
Abstract
Background
Arginase 1 Deficiency (ARG1-D) is an inherited metabolic disease that leads to significant morbidity.
Aims
Despite the recognized burden of disease, information on health care resource utilization (HCRU) among patients with ARG1-D is lacking. We, therefore, sought to evaluate HCRU in ARG1-D relative to non-ARG1-D cohort.
Materials and methods
Patients with ≥2 ICD-10-CM diagnosis codes for ARG1-D were identified (first diagnosis code = index date) using professional fee claims linked with prescription claims. Patients with ARG1-D were matched 1:1 to a comparator cohort of patients with other medical conditions. Matching variables included age, sex, index year, payer type (Medicare, Medicaid, third party) and geographic region.
Results
A total of 77 patients met the inclusion criteria for the ARG1-D cohort, with a median age of 15 years, 52% <18 years, and 52% male. Several concurrent diagnoses were recorded at a higher frequency in the ARG1-D cohort versus the matched comparator (spasticity 7 times higher; developmental delay ∼2 times higher; intellectual disability 5 times higher; and seizures 8 times higher). Emergency room visits occurred twice as often, laboratory tests were performed 1.5 times more often, hospitalization was required 3 times more often, and mean length of stay was longer for patients with ARG1-D than the comparator cohort (2.4 days vs. 0.3 days).
Limitations
A relatively short study period while the burden of ARG1-D increases over a lifetime due to disease progression.
Conclusions
Patients with ARG1-D had significantly greater HCRU compared with those without the disease; they presented with a more extensive comorbidity profile, accessed the health care system more frequently, required more intense monitoring and management, and had more frequent and longer hospitalizations relative to the comparator group. These findings demonstrate a high health burden in ARG1-D that is not mitigated by standard-of-care measures and emphasize the need for improved treatment options.
Introduction
Urea cycle disorders (UCDs) represent a group of inborn errors of metabolism, due to defects in one of the enzymes or transporters that are involved in the metabolism of amino acids and impair the detoxification of ammoniaCitation1. The most unique and least common among UCDs is Arginase 1 Deficiency (ARG1-D)Citation2, an ultra-rare disorder that has been estimated to occur globally in approximately 1 in 357,000 live births with a population prevalence of 1 in 726,000Citation3. ARG1-D is a distinct UCD caused by mutations in the ARG1 gene, which results in diminished or absent arginase 1 enzyme activity and subsequently, accumulation of arginine (hyperargininemia)Citation1. Excessive levels of arginine lead to progressive neurologic manifestations, significant morbidity, and early mortalityCitation2,Citation4,Citation5. Although the clinical presentation of ARG1-D differs by patient, it typically presents in early childhood, with progressive and debilitating manifestations that include motor deficits (e.g. spasticity and difficulty walking), developmental delays, intellectual disabilities, and in some cases, seizuresCitation5–10. Excess ammonia in the blood (hyperammonemia) is usually less frequent and not as severe as in other UCDsCitation1; however, episodes of symptomatic and even severe hyperammonemia may occur and can result in deathCitation4,Citation6,Citation8.
Although all patients show progression of disease over time, the rate of development of manifestations and long-term deterioration of patients with ARG1-D differs between patients, similar to the variability in patient presentation. Many patients develop mobility and motor function deficits, moderate to severe neurological impairments, and reduced quality of life (QoL)Citation1,Citation5,Citation11. As such, ARG1-D results in a significant burden on patients and their caregiversCitation5. Although the effect of reduced QoL in patients with ARG1-D and the impact on the use of health care systems has not been evaluated specifically, studies in analogous patient populations with chronic conditions (associated with reduced QoL) have demonstrated higher health care resource useCitation12–15. In addition, patients with ARG1-D require a range of continuous therapies and ongoing monitoring to control manifestations of the disease and are often hospitalized multiple times, most often related to hyperammonemia, infection, or gastrointestinal-related illness on an annual basisCitation1,Citation4,Citation5.
