
Abstract
Aim
Gaucher disease (GD) is a rare autosomal recessive condition. Type 1 GD (GD1) is the most prevalent form of GD in Western countries; enzyme replacement therapy (ERT) is a treatment option for patients with GD1. To understand the economic value of the GD1 ERT velaglucerase alfa, a budget impact model (BIM) was developed from a United States (US) payer perspective.
Methods
We estimated the budget impact of velaglucerase alfa for a 10-million-member US health plan by comparing the annual total costs of therapy between a scenario using current velaglucerase alfa uptake to a projected scenario with increased velaglucerase alfa uptake. Total drug costs for both scenarios were estimated as the sum of the product of the number of eligible patients on each treatment and the annual per-patient cost of each medication. Average per-patient costs for ERTs were calculated by adding the yearly drug acquisition, drug administration, and site-of-care markup costs. The budget impact was measured over years 1–3.
Results
An estimated 65 patients would receive velaglucerase alfa treatment in year 1, increasing to 90 patients by year 3. Across analyses, cost savings were realized with velaglucerase alfa compared with imiglucerase ($115,909) and taliglucerase alfa ($80,401). An annual total budget savings of $8.67 million could be realized for a hypothetical 10-million-member US health plan with increased velaglucerase alfa uptake. The per-member per-month costs decreased by $0.0241 across years 1–3.
Conclusions
BIM results show that increased velaglucerase alfa uptake for GD1 treatment is cost-saving for US health plans.
PLAIN LANGUAGE SUMMARY
Type 1 Gaucher disease (GD1) is a rare inherited condition. Long-term enzyme replacement therapy (ERT) can reverse and prevent complications. Imiglucerase, taliglucerase alfa, and velaglucerase alfa are 3 ERTs used to treat GD1. In this study, we estimated how increasing uptake of velaglucerase alfa vs. the other ERTs would impact the budget of a hypothetical US healthcare plan. The results show that increased uptake of velaglucerase alfa is cost-saving for US health plans.
Introduction
Gaucher disease (GD) is a rare autosomal recessive condition resulting from mutations in the glucosylceramidase beta (GBA) gene encoding the glucosylceramidase enzymeCitation1. The estimated prevalence of GD is 1 to 2 cases per 100,000 in the general population, with higher rates reported among people of Ashkenazi Jewish descentCitation2. The estimated birth incidence of GD in the general population (including data from North America, Asia, Europe, Africa, and Australia) ranges from ∼0.39 to 5.80 cases per 100,000 birthsCitation2.
Type 1 GD (GD1) is the most prevalent form of GD in Western countries, accounting for 95% of casesCitation1. The prevalence of GD1 is higher in individuals of Ashkenazi Jewish ethnicity, with a birth incidence of ∼1 in 850Citation3,Citation4. Patients with GD1 have normal brain development and no central nervous system involvementCitation1. These patients may exhibit thrombocytopenia, anemia, hepatosplenomegaly, delayed growth in children, and various skeletal abnormalities, although signs and symptoms vary considerably among patientsCitation1.
Key assessments for GD include quantitative volumetric imaging of the liver and spleen, measurement of bone mineral density, and evaluation for skeletal abnormalitiesCitation5. Additionally, there is evidence that elevated levels of the lysosomal storage product and sphingolipid, glucosylsphingosine (lyso-Gb1), a direct metabolite of Gb1, are pathogenic to cells in GD; lyso-Gb1 is now used as a long-term prognostic biomarker in GDCitation6. Another regular biomarker testing is outlined in the GD Delphi consensus publicationCitation5. Past management of GD often included splenectomy for life-threatening thrombocytopenia or hypersplenism. This practice is now quite rare as management of the disease has changed with the advent of enzyme replacement therapy (ERT), which generally reverses GD clinical symptoms, including symptomatic splenomegaly. ERT prevents progressive manifestation of GD and ameliorates GD-associated anemia, thrombocytopenia, organomegaly, bone pain, and bone crisesCitation7.
