
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Aims
To describe clinical complications, treatment use, healthcare resource utilization (HCRU), and costs among patients with transfusion-dependent β-thalassemia (TDT) in the United States.
Materials and methods
Merative MarketScan Databases were used to identify patients with β-thalassemia between 1 March 2010, and 1 March 2019. Patients were eligible for inclusion with ≥1 inpatient claim or ≥2 outpatient claims for β-thalassemia and ≥8 red blood cell transfusions (RBCTs) during any 12-month period after and including the date of the first qualifying β-thalassemia diagnosis code. Matched controls consisted of individuals without β-thalassemia. Clinical and economic outcomes of patients were assessed during ≥12 months of follow-up, defined as the period from the index date (i.e. the first RBCT) to either the end of continuous enrollment in benefits, inpatient death, or 1 March 2020.
Results
Overall, 207 patients with TDT and 1035 matched controls were identified. Most patients received iron chelation therapy (ICT) (91.3%), with a mean of 12.1 (standard deviation [SD] = 10.3) ICT claims per-patient-per-year (PPPY). Many also received RBCTs, with a mean of 14.2 (SD = 4.7) RBCTs PPPY. TDT was associated with higher annual ($137,125) and lifetime ($7.1 million) healthcare costs vs. matched controls ($4183 and $235,000, respectively). Annual costs were driven by ICT (52.1%) and RBCT use (23.6%). Patients with TDT had 7-times more total outpatient visits/encounters, 3-times more prescriptions, and 33-times higher total annual costs than matched controls.
Limitations
This analysis may underestimate the burden of TDT, as indirect healthcare costs (e.g. absenteeism, presenteeism, etc.) were not included. Results may not be generalizable to patients excluded from this analysis, including those with other types of insurance or without insurance.
Conclusions
Patients with TDT have high HCRU and direct healthcare costs. Treatments that eliminate the need for RBCTs could reduce the clinical and economic burden of managing TDT.
Introduction
β-thalassemia is a hereditary hemoglobinopathy that affects approximately 40,000 births worldwide per yearCitation1,Citation2. β-thalassemia primarily impacts individuals in Mediterranean, Middle Eastern, and Southeast Asian countries; however, increased migration from these regions with high carrier rates in the past 50 years has led to an increase in the prevalence of β-thalassemia in the United StatesCitation3–5. Despite awareness of the impact of these migration patterns, the true prevalence of this hemoglobinopathy remains poorly documented in the United StatesCitation6,Citation7.
β-thalassemia is caused by any of >200 mutations in the β-globin gene (HBB)Citation3,Citation8. These mutations lead to reduced (β+) or absent (β0) β-globin chain synthesis and excessive unbound α-globin chain synthesisCitation3,Citation8. In β-thalassemia, unbound α-globin chains are unstable and contribute to the development of clinical complications, including ineffective erythropoiesis, iron overload, and anemia, as well as negative effects on growth, vascular function, organ systems (e.g. the cardiovascular, endocrine, hepatic, and skeletal systems), and mortalityCitation3,Citation8. The clinical severity of the disease reflects many different factors, including the type of mutation in HBB, the extent of α-to-β-globin chain imbalance, ineffective erythropoiesis, anemia, and the number of required red blood cell transfusions (RBCTs)Citation8,Citation9.
A subgroup of patients with β-thalassemia, or those with transfusion-dependent β-thalassemia (TDT), depend on regular RBCTs to manage their clinical complications and risk for mortalityCitation8,Citation9. Iron chelation therapy (ICT) is further required to manage the iron overload stemming from both the disease pathology and RBCTs in patients with TDTCitation8,Citation9. Ultimately, there remains an unmet need to understand the clinical and economic burden associated with managing TDT.