In most cases, manifestations of ARG1-D first appear between the ages of 1 and 3 years. Diagnosis is based on elevated plasma arginine levels, reduced red blood cell arginase activity, or identification of ARG1 variants, after clinical suspicionCitation1. However, delays in diagnosis or misdiagnosis are frequent for several reasonsCitation16. Likely due to its perceived rarity, disease awareness is lower for ARG1-D than other UCDs, which can result in delays in appropriate screening/work-up activities. Further, other more common neurological diseases such as cerebral palsy and hereditary spastic paraplegia, are similar to ARG1-D in initial clinical presentation and may lead to misdiagnosis and/or delayed ARG1-D diagnosisCitation2,Citation17. In areas where newborn screening is available, infants with ARG1-D can be identified in the first days of lifeCitation1; however, there are limitations to these programs, including false-negative results, lack of universal availability, and an absence of standardized diagnostic thresholdsCitation18–20. The current challenges with diagnosis may prevent timely treatment initiation, which may contribute to the progression of the disease and lead to increasing health care resource utilization (HCRU) over time.
Disease management recommendations for ARG1-D are focused on reducing plasma arginine levels while maintaining adequate nutrition to prevent growth impairment, decreasing the risk of hyperammonemia, and managing any hyperammonemic episodesCitation9,Citation21. Management largely relies on a protein-restricted diet to limit arginine intake, often with essential amino acid and vitamin supplementation, under the guidance of a metabolic dietitian. Protein restriction has limited effectiveness, as a minimal amount of arginine comes from dietary intake whereas the majority comes from endogenous protein turnover and de novo synthesisCitation22. In some patients, ammonia scavengers may be used to prevent or manage hyperammonemia episodes, which can be severe and often require hospitalization, but use of scavengers is primarily indicated to reduce nitrogen flux through the urea cycle. Common medical interventions/procedures include physical and occupational therapy, imaging, and laboratory assessments. A subset of patients may require high-cost interventions, including dialysis and/or liver transplantationCitation9,Citation21. There are currently no management strategies or treatments that effectively address high arginine levels to prevent disease progression.
Several studies have demonstrated that the economic burden (high direct and indirect medical costs) of rare diseases in general is significant due to substantial use of health care resourcesCitation23–25. Based on a health care database analysis from 2006 to 2020, Tisdale et al. reported that direct per-patient medical costs for individuals with rare diseases were 3- to 5-fold higher than age-matched comparatorsCitation25. Further, ARG1-D and other UCDs were associated with the highest per-patient per-year costs compared with 12 other rare diseasesCitation25. Beyond this analysis, there is little information on the HCRU of patients with UCDs overall or ARG1-D specifically. Therefore, the objective of this study was to assess and understand the all-cause HCRU in the United States (US) among patients diagnosed with ARG1-D relative to a comparator cohort.
The study findings may have implications on patients, healthcare givers, healthcare providers, and payers in the US. The major practical implication of this study is that it describes HCRU and burden of ARG1-D. This information is important given that all other published studies describe burden of UCDs in general without focusing specifically on ARG1-D. Further exploring claim databases and having different perspectives may help future observational real-world studies to further understand key drivers of HCRU and the potential impact of new treatments for ARG1-D.
Methods
This retrospective, observational, real-world study was conducted using claims data from the IQVIA New Data Warehouse from 1 October 2015 through 31 May 2020. Eligibility criteria were preidentified to include patients in two separate cohorts (ARG1-D and non-ARG1-D). The ARG1-D cohort required ≥2 non-same-day medical claims with the ARG1-D specific ICD-10 diagnosis code (E72.21) at any time during the patient identification period, with the first date considered to be the index date. Eligible patients were required to have linkage between the pharmacy and physician claim databases, and to have continuous medical and pharmacy coverage for six months (180 days) before (pre-index period) and starting on (post-index period) the index date. Claims with a missing diagnosis code or an invalid year of birth or sex were excluded to maintain data quality. Linkage with hospital claims data was not required, as this information was available for only a small subset of patients with this rare disease during the study period. The comparator cohort had the same requirements excluding the diagnosis of ARG1-D, with index dates randomly assigned to replicate the distribution of index dates in the ARG1-D cohort. Notably, the comparator cohort was not intended to represent the healthy US general population because it was developed using the claims database and thus included patients with acute or chronic illnesses requiring utilization of health care resources, resulting in a claim. Therefore, the comparator cohort included a mix of healthy, acute, and chronic patients.