Intravenous (IV) ERT with recombinant human acid ß-glucosidase (imiglucerase; Cerezyme; Genzyme Corporation, Cambridge, MA) was approved in 1994, and for more than two decades has been the standard of care for GD1Citation8,Citation9. Treatment with ERT reverses and prevents many of the manifestations of GD and can prevent disease progressionCitation10. More recently, velaglucerase alfa (VPRIV; Shire Human Genetic Therapies, Inc., Cambridge, MA) and taliglucerase alfa (Elelyso; Pfizer Inc., New York, NY) have been introduced into clinical practiceCitation11,Citation12. These treatments were developed to replace or supplement deficient endogenous enzyme activity in affected individuals and enhance the breakdown of the accumulated substrateCitation8.
Early treatment of GD1 with ERT during childhood can potentially prevent some complications of GD, including progression of irreversible skeletal manifestations. Results from a long-term, phase 3 extension study enrolling patients who were treatment naïve before initiating velaglucerase alfa therapy showed continued improvement in signs and symptoms of GD1 in patients who received more than 2 years of therapyCitation13,Citation14. To understand the economic value of velaglucerase alfa in the treatment of GD1, a budget impact model (BIM) was developed from a United States (US) payer perspective.
Methods
Model framework and assumptions
Model structure
A BIM was developed in Microsoft Excel (Microsoft Corp., Redmond, WA, USA) to estimate the financial impact of increased velaglucerase alfa uptake to a formulary of a hypothetical US payer. The target population was defined as patients diagnosed with GD1 and eligible for treatment with ERT.
The budget impact of velaglucerase alfa was estimated by comparing the annual total costs of therapy between two scenarios over a 3-year time horizon, which is commonly used in US models and is consistent with the budgeting/planning periods of US budget holdersCitation15,Citation16 (). The projected scenario reflects an increase in velaglucerase alfa use. The total drug costs for both scenarios were estimated as the sum of the product of the number of eligible patients for each treatment and the annual per-patient cost of each specific medication. Average per-patient costs for ERTs were calculated by adding the yearly drug acquisition, drug administration, and site-of-care markup costs. The model estimated the budget impact over the course of 1–3 years.
Figure 1. Model flow diagram. Abbreviations. GD1, Gaucher disease type 1; ERT, enzyme replacement therapy.
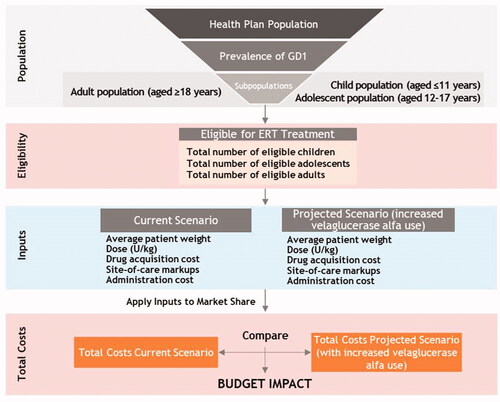
A four-step process was used to calculate the budget impact. First, the total target population using ERT was estimated using epidemiology and treatment use data obtained from the published literature. Second, the number of patients treated with a specific ERT in the current and projected market scenarios was determined. The target population receiving treatment with ERTs was subdivided into specific ERT treatments based on the market share of each treatment in the current and projected scenarios. Third, the number of patients on each ERT was multiplied by the annual per-patient cost of each specific ERT—including drug acquisition, site-of-care markups, and drug administration costs—to calculate total costs in both market scenarios. Lastly, the difference in total costs between the two scenarios was estimated as the difference in total costs between the current scenario and the scenario with increased velaglucerase alfa uptake (i.e. projected scenario).
Model assumptions
The model included the following assumptions:
Patient age distribution, patient weight, dose administered, site-of-care utilization, and markups were assumed equal among all ERTs.
All ERTs were assumed administered 26 times per year.
Markups were assumed different depending on the site of treatment (i.e. home infusion, infusion clinic, hospital outpatient).
The population consisted of patients aged ≥4 years in accordance with product labels.
Separate analyses for children, adolescents, and adults were allowed by the model.
Adverse event and monitoring costs were not assumed different among ERTs, and therefore were not included in the model.
Switching between ERTs during the same year was not explicitly modeled; switches in subsequent years were included as market share changes over time.
In the current scenario, market shares were not assumed to change over time.
Model inputs
An overview of model inputs for the base case analysis is provided in .
Table 1. Base case model inputs.