Previous studies have reported high healthcare resource utilization (HCRU) and healthcare costs for patients living in regions with high carrier rates of TDT, such as TaiwanCitation10 and the United Arab EmiratesCitation11. To our knowledge, however, only two studies have evaluated the economic outcomes associated with TDT in the United States. Their study populations were either limited to adult patients with TDT with commercial insurance coverageCitation12 or included a broad group of patients aged 3 to 52 years and with any form of thalassemia, ≥1 RBCT, and ≥1 claim for deferoxamineCitation13. Moreover, the latter study was conducted from 1997 to 2004 prior to the approval of oral ICTsCitation13. Contemporary and comprehensive data, obtained in patients with TDT with different ages and types of insurance coverage, are needed to fully understand the clinical and economic impact of TDT. Therefore, the primary aim of this retrospective real-world analysis, which used U.S. administrative claims data, was to describe the clinical and economic burden associated with TDT.
Methods
Study design and data source
The study methods for a similarly designed retrospective real-world analysis of patients with sickle cell disease have been previously describedCitation14. The Merative MarketScan Commercial, Medicare Supplemental, and Multi-State Medicaid Databases house inpatient medical, outpatient medical, and outpatient pharmacy data and were used to identify patients with β-thalassemia between 1 March 2010, and 1 March 2019. Approximately 89 million individuals and their dependents with commercial insurance coverage, 4.6 million individuals with Medicare Supplemental coverage, and 20 million individuals with Medicaid coverage were included in the databases during the study period (1 March 2010, to 1 March 2020). This study did not require institutional review board approval due to the de-identified nature of the data.
Study population
The study included patients of any age with a diagnosis code for β-thalassemia (International Classification of Diseases, Ninth or Tenth Revision, Clinical Modification [ICD-9/10-CM]: ICD-9-CM 282.44 and 282.47; ICD-10-CM D56.1 and D56.5) between 1 March 2010, and 1 March 2019. In addition, eligible patients had to have ≥1 non-diagnostic inpatient claim or ≥2 non-diagnostic outpatient claims with service dates within 365 days of each other (). Evidence of TDT was confirmed through documentation of ≥8 RBCTs during any 12-month period after and including the date of the first qualifying diagnosis code for β-thalassemia. RBCTs were identified through current procedural terminology codes (CPT36430 Transfusion, blood or blood components; CPT36440 Venipuncture and transfusion procedures; CPT36450 Exchange transfusion, blood; CPT36455 Exchange transfusion, blood; CPT36456 Exchange transfusion, blood; CPT36512 Therapeutic Apheresis and photopheresis procedures; CPT86999 Transfusion medicine procedures) and ICD-9/10 procedure codes. RBCTs were considered discrete transfusion events if the service dates of the claims occurred ≥3 days apart from each other. The index date was the date of the first RBCT. A minimum of 12 months of continuous enrollment with medical and pharmacy benefits after and including the index date were required to be eligible for inclusion. Patients were excluded if they had ≥2 claims for sickle cell disease or had received a bone marrow transplant or hematopoietic stem cell transplant during the follow-up. All patients were followed for ≥12 months, from the index date to the earliest date of either inpatient death, end of continuous enrollment, or end of the study period (1 March 2020).
Figure 1. Patient attrition.
Abbreviations. BMT, bone marrow transplant; HSCT, hematopoietic stem cell transplant; RBCT, red blood cell transfusion; SCD, sickle cell disease. aClaims must have occurred within 365 days of each other. Only non-diagnostic claims were included; non-diagnostic claims are medical claims in which diagnosis codes reflect confirmed diagnoses rather than suspected diagnoses reported to justify diagnostic tests or procedures. bRBCTs were considered discrete transfusion events if the service dates of the claims occurred ≥3 days apart from each other.

Patients with TDT were matched to up to five individuals without β-thalassemia in the Merative MarketScan Databases to aid in the interpretation of healthcare costs. Matching was based on age, sex, geographic region, payer type, and amount of follow-up data. Matched controls could not have had claims for nonmalignant blood disorders, such as anemia, β-thalassemia, or sickle cell disease and were required to have ≥12 months of continuous enrollment with medical and pharmacy benefits after and including the index date to be eligible for inclusion. Random index dates were assigned to matched controls based on the distribution of index dates among patients with TDT.
