
Abstract
Objective
To describe the historical baseline landscape of cardiovascular drug post-approval activity, including the number and timing of post-approval clinical trials and approved indications. The US Inflation Reduction Act of 2022 (IRA) Drug Price Negotiation Program (DPNP) and its Maximum Fair Prices (MFPs) will affect incentives for investment in post-approval activity such as clinical trials for new indications. While three of the first ten drugs selected for the DPNP and MFP-setting are cardiovascular or antithrombotic drugs, limited attention has been paid to potential cardiovascular drug impacts, and to post-approval innovation.
Methods
For the 65 drugs originally approved by the FDA from 1995 through 2021 for a cardiovascular or antithrombotic indication (60 small molecules and 5 biologics), we develop a novel dataset of industry-sponsored, post-approval clinical trials and FDA-approved label changes for new indications. We analyze their number and timing relative to DPNP drug selection and MFP implementation dates, by drug approval-year cohort.
Results
We find 49% of indications were awarded and 76% of industry-funded clinical trials were completed post-approval, reaching 98% of trials for drugs in the earliest 1995–99 cohort. For the 60 small molecules, 76% of post-approval trials ended five years or more after original drug approval, 65% ended seven or more years after original drug approval (i.e. after potential DPNP selection), and 53% nine or more years after original drug approval (i.e. after potential MFP implementation).
Conclusions
Post-approval FDA indication approvals and clinical trial starts and primary completion dates often occurred after or near new DPNP selection and MFP implementation dates. This has economic consequences for future investment incentives. Post-approval trials for small molecules, longer-duration trials, and larger-enrollment trials, and post-approval indications focused on limited patient populations and older patients could face particular economic challenges.
Introduction
Recent attention on the impact of the US Inflation Reduction Act of 2022 (IRA) on the incentives for manufacturers to invest in post-approval activity has focused on oncology drugs, but cardiovascular (CV) drugs will also be affected. In fact, three of the ten drugs selected for the first round of the new Medicare Drug Pricing Negotiation Program (DPNP) of so-called “Maximum Fair Prices” (MFPs) to be announced by September 1, 2024 and to go into effect January 1, 2026 were first approved for a cardiovascular or antithrombotic indication (i.e. apixaban, sacubitril/valsartan, and rivaroxaban), and others have received CV indications after initial approval.
While these final first-round MFPs will not be announced and become effective until then, descriptive analyses of the historical (pre-IRA) landscape are useful to policymakers and to other researchers even before empirical analyses comparing “pre-IRA” and “post-IRA” activity become available. This descriptive analysis provides valuable information on historical post-approval innovation activity, including the number and timing of post-approval indications and clinical trial starts and ends relative to future DPNP drug selection and MFP implementation timelines. It establishes a baseline for policy-makers to track changes in these metrics over time as the IRA provisions have effects on post-approval investment behavior, and identifies potential variables of interest for future researchers.
Cardiovascular disease remains the leading cause of death in the US, with over 928,000 deaths from heart disease or stroke in 2020, or more than one-quarter of all deathsCitation1. The estimated direct and indirect cost of cardiovascular disease was $407.3 billion for 2018 to 2019, including direct costs to the healthcare system and the indirect costs of lost productivity due to premature mortality, and cardiovascular disease accounts for a higher share of total US health expenditures than any other major diagnostic group. CV drugs will continue to be important therapies for older Americans and represent substantial Medicare spend, due to their widespread usage. An earlier study found that 42% of Medicare beneficiaries aged 65 years and over had at least one heart conditionCitation2. Moreover, hypertension remains the leading preventable cause of death and disability throughout the worldCitation3. Thus, incentives for continued development of the full potential of drugs treating heart disease and preventing cardiovascular complications such as heart attack and stroke are clinically and economically important.
The US Inflation Reduction Act of 2022 (IRA) and the new Drug Pricing Negotiation Program (DPNP)
The IRA contains several provisions affecting the Medicare drug program. The provision that has gained the most attention is the creation of a new Drug Pricing Negotiation Program (DPNP) that requires CMS to set the law’s “Maximum Fair Prices” (MFPs) for certain high Medicare-spend Part D and Part B prescription drugs that have not yet experienced generic or biosimilar competition. Ten Part D drugs will be selected for the first round of MFPs to be effective in 2026, with another 15 Part D drugs in 2027, 15 Part D or Part B drugs in 2028, and 20 Part D or Part B drugs in 2029 and later years.Footnotei By 2031, 100 drugs will have been selected for MFP-setting across both Medicare Part B and Part D; others have estimated that the drugs likely to be selected represented almost half of Part B and Part D drug spending in 2020Citation4.