Model perspectives
The model calculates the budget impact of velaglucerase alfa for GD1 compared to other ERTs from a US payer perspective. All cost inputs were included as expenses to the payer.
Eligible treatment population
Plan size
The model assumed a total health plan size of 10 million members to simulate a large US health plan with a sufficient number of patients, due to the rarity of GD.
Age distribution
The GD1 child, adolescent, and adult age distributions were calculated using the age distributions in an international GD1 patient study and assumed that 7.37% of the GD1 population was aged ≤11 years and 87.94% was aged ≥18 yearsCitation17.
Eligible treatment population
The eligible treatment population was assumed to be 97.18% of the GD1 prevalent population. This was based on the proportion of patients in the US receiving GD1 treatment as asymptomatic patients with mild genetic mutations, such as N370S (i.e. c.1226A > G; p.N409S), may not require treatmentCitation17. The eligible treatment population for the present analysis was defined as the proportion of GD1 patients in the US that received treatment in the Gaucher Outcome Survey International Disease-Specific Registry.
GD1 prevalence
The target population consisted of patients with GD1. A prevalence of 1.33 cases per 100,000 population was incorporated into the model obtained from a systematic literature review of GD epidemiology performed by Nalysnyk et al. in 2015Citation2. Prevalence was assumed constant over the modeled period.
Dosing
Patient weight
The cost of an IV-administered drug can vary by over 100% based on patient weight, as the dosage is individualizedCitation18. To account for differences in the weight of children, adolescents, and adults, the total drug dose was calculated for each age category using the US Centers for Disease Control and Prevention (CDC)-reported average weights by ageCitation19. Average weights were assumed to match the US national average from 1999–2000 to 2015–2016 reported by the CDC in Fryar et al.Citation19. The average weight for children aged ≤11 years was 30.8 kg, for adolescents aged 12–17 years was 63.9 kg, and for adults aged ≥18 years was 83.5 kg. Children aged <4 years were not included in the base case scenario weight calculation.
Dosing for ERT
The number of infusions administered yearly and ERT dosing were based on label-advised doses.
Drug acquisition cost
Wholesale acquisition costs (WACs; year 2021)—i.e. a manufacturer’s list price for sale to direct purchasers or wholesalers, without discounts, rebates, or incentivesCitation20—were used for ERT in the base case modelCitation20. The model was also run using average sales price (ASP), a pricing method for drugs and biologics covered by Medicare Part B that is based on manufacturer-reported data on actual selling prices, inclusive of rebates, discounts, and other price concessionsCitation21. Yearly ERT drug acquisition costs were calculated by multiplying the number of vials used per infusion by the cost per package (the cost for 1 infusion) by the number of annual infusions. The number of vials used per infusion was determined by multiplying the patient weight by the patient dose and dividing the total dose by the number of units per vial.
Site-of-care markup costs
In the US, the cost of an IV-administered drug varies depending on where patients receive their infusions (i.e. home setting, infusion clinic, or hospital outpatient setting) because of the different drug markups applied. Providers seeking reimbursement from insurers for drug administration will include additional costs beyond the drug price; this site-of-care markup tends to be highest with hospital administration. Site-of-care markups were sourced from internal data. We calculated the yearly site-of-care cost by multiplying the yearly drug acquisition cost by the average markup (29%). The average markup was the sum of the products of site-of-care distribution and site-of-care markups.
Market share
The budget impact of velaglucerase alfa was estimated by comparing the total costs between two scenarios. The projected scenario reflects a hypothetical new formulary with greater velaglucerase alfa uptake. In the projected scenario, a market shift was assumed from imiglucerase and taliglucerase alfa to velaglucerase alfa ().
Table 2. ERT market share: current and projected scenarios.
Results
Budget impact velaglucerase alfa
Treated patient population
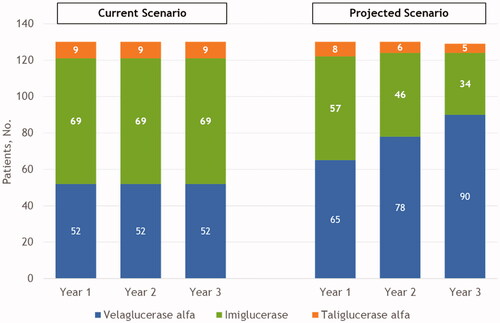
In a hypothetical 10-million-member US health plan for the projected scenario, 65 patients were estimated to be treated with velaglucerase alfa in year 1, 78 patients in year 2, and 90 patients in year 3 ().