Study outcomes
Clinical outcomes, including clinical complications and treatment use, were evaluated during the variable-length follow-up. Clinical complications were identified using non-diagnostic claims, and treatment use was determined by the presence and total number of medical and pharmacy claims for key treatments of interest (e.g. ICT). Only clinical complications and treatment use documented in >10% of the overall cohort are reported here. Rates of RBCT and ICT use were characterized separately.
Economic outcomes, including the annual rates of inpatient admissions, outpatient visits/encounters, and outpatient prescriptions, as well as annual and lifetime healthcare costs, were also determined during the variable-length follow-up. Total outpatient visits/encounters were counted by identifying the number of unique dates with a claim for a particular healthcare provider type (i.e. emergency department visit, physician office visit, laboratory encounter, or “other” outpatient visit/encounter [i.e. defined as a claim for a non-office setting visit]). For example, an emergency department claim and a physician’s office claim on the same date would count as two outpatient visits/encounters, whereas two physician’s office claims on the same date would count as one outpatient visit/encounter. Emergency department visits that resulted in an inpatient admission were counted as inpatient admission.
Adjudicated claims (e.g. insurer and health plan payments) and patient cost-sharing (e.g. co-payments, deductibles, and co-insurance)Citation15,Citation16 were utilized to calculate per-patient-per-year (PPPY) healthcare costs. Using the Medical Care Component of the Consumer Price Index, costs were adjusted for inflation to 2020 U.S. dollars and medical pricesCitation17. Lifetime costs were calculated through patients’ estimated life expectancies (LEs) and in pre-specified age categories (0 to 11 years, 12 to 17 years, 18 to 35 years, and ≥36 years) as follows:
In lifetime costs calculations, it was assumed that a patient lived to age 50; adjustments were not made for expected mortality.
Statistical analysis
Clinical and economic outcomes for patients with TDT were summarized using descriptive statistics, while differences in HCRU and healthcare costs between patients with TDT and matched controls were evaluated using comparative analyses. Chi-squared tests were used to determine the statistical significance of differences for categorical variables, and t-tests were used for continuous variables. Clinical complications were evaluated in pre-specified age groups (0 to 11 years, 12 to 35 years, ≥36 years). A p-value of < .05 was considered statistically significant.
Results
Demographics
In total, 4504 patients in the MarketScan Databases had claims for β-thalassemia between 1 March 2010, and 1 March 2019. After the inclusion and exclusion criteria were applied and patients were matched to controls from the general population, 207 patients were identified as having TDT and 1035 individuals identified as matched controls (). The mean age of patients was 21.9 years (standard deviation [SD] = 16.4 years; range: 0–85 years) and most were female (57.5%) and included in the Commercial Database (80.7%). Both groups were balanced on matched characteristics after matching criteria were applied ().
Table 1. Baseline demographics and payer typesa.
Clinical complications and treatment use
The most common clinical complications (documented in >10% of the patient population) were iron overload (87.9%), endocrine complications (e.g. diabetes mellitus, osteopenia, or osteoporosis [41.5%]), acute infections (30.4%), hepatobiliary complications (e.g. gallstones or hepatitis [20.8%]), cardiopulmonary complications (e.g. arrhythmia or heart failure [18.4%]), and splenomegaly (17.4%) (). The prevalence of endocrine, hepatobiliary, and cardiopulmonary complications generally increased with patient age (Supplementary Table 1). Further analysis by prespecified age subgroups showed that a considerable proportion of patients with TDT in the youngest age group (0 to 11 years) experienced clinical complications, with the most common being iron overload (93.1%), acute infections (34.7%), and splenomegaly (30.6%) (Supplementary Table 1).
Table 2. Clinical complications associated with TDTa,b.