The group of patients with ARG1-D was propensity score-matched on a one-to-one basis to the non-ARG1-D comparator cohort to adjust for observable selection bias and potential baseline differences between cohorts. A Greedy Nearest Neighbor matching algorithm was used without replacement at a ratio of 1:1 using caliper widths of 0.01 of the standard deviation of the logit of the propensity score. Matching variables included age group, sex, index year, payer type (Medicare, Medicaid, third party) and geographic region (Northeast, Midwest, South, West). All patients in the comparator group who were not successfully matched were excluded from further analysis.
The data collection process was standardized and structured to reduce observation bias. The demographics included age, both continuous and categorical, sex, geographic region, and payer type. Baseline clinical characteristics were evaluated during the fixed pre-index period (180 days). Clinical characteristics captured included Charlson Comorbidity Index (a method of categorizing comorbidities of patients based on the International Classification of Diseases diagnosis codes)Citation26, frequent comorbidities as well as select comorbidities of interest, symptoms and manifestations of interest, frequent medical procedures as well as select medical procedures of interest, specific medical devices and mobility aids, and targeted concomitant medications of interest (nitrogen scavengers, antispasmodic agents, anti-epileptics, medical foods, and psychiatric medications).
Pre- and post-index all-cause HCRU included pharmacy utilization (≥1 pharmacy claims for specific categories, ARG1-D related medications, and any medication usage), outpatient services (emergency room [ER] visits, physician office visits, procedures, and lab and pathology assessments), and inpatient services among the subset of patients with this database linkage who had ≥1 inpatient stay.
This study used claims data from 1 October 2015 (the first date of ICD-10 implementation) through 31 May 2020 (the last date the data were available when the study was designed). To have enough window to capture patients with ARG1-D (an ultra-rare disease) and to allow an analysis window of 6 months before with a 6-month follow-up period after the index date, patients were identified based on index dates from 1 April 2016 through 30 November 2019. The analytic file was created using SAS v9.4 (SAS Institute Inc., Cary, NC). Descriptive and adjusted analyses were conducted to evaluate and compare the study measures. For categorical variables, frequency and percentage were evaluated using a Chi-square test or McNemar’s test. For the continuous variables, mean, median, and standard deviation (SD) were reported using an independent sample t-test, Wilcoxon rank-sum test, paired t-test, or Wilcoxon signed-rank test, as appropriate.
Results
Using the claims-based algorithm for ARG1-D, a total of 138 patients were identified, of which 77 met all inclusion criteria. The other 61 patients were excluded due to lack of database linkage (n = 27) or non-continuous insurance enrollment requirements (n = 34). Of the final sample, 24/77 prevalent patients had a diagnosis code for ARG1-D identified during the pre-index period. Linkage to the hospital claims database was available for 17 patients in the ARG1-D cohort and 16 patients in the comparison cohort.
Baseline demographic characteristics
Demographic characteristics were well balanced between the two cohorts (ARG1-D cohort: 52% male, mean age ± SD of 25.6 ± 23.9 years, median age of 15 years; comparator cohort: 52% male, mean age ± SD of 26.3 ± 24.1 years, median age of 15 years). In each cohort, the most common age group was 2–9 years (26.0%) and 52% of patients <18 years of age ().
Table 1. Baseline demographic characteristics.
Baseline clinical characteristics
There was a high degree of variability in diagnosis codes in the comparator cohort, and no frequency trends toward any one diagnosis were observed. The mean ± SD of the Charlson Comorbidity Index score was higher for patients with ARG1-D than the comparator cohort (1.1 ± 2.2 vs. 0.3 ± 0.7). However, there was no statistically significant difference among cohort subgroups based on specific Charlson Comorbidity Index scores as the non-ARG1-D cohort had a variety of diagnoses (i.e. around 20% of the patients had Charlson Comorbidity Index score ≥1).
The occurrence of specific manifestations was more frequent and greater in magnitude in the ARG1-D cohort compared with the comparator group. For example, in patients with ARG1-D, spasticity (7 times higher), developmental delay (∼2 times higher), intellectual disability (5 times higher), and seizures (8 times higher) were multiple-fold more frequent compared with patients without ARG1-D (). The overall occurrence of other comorbidities was also more common among patients with ARG1-D (i.e. liver disease [13 times], epilepsy [7.5 times], cirrhosis/fibrosis [6 times]) compared with patients without ARG1-D ().