The MFPs must be offered to all eligible Medicare purchasers and beneficiaries, including Part B purchasers such as physician organizations, and Part D plans. The law sets a maximum ceiling price for the MFP, defined as the lesser of the weighted average net price paid after price concessions in Medicare Part D or Part B, or a percentage of the average non-federal average manufacturer price (or “non-FAMP”) in commercial markets, which varies by the number of years from the drug’s US approval.Footnoteii However, with only a very limited exception, it does not set a minimum floor price, and the final prices that will result are yet unknown.Footnoteiii Rather, CMS is directed to “develop and use a consistent methodology and process… that aims to achieve the lowest maximum fair price for each selected drug,”Footnoteiv and in doing so to consider a number of factors, including the cost of therapeutic alternatives, comparative effectiveness, unmet medical need, the degree to which manufacturers have “recouped the cost of R&D,” the costs of production and distribution, and prior federal financial support. The Congressional Budget Office previously estimated that MFPs will reflect an average 50% reduction in the price paid by MedicareCitation5.
Drugs are selected for the DPNP process after seven years following FDA approval of the first indication in the case of small molecule drugs (i.e. drugs approved and marketed under the Federal Food, Drug, and Cosmetic Act) and eleven years in the case of biologics (i.e. drugs approved and marketed under the Public Health Service Act). The MFPs that CMS determines then become effective nine years after initial US drug approval for small molecules, and thirteen years after approval for biologics. These prices will become public and may affect the prices paid by other payers, for instance through State Prescription Drug Affordability Boards (PDABs) created in some states that have authority to set maximum payment levels for drugs covered by both public and commercial plans and where the new Medicare MFPs will affect the upper payment limits they set. MFPs for the first set of ten Medicare Part D drugs selected by CMS will be set by September 1, 2024 and will be effective January 1, 2026. Manufacturers who reject CMS’s final MFP offer as part of the DPNP process face unsustainably large financial penalties, so the CBO and others assume that all manufacturers will participate in the DPNP and accept the CMS MFPs.Footnotev
As a result of these timelines, the law introduces new considerations into a manufacturer’s calculus relating to future investments in post-approval activity. Currently, small molecule drugs are expected to lose share and sales rapidly following launch of an equivalent generic drug. For all drugs experiencing first generic drug entry in 2017–2019, the retained branded drug share reached 23% twelve months after generic entryCitation6. For these drugs, the period between US branded drug launch and first generic drug entry averaged 13.0 years for all drugs and 14.1 years for drugs with sales of more than $250 million (in 2008 dollars) in the year prior to first generic entry. This period of time (the “market exclusivity period” or MEP) has ranged between an average of 12.2 and 14.6 years for higher-sales small molecule drugs since 1995–1996, and represents the commercial life during which manufacturers expect to earn US revenues and a return on successfully-developed drugs. For biologics, experience under the US biosimilar pathway is less mature, so there is more uncertainty for manufacturers about how long the typical period before biosimilar entry will be, and what the expected retained brand share after biosimilar entry will be.
Because the new MFPs will take effect and reduce revenues prior to the expected end of the MEP (or at nine years for small molecule MFPs, versus the average of approximately 13 to 14 years for generic drug entry), they will have the effect of reducing the expected value of post-approval investments, sometimes substantially. As a result, some post-approval activity that would otherwise be justified economically will no longer be.
Methods
Following prior methods, we examine two measures of post-approval activity: additional approved on-label indications, and additional post-approval clinical trial activityCitation7.
We investigate the extent and timing of post-approval activity for drugs originally approved in the US as NMEs (new molecular entities) or through Biologic License Applications (BLAs) between January 1, 1995 and December 31, 2021. We restrict the list of drugs to those for which the first US-approved indication was for cardiovascular disease or antithrombosis.Footnotevi This requirement excludes drugs which were not originally approved as cardiovascular agents but which later demonstrated cardiovascular benefits and received a cardiovascular indication, such as GLP-1 receptor agonist drugs, which were originally approved for glycemic control and which have received intense clinical interest recently. We also exclude drugs that will not be subject to the DPNP, radiodiagnostic agents or those used to enhance diagnostic imaging, plasma-derived products, and those indicated only as parts of interventional procedures, or that have been withdrawn from the US.
This yields a list of 65 drugs. For each drug, we record the date of original approval (as reflected in its Drugs@FDA record), and whether the drug is a small molecule or a biologic (as defined by the regulatory approval path for the drug, which determines the year when MFPs may be effective). The sample was overwhelmingly composed of small molecules; only 5 of the drugs are biologics and the remaining 60 are small molecules.
In our analysis, we define the term “indication” to mean the condition for which a drug would be used, including any restrictions on the patient population, co-therapy conditions, or other factors reflected in the label. Following this definition, we distinguish an indication from a disease or condition. An example of an indication in the data is “Prophylaxis of venous thromboembolism (VTE) in acutely ill medical patients.”