Patients had a mean of 14.2 discrete RBCTs PPPY (SD = 4.7), and the average time between transfusions was 27.7 days (SD = 9.3). Most patients were also receiving ICT (91.3%), and these patients had an average of 12.1 ICT claims PPPY (SD = 10.3) ().
Table 3. Treatment use among patients with TDTa.
HCRU and costs
Compared to matched controls, patients with TDT had significantly higher HCRU across all inpatient and outpatient HCRU variables assessed (). A higher proportion of patients with TDT than matched controls had ≥1 inpatient admission (28.5% vs. 7.1%; p < .0001) () and emergency department visit (55.6% vs. 38.0%; p < .0001) in the variable-length follow-up. Total mean (SD) outpatient visits/encounters PPPY (81.8 [41.4] vs. 11.4 [16.2]; p < .0001) were higher for patients with TDT than for matched controls (). Patients with TDT also had substantially higher total annual healthcare costs PPPY than matched controls ($137,125 vs. $4183; p < .0001); the overall cost differences between the two cohorts were primarily driven by outpatient medical and pharmacy costs ().
Figure 2. Annual healthcare costs per patient with TDT and matched controlsa.
Abbreviations. PPPY, Per-patient-per-year; SD; Standard deviation; TDT, Transfusion-dependent β-thalassemia. aMean costs of outpatient visits/encounters for patients with TDT included those for emergency department visits ($1364 [SD = $9041]), physician office visits ($2444 [SD = $1810], laboratory encounters ($15,537 [SD = $16,994]), and other outpatient visits/encounters ($42,320 [SD = $35,966]).
![Figure 2. Annual healthcare costs per patient with TDT and matched controlsa.Abbreviations. PPPY, Per-patient-per-year; SD; Standard deviation; TDT, Transfusion-dependent β-thalassemia. aMean costs of outpatient visits/encounters for patients with TDT included those for emergency department visits ($1364 [SD = $9041]), physician office visits ($2444 [SD = $1810], laboratory encounters ($15,537 [SD = $16,994]), and other outpatient visits/encounters ($42,320 [SD = $35,966]).](/cms/asset/1a09df1d-bd98-4f3e-9efb-083cd86ec0c4/ijme_a_2235928_f0002_b.jpg)
Table 4. Annual HCRU for patients with TDT and matched controlsa.
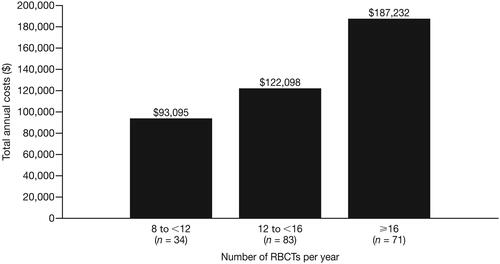
In patients with TDT, the mean annual cost of RBCTs was $32,325 PPPY (SD = $28,285); RBCT-related costs are included in total inpatient and outpatient costs, which varied depending on the setting where the transfusion occurred. In addition, the mean annual cost of ICT was $71,506 PPPY (SD = $60,050). Total annual healthcare costs were higher in patients with greater numbers of RBCTs: patients who received 8 to <12 RBCTs per year (n = 34) had total annual healthcare costs of $93,095, and patients who received ≥16 RBCTs (n = 71) per year had total annual healthcare costs of $187,232 ().
Figure 3. Total annual healthcare costs by annual number of RBCTs in patients with TDT. Abbreviations. RBCT, Red blood cell transfusion; TDT, Transfusion-dependent β-thalassemia.
Total annual healthcare cost data were extrapolated to evaluate the costs incurred by patients with TDT over their lifetime. Projected lifetime healthcare costs were estimated to be $7.1 million for patients with TDT at age 50 compared to $235,000 for matched controls (). After age 36, each additional year of life added $152,482 to the total lifetime costs for patients with TDT and $7258 to those for matched controls.