Figure 1. Prevalent manifestations. Abbreviation. ARG1-D, Arginase 1 Deficiency.
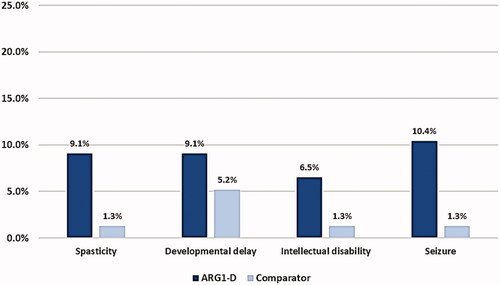
Table 2. Baseline clinical characteristics.
In the pre-index period, the ordering of laboratory tests for complete blood counts was reported approximately 2 times more frequently in the ARG1-D cohort, compared with the non-ARG1-D cohort (p < .05). Physical and occupational therapy was accessed nearly 2 times more frequently and CT scans were obtained 10 times more frequently in the ARG1-D cohort, compared with the comparison cohort (). As expected, medications commonly used in the management of ARG1-D were prescribed more frequently in this cohort. Anti-epileptics were prescribed around 3 times more frequently in patients with ARG1-D (p = .0002). Prescription rates for psychiatric medications (1.5 times higher), the antispasmodic medication baclofen (10 times higher, p < .01), and medical foods (5 times higher, p < .05) were more common in the ARG1-D cohort (). Assistive devices are commonly prescribed for patients with mobility deficits, such as patients with ARG1-D. Use of medical devices and aids, including ankle-foot orthoses, arm crutches, wheelchair, and canes/walking aids, was numerically higher in the ARG1-D cohort, but did not reach statistical significance.
All-cause health care resource utilization
Inpatient and outpatient visits are frequently needed to manage serious medical conditions. In the post-index period, around 20% of the patients had the linkage to hospital data (i.e. three patients in the disease cohort and one in the comparator cohort with an inpatient stay). Patients with ARG1-D were hospitalized nearly 3 times more often and the mean length of stay was longer compared with patients without ARG1-D, although this trend was not statistically significant. The proportion of patients with emergency room visits in the ARG1-D cohort was nearly double that for patients without ARG1-D (p < .05). Laboratory/pathology claims were 1.5 times more frequent with ARG1-D cohort (p < .01) (). Utilization of ammonia scavengers, including glycerol phenylbutyrate, sodium phenylbutyrate, or sodium benzoate/sodium phenylacetate were reported in a small percentage of ARG1-D patients post-index (). There was no trend suggesting use of any one medication class in the comparator group.
Table 3. All-cause health care resource utilization.
Discussion
To our knowledge, this is the first study to describe HCRU and burden of ARG1-D. Some studies have examined the clinical and economic burden in very specific populations such as in Alzheimer’s diseaseCitation27,Citation28 and more generally as a component of a broader condition such as intoxication-type inborn errors of metabolism (IT-IEM)Citation29–31, but none have focused specifically on the HCRU associated with ARG1-D. Our results suggest substantial HCRU and treatment burden in ARG1-D, with higher rates of hospitalization, common comorbidities, medication use, and potentially debilitating manifestations (spasticity, developmental delay, intellectual disability, and seizures) reported in the ARG1-D cohort compared with the comparator group.