Figure 2. The treated patient population in the current and projected scenarios.
Yearly cost per patient
Regardless of age group, treatment with velaglucerase alfa was cost saving compared to imiglucerase ($115,909 annual cost savings per patient) and taliglucerase alfa ($80,401 annual cost savings per patient) (). Cost savings were driven by the drug acquisition costs as administration costs were similar among the ERTs.
Figure 3. Yearly cost per patient with GD1 by age group and ERT therapya. aDetermined using wholesale acquisition costs (year 2021). bWeighted average of the yearly cost per patient, weighted by the proportion of children (N = 10), adolescents (N = 6), and adults (N = 114). Weighted averages were not calculated for drug acquisition, administration, or site-of-care markup costs. Abbreviations. ERT, enzyme replacement therapy; GD1, type 1 Gaucher disease; USD, United States dollar.
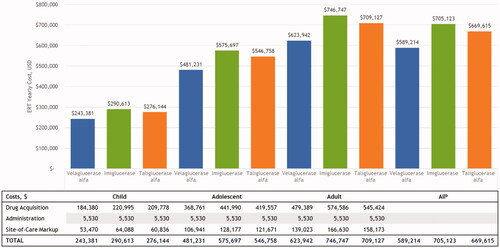
When ASP as opposed to WAC was incorporated into the model, the average annual treatment cost per patient did not change substantially (velaglucerase alfa, $576,123; imiglucerase, $719,625; taliglucerase alfa, $679,854); the cost savings with velaglucerase alfa were $143,502 compared with imiglucerase and $103,731 compared with taliglucerase alfa. Average annual treatment costs per patient for velaglucerase alfa were $238,046 for children, $470,562 for adolescents, and $610,072 for adults.
Budget impact
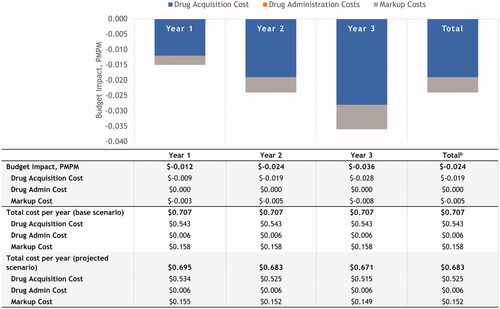
Results from the BIM showed that for a hypothetical 10-million-member US health plan, an increase in the uptake of velaglucerase alfa decreased the annual total budget impact by $8.67 million and the per-member per-month costs by $0.0241 across the first 3 years ().
Figure 4. PMPM budget impact for payers with current vs. projected velaglucerase alfa uptake scenarioa. aDetermined using wholesale acquisition costs (year 2021). Costs are presented in United States dollars. bAverage of years 1–3. Abbreviation. PMPM, per-member per-month.
Discussion
Patients with GD have a lysosomal enzyme deficiency resulting in a buildup of glucocerebrosideCitation1. For patients with symptomatic disease, long-term treatment with either ERT or substrate reduction therapy is neededCitation8. ERT provides the enzymes needed to break down glucocerebrosides whereas substrate reduction therapy blocks the production of glucocerebrosides. Currently, several formulations of ERT are administered intravenously, usually once every 2 weeks. The three commercially available formulations of ERTs are not biosimilar; similarities and dissimilarities between imiglucerase, velaglucerase alfa, and taliglucerase alfa have been presented earlierCitation22. Of the available ERTs, only velaglucerase alfa is produced by gene activation via a human cell lineCitation22. The resulting protein produced is identical in amino acid sequence to the naturally occurring enzyme. Differences in glycosylation patterns and manufacturing process from a human cell line may explain the lower immunogenicity of velaglucerase alfa, contributing to its improved efficacyCitation22. More recently, studies have shown a steeper and faster decrease of lyso-Gb1 levels, a key GD-specific biomarker in velaglucerase alfa as compared to other ERTsCitation6.
