependent kinases. Oncogene. 2014;33(15):1890–1903.
- Wong M, Bierman Y, Pettaway C, et al. Comparative analysis of p16 expression among African American and European American prostate cancer patients. Prostate. 2019;79(11):1274–1283.
- Xiao GQ, Nguyen E, Unger PD, et al. Comparative expression of immunohistochemical biomarkers in cribriform and pattern 4 non-cribriform prostatic adenocarcinoma. Exp Mol Pathol. 2020;114:104400.
- Mukhopadhyay NK, Cinar B, Mukhopadhyay L, et al. The zinc finger protein ras-responsive element binding protein-1 is a coregulator of the androgen receptor: implications for the role of the ras pathway in enhancing androgenic signaling in prostate cancer. Mol Endocrinol. 2007;21(9):2056–2070.
- Weber MJ, Gioeli D. Ras signaling in prostate cancer progression. J Cell Biochem. 2004;91(1):13–25.
- Bakin RE, Gioeli D, Sikes RA, et al. Constitutive activation of the Ras/mitogen-activated protein kinase signaling pathway promotes androgen hypersensitivity in LNCaP prostate cancer cells. Cancer Res. 2003;63(8):1981–1989.
- Moon DO, Kim BY, Jang JH, et al. K-RAS transformation in prostate epithelial cell overcomes H2O2-induced apoptosis via upregulation of gamma-glutamyltransferase-2. Toxicol in Vitro. 2012;26(3):429–434.
- National Cancer Intitute [Internet]. GDC data portal. 2020 [cited 2020 May 2]. Available from: https://portal.gdc.cancer.gov/genes/ENSG00000157764?canDistTable_size=100.
- Yao Z, Yaeger R, Rodrik-Outmezguine VS, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017;548(7666):234–238.
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–5510.
- Shorning BY, Dass MS, Smalley MJ, et al. The PI3K-AKT-mTOR pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. IJMS. 2020;21(12):4507.
- Gerlinger M, Catto JW, Orntoft TF, et al. Intratumour heterogeneity in urologic cancers: from molecular evidence to clinical implications. Eur Urol. 2015;67(4):729–737.
- Tolkach Y, Kristiansen G. The heterogeneity of prostate cancer: a practical approach. Pathobiology. 2018;85(1–2):108–116.
- Traish AM, Morgentaler A. Epidermal growth factor receptor expression escapes androgen regulation in prostate cancer: a potential molecular switch for tumour growth. Br J Cancer. 2009;101(12):1949–1956.
- Hernes E, Fossa SD, Berner A, et al. Expression of the epidermal growth factor receptor family in prostate carcinoma before and during androgen-independence. Br J Cancer. 2004;90(2):449–454.
- Schlomm T, Kirstein P, Iwers L, et al. Clinical significance of epidermal growth factor receptor protein overexpression and gene copy number gains in prostate cancer. Clin Cancer Res. 2007;13(22):6579–6584.
- Shuch B, Mikhail M, Satagopan J, et al. Racial disparity of epidermal growth factor receptor expression in prostate cancer. J Clin Oncol. 2004;22(23):4725–4729.
- Di Lorenzo G, Tortora G, D'Armiento FP, et al. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin Cancer Res. 2002;8(11):3438–3444.
- Hashmi AA, Hashmi SK, Irfan M, et al. Prognostic utility of epidermal growth factor receptor (EGFR) expression in prostatic acinar adenocarcinoma. Appl Cancer Res. 2019;39(1):2.
- Visakorpi T, Kallioniemi OP, Koivula T, et al. Expression of epidermal growth factor receptor and ERBB2 (HER-2/Neu) oncoprotein in prostatic carcinomas. Mod Pathol. 1992;5(6):643–648.
- Nastały P, Stoupiec S, Popęda M, et al. EGFR as a stable marker of prostate cancer dissemination to bones. Br J Cancer. 2020;123(12):1767–1038.
- de Muga S, Hernández S, Agell L, et al. Molecular alterations of EGFR and PTEN in prostate cancer: association with high-grade and advanced-stage carcinomas. Mod Pathol. 2010;23(5):703–712.
- Kankaya D, Sertcelik A, Kaygusuz G, et al. Her-2/Neu oncogene expression in prostate carcinoma: evaluation of gene amplification by FISH method. Turk J Pathol. 2008;24(2):76–83.
- Minner S, Jessen B, Stiedenroth L, et al. Low level HER2 overexpression is associated with rapid tumor cell proliferation and poor prognosis in prostate cancer. Clin Cancer Res. 2010;16(5):1553–1560.
- Sharifi N, Salmaninejad A, Ferdosi S, et al. HER2 gene amplification in patients with prostate cancer: evaluating a CISH-based method. Oncol Lett. 2016;12(6):4651–4658.
- Signoretti S, Montironi R, Manola J, et al. Her-2/neu expression and progression toward androgen independence in human prostate cancer. J Natl Cancer Inst. 2000;92(23):1918–1925.
- Gu K, Mes-Masson AM, Gauthier J, et al. Overexpression of HER2/neu in human prostate cancer and benign hyperplasia. Cancer Lett. 1996;99(2):185–189.
- Neto AS, Tobias-Machado M, Wroclawski ML, et al. Her-2/neu expression in prostate adenocarcinoma: a systematic review and meta-analysis. J Urol. 2010;184(3):842–850.
- Ziada A, Barqawi A, Glode LM, et al. The use of trastuzumab in the treatment of hormone refractory prostate cancer; phase II trial. Prostate. 2004;60(4):332–337. 2004
- Konishi N, Hiasa Y, Tsuzuki T. Detection of RB, p16/CDKN2 and p15ink4B gene alterations with immunohistochemicals studies in human prostate carcinomas. Int J Oncol. 1996;8(1):107–112.
- Kudahetti SC, Fisher G, Ambroisine L, et al. Immunohistochemistry for p16, but not Rb or p21, an independent predictor of prognosis in conservatively treated, clinically localized prostate cancer. Pathology. 2010;42(6):519–523.
- Remo A, Pancione M, Zanella C, et al. p16 expression in prostate cancer and nonmalignant lesions: novel findings and review of the literature. Appl Immunohistochem Mol Morphol. 2016;24(3):201–206.
- Iemelynova AA, Grygorenko VM, Cheremuha SV, et al. Correlation between histological type and immunohistochemical profile of prostate cancer and gleason scale gradation. Exp Oncol. 2009;31(4):246–249.
- Vlachostergios PJ, Karasavvidou F, Kakkas G, et al. Lack of prognostic significance of p16 and p27 after radical prostatectomy in hormone-naive prostate cancer. J Neg Res Biomed. 2012;11(1):2–7.
- Boldrini L, Manassero F, Selli C, et al. "p16 and its putative interplay with metabolic factors in prostate cancer: an analysis based on public TCGA data". World Acad Sci J. 2020;2(1):9–13.
- Liu D, Shoag JE, Poliak D, et al. Integrative multiplatform molecular profiling of benign prostatic hyperplasia identifies distinct subtypes. Nat Commun. 2020;11(1):1987.