Eligibility criteria for patients to be included in this study were preidentified to avoid selection bias. In addition, this study used propensity score matching, a statistical technique used to reduce selection bias, to match patients with ARG1-D on a one-to-one basis with non-ARG1-D comparatorsCitation32. We used this technique to control for potential confounding variables (age, sex, index year, payer type, and geographic region) while estimating HCRU for ARG1-D patients. Nonetheless, actual HCRU associated with ARG1-D may be underestimated in our findings. First, the global prevalence of ARG1-D may be higher than currently reported because of low disease awareness, limited newborn screening, and misdiagnosis (e.g. as cerebral palsy) or delays in diagnosisCitation15,Citation33. These challenges can lead to a diagnosis occurring later in the disease course, and therefore delays in treatment that lead to unchecked progression of spasticity (unlike cerebral palsy), cognitive dysfunction, and mobility issues that in turn increases disease burden and HCRU. Diaz et al. reviewed the literature and concluded that delays in diagnosis are associated with poor clinical outcomes in patients with ARG1-DCitation34. While Diaz et al. reported high proportions of patients with spasticity (84%)Citation34, the current study reported a lower rate of spasticity (9%) which is likely attributed to the fact that patients with ARG1-D in this claims analysis were identified based on first diagnosis code as the index date and could have been relatively early in their disease course or spasticity could be underreported in the database. Even though most patients with ARG1-D were newly diagnosed, it is not clear how far along in their disease they were (e.g. they could have been very early or pre-symptomatic if diagnosed by newborn screening). During the patient identification period, the first date with a diagnosis code for ARG1-D was considered to be the index date. However, of the 77 patients included, 24 also had a diagnosis code for ARG1-D identified before the patient identification period (i.e. greater HCRU), and 53 patients were identified in the earlier course of their disease (i.e. no diagnosis code for ARG1-D identified before the patient identification period) and likely with less disease progression/burden (i.e. less HCRU) than the general ARG1-D population. In addition, this study was conducted over a relatively short period with limited collection of longitudinal data, and therefore is not able to reflect the increasing burden of disease as ARG1-D progresses over a span of years. In the present study, there was a larger proportion of younger patients in the ARG1-D cohort (median age, 15 years; 26% aged 2–9 years), and manifestations are known to worsen progressively over time. In a previous analysis, Bin Sawad (2021) systematically reviewed published case reports and described the natural history of ARG1-DCitation35. That study found a higher proportion of patients with common ARG1-D clinical manifestations than the current analysis (spasticity: 69%, developmental delay: 37%, intellectual disability: 55%, and seizures: 50%)Citation35, further suggesting that the real-world burden and HCRU associated with ARG1-D may be greater than identified in the current study. Importantly, the occurrence of these manifestations may also have been underestimated in the previous analysis, as case reports do not often include comprehensive diagnostic/clinical characterization and follow-up; therefore, lack of mention of a given manifestation or intervention in case reports is not necessarily evidence of its absence.
The negative impact of ARG1-D on mental health was also observed in this study. Although the prevalence of diagnosis codes for disorders such as depression and anxiety was relatively low in this cohort, psychiatric medications were prescribed in approximately one-third of patients. This rate is considerably higher than observed for matched comparators. Additionally, patients in the ARG1-D cohort had more ER visits and laboratory/pathology utilization than the comparator cohort. However, they had a lower rate of physician office visits, possibly indicating challenges with transportation (e.g. due to mobility impairments or other health-related limitations) and/or a need to access competing or other health services (e.g. home care) that would make doctor visits less frequent.
There are some limitations to this study, given the retrospective nature of the analysis, data source, study duration, and patient population. Claims databases are limited by their intended purpose of reimbursement vs research use, and as such may not be generalizable. Similarly, the patient population consisted of individuals enrolled in managed care plans in the US (commercial, Medicare, Medicaid) and may not be applicable to other populations (e.g. those in the US with other forms of health insurance, or the uninsured, and those in other countries with different healthcare models). As a result, key outcome measures such as disease manifestations and medication use may be underrepresented because every code may not be recorded during our study period.
While a claim was required to be present in the database itself and in the comparator cohort, that claim could have been for a physician office visit, a simple pharmacy claim, an event requiring treatment or an inpatient admission. So, we cannot draw conclusions about the relative health of the comparator cohort. However, the non-ARG1-D cohort in this study appears to not represent a healthy population. Although the percentage of patients in the comparator cohort with ≥1 ER visit (18%) or hospitalization (6%) is similar to rates reported for the general population (20–27% and 7%, respectively) Citation36,Citation37, there were important differences compared with the general population. Among the US general population, approximately 45% of individuals filled ≥1 prescriptions in the period 2015–2018Citation38 and an estimated 30% reporting having visited or consulted a primary care physician in a given yearCitation39. In our study’s comparator cohort, 83% had ≥1 prescription filled and 75% of patients had ≥1 physician office visits, which translates to rates approximately double the general population. These observations can be attributed to the fact that the individuals included in our comparator cohort had significant health concerns and required health care interventions on an outpatient and regular basis. As such, the relative burden of ARG1-D compared with the general population may be even greater than observed in this study because of the use of a non-healthy comparator cohort.