Figure 4. Lifetime healthcare costs for patients with TDTa and matched controlsb.
Abbreviations: K, Thousand; M, million; TDT, Transfusion-dependent β-thalassemia.
aAnnual costs for patients with TDT per age group were $99,931 (age 0 to 11 years [n = 72]); $129,259 (age 12 to 17 years [n = 24]); $168,848 [age 18 to 35 years [n = 71]); and $152,482 (age ≥36 years [n = 40]). bAnnual costs for controls per age group were $1,927 (age 0 to 11 years [n = 350]); $2,803 (age 12 to 17 years [n = 130]); $5,181 (age 18 to 35 years [n = 355]); and $7,258 (Age ≥36 years [n = 200]).
![Figure 4. Lifetime healthcare costs for patients with TDTa and matched controlsb.Abbreviations: K, Thousand; M, million; TDT, Transfusion-dependent β-thalassemia.aAnnual costs for patients with TDT per age group were $99,931 (age 0 to 11 years [n = 72]); $129,259 (age 12 to 17 years [n = 24]); $168,848 [age 18 to 35 years [n = 71]); and $152,482 (age ≥36 years [n = 40]). bAnnual costs for controls per age group were $1,927 (age 0 to 11 years [n = 350]); $2,803 (age 12 to 17 years [n = 130]); $5,181 (age 18 to 35 years [n = 355]); and $7,258 (Age ≥36 years [n = 200]).](/cms/asset/73d0abfc-5010-4b4c-ae99-b1ef3a5447b6/ijme_a_2235928_f0004_b.jpg)
Discussion
This study used administrative claims data to evaluate real-world clinical complications, treatments, HCRU, and healthcare costs in patients with TDT in the United States. During the variable-length follow-up, patients experienced substantial clinical complications and required regular supportive care with both RBCTs and ICT. HCRU, annual healthcare costs, and lifetime healthcare costs were significantly higher for patients with TDT than for matched controls; the increased costs for patients with TDT were largely driven by their higher outpatient medical and pharmacy costs and the costs for regular RBCTs and ICT.
The extent of iron overload observed in this patient population is notable. Chronic iron overload can arise from the pathophysiology of the disease, as well as from the use of regular RBCTs, and is associated with high morbidity and mortalityCitation9. ICT is recommended for the management of chronic iron overload in patients with TDTCitation9, with certain chelators demonstrating long-term safety and efficacy in patients as young as 2 years oldCitation18. Consequently, the high prevalence of iron overload (87.9%) observed in patients of all ages, despite frequent claims for ICT, suggests that many patients may still experience elevated iron levels while being treated with ICT. Further, the numerically higher prevalence of iron overload in younger (0 to 11 years: 93.1%) vs. older patients (≥36 years: 82.5%) suggests that better management of this clinical complication may be warranted, particularly in pediatric patients with TDT.
In addition to iron overload, infections and hepatobiliary, endocrine, and cardiopulmonary complications were also common in patients with TDT. Hepatobiliary, endocrine, and cardiopulmonary complications increased in prevalence with patient age and were most prevalent in patients aged ≥36 years, demonstrating the chronic and progressive nature of TDT. These complications are thought to be associated with iron overload resulting from disease pathophysiology and frequent RBCTs over these patients’ lifetimesCitation3. The prevalence of iron overload and these associated clinical complications, despite patients receiving the best available care (e.g. frequent RBCTs and ICT), indicates an unmet need for additional effective therapies for TDT.