The goal of GD treatment is to achieve major hematologic, organomegaly, and skeletal manifestation goals within 1–2 years of treatment initiationCitation23. ERT is considered a first-line treatment for patients with GD1 as ERT may reverse inflammation and the hematologic effects of GD through supplementation of the defective/deficient enzymeCitation24. Early initiation of ERT therapy may delay and prevent complications and, therefore, ERT should be initiated early in the disease courseCitation25.
Among patients with GD1 who are treated with ERT, results from this BIM show that velaglucerase alfa was cost-saving compared to imiglucerase and taliglucerase alfa. The estimated annual cost savings per patient was $115,909 compared with imiglucerase and $80,401 compared with taliglucerase alfa. Overall, the model estimated an ∼$8.7 million cost savings annually with increased velaglucerase alfa uptake for a hypothetical 10-million-member US health plan. The estimated cost savings were driven primarily by drug acquisition costs as administration costs were similar among all ERTs. Results were similar when the model incorporated ASP compared with WAC.
Limitations
As real-world use of ERT in GD1 may vary, our results may not be generalizable to all settings. Although baseline drug utilization data were based on the product label and published literature, actual drug utilization may be substantially different due to differences in standards of care and the use of other interventions that impact each patient population. We found that markup costs were a significant contributor to the annual cost of care per patient. As we sourced site-of-care markups from internal data, site-of-care markups may vary across the different institution and payer settings. We found that drug acquisition costs were the main driver of cost savings for velaglucerase alfa compared to other ERTs. As these costs can be affected by rebates and discounts, results may differ for specific payers or by locality. This model used a rudimentary forecast of potential market share shift based on assumptions and therefore may not accurately reflect changes in market share. Finally, adverse event and monitoring costs were assumed to be similar among ERTs and therefore were not included in the model. Since ERTs for patients with GD1 rarely result in adverse eventsCitation8, any potential bias due to this assumption is likely minimal.
Conclusions
Results from this budget impact analysis suggest potential cost savings for a 10-million-member US health plan with increased use of velaglucerase alfa for GD1. The potential cost-saving estimates may vary depending on drug pricing, market shift to velaglucerase alfa, and treatment and patient dosage assumptions.
Transparency
Declaration of funding
Takeda provided funding for this study.
Declaration of financial/other relationships
SF is a full-time employee of Takeda and a stockholder of Takeda Pharmaceuticals Company Limited. Takeda provided medical writing support in relation to this work.
TJI is a former full-time employee of OPEN Health. OPEN Health received funding from Takeda for medical writing support in relation to this work.
GP is a former full-time employee of OPEN Health. OPEN Health received funding from Takeda for medical writing support in relation to this work.
EW is a full-time employee of Takeda and a stockholder of Takeda Pharmaceuticals Company Limited. Takeda provided medical writing support in relation to this work.
RRP is a full-time employee of Takeda and a stockholder of Takeda Pharmaceuticals Company Limited. Takeda provided medical writing support in relation to this work.
MB is a full-time employee of Takeda and a stockholder of Takeda Pharmaceuticals Company Limited. Takeda provided medical writing support in relation to this work.
A reviewer on this manuscript has disclosed that they have acted as an adviser and speaker for all three companies but have no formal or ongoing affiliations. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Author contributions
SF, TJI, and MB: concept and design. SF, TJI, GP, EW, RRP, and MB: analysis and interpretation of the data, drafting of the paper or revising it critically for intellectual content, and final approval of the version to be published. All authors agree to be accountable for all aspects of this work.
Acknowledgements
Medical writing support was provided by Beth Lesher, PharmD, BCPS, from OPEN Health, Bethesda, MD, and funded by the study sponsor.
References
- Martins AM, Valadares ER, Porta G, et al. Recommendations on diagnosis, treatment, and monitoring for Gaucher disease. J Pediatr. 2009;155(4 Suppl):S10–S18.
- Nalysnyk L, Rotella P, Simeone JC, et al. Gaucher disease epidemiology and natural history: a comprehensive review of the literature. Hematology. 2017;22(2):755–73.
- Zimran A, Elstein D. Gaucher disease and related lysosomal storage diseases. In: Kaushansky K, Lichtman M, Prchal J, editors. Williams hematology. 9th ed. New York (NY): McGraw-Hill; 2016.
- Zimran A, Gelbart T, Westwood B, et al. High frequency of the Gaucher disease mutation at nucleotide 1226 among Ashkenazi Jews. Am J Hum Genet. 1991;49(4):855–859.