The number of patients included in the study is relatively small, but this fact reflects the rarity of the ARG1-D; there are only approximately 250 patients in the USCitation40. Therefore, the 77 patients in our cohort could represent nearly 30% of the total estimated US population with ARG1-D. In addition, even with this small sample size, statistical significance was demonstrated in many outcome measures (e.g. morbidity, manifestations). In addition to limited sample size, the relatively short follow-up period (6 months) after diagnosis (index date) and a comparator group containing a non-healthy population (that may include a high number of patients with mobility issues) may explain the numerically higher but non-statistically significant rates for use of medical devices and aids among the two cohorts. Growth and development in children may affect their need for assistive devices but the duration of the study is not sufficient to reflect these changes; similarly, this follow-up duration is likely too short to reflect progression of manifestations associated with ARG1-D, such as spasticity, that may impact the need for assistive devices. There was a higher frequency of hospitalizations in the ARG1-D cohort, which can be attributed to the nature of the underlying disease and comorbidities; however, the numbers of subjects requiring a hospital stay in both cohorts were small and the conclusions that can be drawn from this observation are limited.
Lastly, while any diagnosis or procedure code may have been incorrectly coded, this risk was mitigated by the requirement that patients had to have at least two claims for ARG1-D. The presence of a dispensed medication does not indicate that the medication was administered or that it was taken correctly as prescribed, but these are recognized limitations of using real-world data. Only patients with the proxy for continuous eligibility with medical and pharmacy benefits were included, and thus patients who utilized health care during the outcome period for six months starting on the index date were not included in the sample, nor were patients without linkage between the pharmacy and physician claims databases. The number of claims for ARG1-D-related medications may have been impacted by the selection criteria used to define the sample, as well as the fact that not all specialty pharmacies were represented in the database. Similarly, inpatient data were likely underreported in this sample due to low representation in the hospital claim database.
A discussion of HCRU would not be complete without a mention of the impact on patients and their families and their indirect costs and time and QoL. Patient and caregiver burden can be extremely high with ARG1-D for direct care given the multiple hospitalizations and outpatient physician visits to manage the disease itself as well as the various associated manifestations. As these patients have functional difficulties with mobility, transportation challenges can add to the burden for caregivers. Multiple weekly trips to occupational and/or physical therapy add to the commute for lengthy appointments. Frequent blood draws and imaging visits are also time consuming, complicated, and can be painful. Obtaining, keeping track of, and administering multiple medications with various doses at different times of day is also challenging and demanding. Getting patients assessed, fitted for, and trained in the correct use of medical devices is also onerous, particularly for growing children who will need to be refitted on an ongoing basis. It is important to understand the costs and challenges of living with and managing this disease and what can be done to decrease the burden for those affected by ARG1-D.
Conclusions
In this retrospective claims study of patients with ARG1-D, patients presented with a substantial comorbidity profile and considerable treatment burden with significant HCRU despite representing a relatively young, newly diagnosed cohort, being followed for a relatively short amount of time, and being treated with current standard of care. Patients with ARG1-D had a statistically higher percentage of hospitalizations, ER visits, therapy claims, medication usage and a higher mean number of imaging/laboratory/pathology tests. This increased level of utilization appeared to balance the lower number of physician office visits. Despite the fact that the index date represented the date of first diagnosis in two-thirds of the ARG1-D cohort indicating that they may have been relatively early in their disease course, patients with ARG1-D still had significantly higher HCRU during the 6-month study period compared with a matched cohort of patients without ARG1-D (who were, however, already using health care resources). The comorbidity profile of patients with ARG1-D was associated with a substantial health care resource use and cost burden prior to diagnosis.
This study is limited by not measuring components like direct medical costs, indirect costs, loss productivity, and QoL of patients with ARG1-D. Future research is warranted to further understand key drivers of HCRU and the potential impact of new treatments that could effectively lower arginine levels to therapeutic guidelines or even normal levels, to prevent, delay, improve, or reverse disease manifestations, with the goal of significantly reducing the health and economic and treatment burden on health care systems and among patients with ARG1-D and their families.