Total annual healthcare costs for patients with TDT were $137,125, which is consistent with the results of the only other empirical assessment of economic outcomes associated with TDT in the United States based on administrative claims dataCitation12. In both our study and the Weiss et al.Citation12 studies, the higher total annual healthcare costs for patients with TDT reflected their more frequent outpatient visits/encounters and higher pharmacy costs compared to matched controls. The positive relationship observed between total annual healthcare costs and the number of RBCTs further exemplifies the contribution of treatment- and medication-specific costs to the overall economic burden of TDT and represents a novel finding. In addition, this study is the first to use administrative claims data to estimate the lifetime healthcare costs for patients with TDT. Lifetime costs for this patient population were estimated to be $7.1 million, which exceeds previous estimates obtained with economic modeling approaches (∼$5.4 million) due to differences between studies in life expectancy estimates (50 vs. 39 years) and claims methodology, which did not account for TDT expected mortality over timeCitation19. Annual healthcare costs also increased across age groups, with patients aged ≥36 years having numerically higher annual healthcare costs than patients aged 0 to 11 years ($152,482 vs. $99,931, respectively). The notion that lifetime costs could reach $7 million by age 50 highlights the unmet need for effective, cost-saving therapies that are available as early as possible in a patient’s disease course.
There are several limitations in this study that should be noted. First, this study relied on administrative claims data collected for reimbursement purposes and is therefore subject to potential misclassification bias. Second, the reliance on only direct costs and exclusion of several variables from our analyses (e.g. the cost of blood donations and blood testing or matching, the time spent undergoing RBCTs, and the impact of indirect factors such as absenteeism or work productivity loss) likely led to an underestimation of the economic burden associated with regular RBCTs in patients with TDT. Inversely, the lifetime healthcare cost calculation utilized a simplifying assumption that patients would be transfusion-dependent from birth until death and could have therefore overestimated lifetime healthcare costs in this patient population; consideration of age-specific annual costs, as done in other claims-based studies of patients with hemoglobinopathiesCitation20, may have mitigated some of this limitation. Third, any potential impact of death, transition to long-term disability coverage, or the use of recently approved therapies for TDT (e.g. luspatercept) were not considered; patients who did not meet the inclusion/exclusion criteria of this study or who received newer disease-modifying therapies may have had different clinical outcomes and costs than the patients included in this study. Fourth, we appreciate that our case definition of β-thalassemia is inconsistent with the disease definition that is currently used in clinical practice. Although previous claims-based studies and clinical trials have employed similar definitions to oursCitation12,Citation21, there are ultimately no validated algorithms for the claims-based diagnosis of β-thalassemia, and positive predictive values for ICD-9/10 codes are unknown. Nevertheless, we expected a high specificity for identifying TDT, as this analysis required additional criteria to be met with respect to RBCTs. Based on published literature, approximately one-third of patients with β-thalassemia require regular RBCTs and are therefore categorized as having TDTCitation6. As a result, our case definition may have led to increased identification and potential misclassification of patients with β-thalassemia and a subsequently lower proportion of patients with TDTCitation12,Citation21. This study utilized the Medical Care Component of the Consumer Price Index to adjust for inflation to 2020 U.S. dollars, a methodology that previous studies involving the MarketScan Databases have usedCitation22–24; there are also alternative methodologies to inflate U.S. medical pricesCitation25. Finally, although this study included a broad population of patients with commercial insurance coverage, Medicare Supplemental coverage, and Medicaid eligibility in the United States, these results may not be generalizable to patients with other payer types, those who are uninsured, and individuals outside the United States. Additionally, the Merative MarketScan Medicare Supplemental Database is not representative of all individuals with Medicare Supplemental coverage, as it only includes retirees whose employers provided Medicare Supplemental coverage.
Conclusions
Despite that patients with TDT receive supportive care, many continue to experience iron overload. Patients with TDT have significant HCRU and costs driven by outpatient and pharmacy costs associated with their regular use of RBCTs and ICT. Future disease-modifying therapies that eliminate the need for RBCTs and effectively address iron overload could significantly improve the clinical and economic outcomes in this patient population.