- Kishnani PS, Al-Hertani W, Balwani M, et al. Screening, patient identification, evaluation, and treatment in patients with Gaucher disease: results from a Delphi consensus. Mol Genet Metab. 2022;135(2):154–162.
- Dinur T, Grittner U, Revel-Vilk S, et al. Impact of long-term enzyme replacement therapy on glucosylsphingosine (Lyso-Gb1) values in patients with type 1 Gaucher disease: statistical models for comparing three enzymatic formulations. IJMS. 2021;22(14):7699.
- Weinreb NJ, Charrow J, Andersson HC, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002;113(2):112–119.
- Zimran A, Elstein D. Management of Gaucher disease: enzyme replacement therapy. Pediatr Endocrinol Rev. 2014;12(Suppl 1):82–87.
- CEREZYME [Prescribing information] [cited 2021 Jan 25]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2005/20367s066lbl.pdf
- Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004;41(4 Suppl 5):4–14.
- VPRIV [Prescribing information] [cited 2021 Jan 25]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022575lbl.pdf
- ELELYSO [Prescribing information] [cited 2021 Jan 25]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022458lbl.pdf
- Zimran A, Elstein D, Gonzalez DE, et al. Treatment-naïve Gaucher disease patients achieve therapeutic goals and normalization with velaglucerase alfa by 4years in phase 3 trials. Blood Cells Mol Dis. 2018;68:153–159.
- Zimran A, Gonzalez-Rodriguez DE, Abrahamov A, et al. Long-term safety and efficacy of taliglucerase alfa in pediatric Gaucher disease patients who were treatment-naïve or previously treated with imiglucerase. Blood Cells Mol Dis. 2018;68:163–172.
- Mauskopf J, Earnshaw S. A methodological review of US budget-impact models for new drugs. Pharmacoeconomics. 2016;34(11):1111–1131.
- Sullivan SD, Mauskopf JA, Augustovski F, et al. Budget impact analysis-principles of good practice: report of the ISPOR 2012 budget impact analysis good practice II task force. Value Health. 2014;17(1):5–14.
- Deegan P, Fernandez-Sasso D, Giraldo P, et al. Treatment patterns from 647 patients with Gaucher disease: an analysis from the Gaucher outcome survey. Blood Cells Mol Dis. 2018;68:218–225.
- Nalysnyk L, Sugarman R, Cele C, et al. Budget impact analysis of eliglustat for the treatment of Gaucher disease type 1 in the United States. J Manag Care Spec Pharm. 2018;24(10):1002–1008.
- Fryar CD, Kruszon-Moran D, Gu Q, et al. Mean body weight, height, waist circumference, and body mass index among adults: United States, 1999–2000 through 2015–2016. Natl Health Stat Report. 2018;(122):1–16.
- Academy of Managed Care Pharmacy (AMCP). Managed care glossary 2022 [cited 2022 May 3]. Available from: https://www.amcp.org/about/managed-care-pharmacy-101/managed-care-glossary
- Academy of Managed Care Pharmacy (AMCP). AMCP guide to pharmaceutical payment methods; Executive summary version 3.0 2013 [cited 2022 May 4]. Available from: https://www.amcp.org/sites/default/files/2019-03/MM%20edits-Executive_Summary%20%28final-mar27%29_MB.pdf
- Revel-Vilk S, Szer J, Mehta A, et al. How we manage Gaucher disease in the era of choices. Br J Haematol. 2018;182(4):467–480.
- Biegstraaten M, Cox TM, Belmatoug N, et al. Management goals for type 1 Gaucher disease: an expert consensus document from the European Working Group on Gaucher disease. Blood Cells Mol Dis. 2018;68:203–208.
- European Medicines Agency. Gaucher disease: a strategic collaborative approach from EMA and FDA. London, United Kingdom; 2014 [cited 2021 Sep 29]. Available from: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/gaucher-disease-strategic-collaborative-approach-european-medicines-agency-food-drug-administration_en.pdf
- Limgala RP, Ioanou C, Plassmeyer M, et al. Time of initiating enzyme replacement therapy affects immune abnormalities and disease severity in patients with Gaucher disease. PLOS One. 2016;11(12):e0168135.