Transparency
Declaration of funding
This study was sponsored by Aeglea BioTherapeutics.
Declaration of financial/other interests
At time of writing, AB, JJ, and MB are employees of Aeglea BioTherapeutics. MH, JY, and YW are employees of IQVIA. GD is employed at the Division of Medical Genetics and Genomics in the Department of Genetics and Genomic Sciences at the Icahn School of Medicine at Mount Sinai and has served as an advisor/consultant and clinical trial investigator for Aeglea BioTherapeutics.
Author contributions
AB, JJ, MB, and GD conceptualized and designed the study. MH, JY, and YW collected and analyzed the data. All authors interpreted the data, critically revised the manuscript, and approved the final version for publication.
Peer reviewer disclosures
Peer reviewers on this manuscript have received an honorarium from JME for their review work but have no other relevant financial relationships to disclose.
Acknowledgements
The authors would like to thank Linda Neuman and Michael Hanley (employees of Aeglea BioTherapeutics) for reviewing the manuscript and providing their feedback, comments, and suggestions. The authors also would like to acknowledge writing assistance from Katharine Coyle, who is an employee of IQVIA.
References
- Summar ML, Koelker S, Freedenberg D, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013;110(1–2):179–180.
- Sin YY, Baron G, Schulze A, et al. Arginase-1 deficiency. J Mol Med. 2015;93(12):1287–1296.
- Catsburg C, Anderson S, Upadhyaya N, et al. Arginase 1 deficiency: using genetic databases as a tool to establish global prevalence. Orphanet J Rare Dis. 2022;17(1):1–7.
- Asrani KH, Cheng L, Cheng CJ, et al. Arginase I mRNA therapy – a novel approach to rescue arginase 1 enzyme deficiency. RNA Biol. 2018;15(7):914–922.
- Enns GM, Porter MH, Francis-Sedlak M, et al. Perspectives on urea cycle disorder management: results of a clinician survey. Mol Genet Metab. 2019;128(1–2):102–108.
- Iyer R, Jenkinson CP, Vockley JG, et al. The human arginases and arginase deficiency. J Inherit Metab Dis. 1998;21(Suppl 1):86–100.
- Morris SM. Jr. Arginases and arginine deficiency syndromes. Curr Opin Clin Nutr Metab Care. 2012;15(1):64–70.
- GARD. Arginase deficiency. [cited 2021 Dec 7]. https://rarediseases.info.nih.gov/diseases/5840/arginase-deficiency.
- Haberle J, Burlina A, Chakrapani A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: first revision. J Inherit Metab Dis. 2019;42(6):1192–1230.
- Sun A, Crombez EA, Wong D, et al. Arginase deficiency. In: Adam MP, Ardinger HH, Pagon RA editors. GeneReviews((R)). Seattle (WA): University of Washington, Seattle; 2000. [updated 2021].
- Blumenfeld AM, Varon SF, Wilcox TK, et al. Disability, HRQoL and resource use among chronic and episodic migraineurs: results from the international burden of migraine study (IBMS). Cephalalgia. 2011;31(3):301–315.
- Dhamane AD, Witt EA, Su J. Associations between COPD severity and work productivity, health-related quality of life, and health care resource use: a cross-sectional analysis of national survey data. J Occup Environ Med. 2016;58(6):e191-197–e197.
- Germain N, Kymes S, Löf E, et al. A systematic literature review identifying associations between outcomes and quality of life (QoL) or healthcare resource utilization (HCRU) in schizophrenia. J Med Econ. 2019;22(5):403–413.
- Pawaskar M, Iglay K, Witt EA, et al. Impact of the severity of hypoglycemia on health – related quality of life, productivity, resource use, and costs among US patients with type 2 diabetes. J Diabetes Complications. 2018;32(5):451–457.
- Huemer M, Carvalho DR, Brum JM, et al. Clinical phenotype, biochemical profile, and treatment in 19 patients with arginase 1 deficiency. J Inherit Metab Dis. 2016;39(3):331–340.
- Diez-Fernandez C, Rüfenacht V, Gemperle C, et al. Mutations and common variants in the human arginase 1 (ARG1) gene: impact on patients, diagnostics, and protein structure considerations. Hum Mutat. 2018;39(8):1029–1050.