Transparency
Declaration of financial/other relationships
CU, YY, UM, and NL are employees of Vertex Pharmaceuticals Incorporated and may hold stock/stock options. KAE and TL are employees of Merative and may hold stock/stock options. JM was an employee of Merative at the time of this analysis and is now employed by Veradigm. BA has received research funding from the American Society of Hematology, Connecticut Department of Public Health, Forma Therapeutics, Global Blood Therapeutics, Hemanext, HRSA, Imara, Novartis, and PCORI, and has served as an advisory board member or consultant for Agios, Aruvant, Bayer, bluebird bio, CRISPR Therapeutics AG, CVS/Accordant, Cyclerion, Emmaus, Forma Therapeutics, GBT, Genentech, Hemanext, Novartis, NovoNordisk, Roche, Sanofi, TerSera, Terumo, and Vertex Pharmaceuticals Incorporated.
Author contributions
All authors met the International Committee of Medical Journal Editors (ICMJE) authorship criteria. Neither honoraria nor payments were made for authorship. CU, KAE, YY, TL, JM, and BA were responsible for the study conception and design. KAE, TL, and JM were responsible for data acquisition. All authors participated in data analysis or interpretation, drafted or critically revised the manuscript for intellectual content, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Previous presentations
Portions of these data were previously presented at the Professional Society for Health Economics and Outcomes Research Annual Meeting (ISPOR) 2022, National Harbor, MD, USA, 15–18 May 2022, European Hematology Association (EHA) 2022 Hybrid Congress, Vienna, Austria, 9–17 June 2022, and Academy of Managed Care Pharmacy (AMCP) Annual Meeting 2022, National Harbor, MD, USA, 11–14 October 2022.
Supplemental Material
Download PDF (68.1 KB)Acknowledgements
The authors would like to thank Paula J. Smith of Merative for providing programming services and Ciara Silverman, PharmD, of Vertex Pharmaceuticals Incorporated for her support in drafting this manuscript and performing data analysis. Medical writing and editing support were provided by Brittany Y. Jarrett, PhD, Natalie Prior, PhD, and Nicholas Strange of Complete HealthVizion, IPG Health Medical Communications, Chicago, IL, USA, funded by Vertex Pharmaceuticals Incorporated.
Data availability statement
This study used data available from Merative. Restrictions apply to the availability of these data, which were used under a licensing agreement.
Additional information
Funding
References
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–487. doi: 10.2471/blt.06.036673.
- Taher AT, Musallam KM, Cappellini MD. Beta-thalassemias. N Engl J Med. 2021;384(8):727–743. doi: 10.1056/NEJMra2021838.
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11. doi: 10.1186/1750-1172-5-11.
- Kattamis A, Forni GL, Aydinok Y, et al. Changing patterns in the epidemiology of beta-thalassemia. Eur J Haematol. 2020;105(6):692–703. doi: 10.1111/ejh.13512.
- Sayani FA, Kwiatkowski JL. Increasing prevalence of thalassemia in America: implications for primary care. Ann Med. 2015;47(7):592–604. doi: 10.3109/07853890.2015.1091942.
- Lal A, Wong T, Keel S, et al. The transfusion management of beta thalassemia in the United States. Transfusion. 2021;61(10):3027–3039. doi: 10.1111/trf.16640.
- Lal A, Wong TE, Andrews J, et al. Transfusion practices and complications in thalassemia. Transfusion. 2018;58(12):2826–2835. doi: 10.1111/trf.14875.
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155–167. doi: 10.1016/S0140-6736(17)31822-6.
- Cappellini MD, Farmakis D, Porter J, et al. 2021 Guidelines for the management of transfusion dependent thalassaemia (TDT). Nicosia (Cyprus): Thalassaemia International Federation; 2021.
- Tang CH, Furnback W, Wang BCM, et al. Relationship between transfusion burden, healthcare resource utilization, and complications in patients with beta-thalassemia in Taiwan: a real-world analysis. Transfusion. 2021;61(10):2906–2917. doi: 10.1111/trf.16636.
- Alshamsi S, Hamidi S, Narci HO. Healthcare resource utilization and direct costs of transfusion-dependent thalassemia patients in Ddubai, United Arab Emirates: a retrospective cost-of-illness study. BMC Health Serv Res. 2022;22(1):304. doi: 10.1186/s12913-022-07663-6.