- Morales JA, Sticco KL. Arginase deficiency. StatPearls. Treasure Island (FL): StatPearls Publishing; 2022.
- Rajabi F. Updates in newborn screening. Pediatr Ann. 2018;47(5):e187–e190.
- Therrell BL, Currier R, Lapidus D, et al. Newborn screening for hyperargininemia due to arginase 1 deficiency. Mol Genet Metab. 2017;121(4):308–313.
- Haberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. J Inherit Metab Dis. 2012;1:S4.
- Schlune A, Vom Dahl S, Häussinger D, et al. Hyperargininemia due to arginase I deficiency: the original patients and their natural history, and a review of the literature. Amino Acids. 2015;47(9):1751–1762.
- Diaz GA, Krivitzky LS, Mokhtarani M, et al. Ammonia control and neurocognitive outcome among urea cycle disorder patients treated with glycerol phenylbutyrate. Hepatology. 2013;57(6):2171–2179.
- Everylife Foundation. The national burden of rare disease study. 2021. [2021 Dec 8]. https://everylifefoundation.org/burden-study/.
- Navarrete-Opazo AA, Singh M, Tisdale A, et al. Can you hear us now? The impact of health-care utilization by rare disease patients in the United States. Genet Med. 2021;23(11):2194–2201.
- Tisdale A, Cutillo CM, Nathan R, et al. The IDeaS initiative: pilot study to assess the impact of rare diseases on patients and healthcare systems. Orphanet J Rare Dis. 2021;16(1):1–8.
- Charlson ME, Pompei P, Ales KL, et al. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373–383.
- Polis B, Samson AO. Role of the metabolism of branched-chain amino acids in the development of Alzheimer's disease and other metabolic disorders. Neural Regen Res. 2020;15(8):1460–1470.
- Bösch F, Landolt MA, Baumgartner MR, et al. Health-related quality of life in paediatric patients with intoxication-type inborn errors of metabolism: analysis of an international data set. J Inherit Metab Dis. 2021;44(1):215–225.
- Zeltner NA, Baumgartner MR, Bondarenko A, et al. Development and psychometric evaluation of the MetabQoL 1.0: a quality of life questionnaire for paediatric patients with intoxication-type inborn errors of metabolism. JIMD Rep. 2017;37:27–35.
- Zeltner NA, Landolt MA, Baumgartner MR, et al. Living with intoxication-type inborn errors of metabolism: a qualitative analysis of interviews with paediatric patients and their parents. JIMD Rep. 2017;31:1–9.
- Vasquez-Loarte T, Thompson JD, Merritt JL. Considering proximal urea cycle disorders in expanded newborn screening. Int J Neonatal Screen. 2020;6(4):77.
- Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res. 2011;46(3):399–424.
- National Organization of Rare Disorders. Rare disease database: arginase-1 deficiency. 2020. [cited 2021 Dec 16]. https://rarediseases.org/rare-diseases/arginase-deficiency/.
- Diaz GA, Longo N, Bubb G, et al. Delays in diagnosis are associated with poor clinical outcomes in patients with arginase 1 deficiency. Ann Neurol. 2019;86:S137–S137.
- Bin Sawad A, Pothukuchy A, Badeaux M, et al. Natural history of arginase 1 deficiency and the unmet needs of patients: a systematic review of case reports. JIMD Rep. 2022. DOI:10.1002/jmd2.12283
- Statista. [cited 2022 Feb 3]. https://www.statista.com/statistics/184432/us-population-with-emergency-room-visits-by-age/.
- Statista. [cited 2022 Feb 3]. https://www.statista.com/statistics/184447/us-population-with-a-hospitalization-by-age/#:∼:text=Hospital%20Stays&text=In%202018%2C%20around%20seven%20percent,hospital%20is%20currently%205.4%20days.
- Statista. [cited 2022 Feb 2]. https://www.statista.com/statistics/492239/us-population-with-usage-of-prescription-drugs-within-one-month/.
- Statista. [cited 2022 Feb 2]. https://www.statista.com/statistics/916781/primary-care-physician-visit-frequency-among-adults-us/.
- Aeglea data on file.