- Weiss M, Parisi Jun M, Sheth S. Clinical and economic burden of regularly transfused adult patients with beta-thalassemia in the United States: a retrospective cohort study using payer claims. Am J Hematol. 2019;94(5):E129–E132. doi: 10.1002/ajh.25429.
- Delea TE, Hagiwara M, Thomas SK, et al. Outcomes, utilization, and costs among thalassemia and sickle cell disease patients receiving deferoxamine therapy in the United States. Am J Hematol. 2008;83(4):263–270. doi: 10.1002/ajh.21049.
- Udeze C, Evans KA, Yang Y, et al. Economic and clinical burden of managing sickle cell disease with recurrent vaso-occlusive crises in the United States. Adv Ther. 2023;40(8):3543–3558. doi: 10.1007/s12325-023-02545-7.
- Tan L, Reibman J, Ambrose C, et al. Clinical and economic burden of uncontrolled severe noneosinophilic asthma. Am J Manag Care. 2022;28(6):e212–e220. doi: 10.37765/ajmc.2022.89159.
- Zeiger R, Sullivan P, Chung Y, et al. Systemic corticosteroid-related complications and costs in adults with persistent asthma. J Allergy Clin Immunol Pract. 2020;8(10):3455–3465.e13. doi: 10.1016/j.jaip.2020.06.055.
- BLS.gov [Internet]. Washington (DC): U.S. Bureau of Labor Statistics; c2023 [cited 2023 Mar 14]. Available from: https://www.bls.gov/cpi/factsheets/medical-care.htm.
- Vichinsky E, El-Beshlawy A, Al Zoebie A, et al. Long-term safety and efficacy of deferasirox in young pediatric patients with transfusional hemosiderosis: results from a 5-year observational study (ENTRUST). Pediatr Blood Cancer. 2017;64(9):8. doi: 10.1002/pbc.26507.
- Udeze C, Maruszczyk K, Atter M, et al. Projected lifetime economic burden of transfusion dependent beta-thalassemia in the United States. HemaSphere. 2022;6:2208–2209. doi: 10.1097/01.HS9.0000852180.45597.87.
- Johnson KM, Jiao B, Ramsey SD, et al. Lifetime medical costs attributable to sickle cell disease among nonelderly individuals with commercial insurance. Blood Adv. 2023;7(3):365–374. doi: 10.1182/bloodadvances.2021006281.
- Locatelli F, Thompson AA, Kwiatkowski JL, et al. Betibeglogene autotemcel gene therapy for non-β(0)/β(0) genotype β-thalassemia. N Engl J Med. 2022;386(5):415–427. doi: 10.1056/NEJMoa2113206.
- Manjelievskaia J, Boytsov N, Brouillette MA, et al. The direct and indirect costs of adult atopic dermatitis. J Manag Care Spec Pharm. 2021;27(10):1416–1425. doi: 10.18553/jmcp.2021.27.10.1416.
- Varnado OJ, Manjelievskaia J, Wenyu Y, et al. Health care resource utilization and costs associated with treatment among patients initiating calcitonin gene-related peptide inhibitors vs other preventive migraine treatments in the United States. J Manag Care Spec Pharm. 2022;28(8):818–829. doi: 10.18553/jmcp.2022.28.8.818.
- Fenske DC, Ding Y, Morrow P, et al. Comparing the burden of illness in patients with alopecia areata vs atopic dermatitis in the US population from a payer perspective. J Manag Care Spec Pharm. 2023;29(4):409–419. doi: 10.18553/jmcp.2023.29.4.409.
- AHRQ.gov [Internet]. Rockville (MD): Agency for Healthcare Research and Quality; c2023 [cited 2023 May 19]. Available from: https://meps.ahrq.gov/about_meps/Price_Index.shtml.