For the analysis of post-approval indications, we constructed a novel data set consisting of the labeled indications for each drug in the sample (as available on the Drugs@FDA website on March 27, 2023). We worked backwards from the current label, and compared each label against all the previous other labels for that drug in the Drugs@FDA record to identify the earliest date when a given indication appearing on the current label was first approved as a change to the label. We restricted our review to the “Indications and Usage” section of the label. We considered a distinct indication to be the lowest level described, which often appears formatted as bulleted text, or with an outline-format number after it to identify separate distinct indications (e.g. 1.1, 1.2, 1.3). Manual review resolved any apparent duplicate potential first-approval dates that exhibited only minor text or formatting differences between them. If multiple indications appeared on the drug’s original label at the time of approval, each was assigned the same approval date (i.e. the original approval date), and was considered an original, rather than post-approval, indication. Using this data, we define “post-approval indications” as those with approval dates after the original US launch date, and distinguish them from “original indications,” those reflected on the drug’s earliest-approved label (i.e. on the original US launch date).
For each indication, the time from the drug’s original approval to the indication approval was calculated by subtracting the date of original approval from the date a given indication was approved, as described above. Our sample includes a total of 146 indications, 72 of which are post-approval indications.
We characterize each post-approval indication according to whether it is for treatment, prevention, or both, following a keyword search, and manually confirming the results. We characterize the disease for each indication according to one of the fifty-three possible categories for cardiovascular and antithrombotic drugs reflected in a proprietary R&D pipeline database (of which sixteen were reflected among the post-approval indication data).Footnotevii We identified pediatric indications according to a keyword search and manually confirmed the results. Summary counts appear in .
Table 1. Post-approval indications.
For the analysis of clinical trials, we rely on data reflected in ClinicalTrials.gov. We downloaded the list of all NCT (National Clinical Trial) codes for each drug as of April 14, 2023 by searching according to the generic name of the drugs in the sample. We also downloaded information on the study’s title, phase, status as of the download date, sponsor, collaborator(s), funder type (industry or not), study type (interventional or not), number of enrollees, permissible study participants by sex and age, start date, primary completion date (i.e. “the date that the final subject was examined or received an intervention for the purposes of final collection of data for the primary outcome”), end date, whether study results were available, study design, and the lists of interventions and outcome measuresCitation8.
We restrict the clinical trial sample for analysis to interventional studies funded by industry, excluding those that have a study status of “withdrawn”, finding that such studies typically were not begun. We further restrict the sample to those for which the listed interventions contain either the generic or brand name of the drug, and eliminated studies where we determined the drug appeared only in a comparator arm, rather than being the focus of the trial. Finally, we exclude trials where the sponsor of the trial was not the current NDA or BLA holder, or was not recognized as the drug’s originator or a licensee.
We perform our analysis on the remaining 1,689 trials that met all four sets of these screens (i.e. drug-level, study-level, study arm-level, and study sponsor-level requirements), which included 1,286 post-approval trial starts.
We tally the number of qualifying clinical trials for each drug, and calculate the duration of each trial by subtracting the trial’s primary completion date from its start date, in days. The time from original approval to clinical trial start is calculated as the difference between the drug’s original approval date (as reflected in the Drugs@FDA data described above), and the trial start date as reflected in the ClinicalTrials.gov record. We define “post-approval trials” as those with trial start dates after the original US launch date. We define pediatric trials as those where the term “child” appears in the Age field in the ClinicalTrials.gov field (e.g. “18 months to 36 years (child, adult)”).
Not all studies had all data elements recorded in the ClinicalTrials.gov record, and as a result, the sample sizes for the various clinical trial-related calculations varies. The number of studies for a given analysis was limited by the lowest number of entries for any of its required elements (i.e. blank values for a necessary element of the calculation, such as trial start date, eliminated the drug from that reported result).
For additional detail, see the Online Supplement.
Results
Post-approval activity represents approximately half of all indications and three-quarters of industry-funded clinical trials
We find that 49% of all indications and 76% of all industry-funded clinical trials for the cardiovascular drugs in the sample represent post-approval activity, versus original approved indications and pre-approval industry-funded trial starts, according to our definitions ().
Table 2. Drugs, indications, and industry-funded clinical trials by approval-year cohort.
The level of post-approval activity varied by drug, from a low of no additional post-approval indications to a high of 9 additional post-approval indications, and from a low of no additional post-approval clinical trial starts to a high of 141 additional post-approval clinical trial starts. Post-approval indications were concentrated among fewer drugs; just over half of the drugs (54%) have shown no additional indications thus far (). For clinical trial starts, fewer drugs, or only 11%, have had no post-approval clinical trial starts reflected thus far ( and ).
Figure 1. Post-approval indications per drug. Source: Authors’ calculations of Drugs@FDA data. Notes. Post-approval indications have approval dates after US drug approval date.
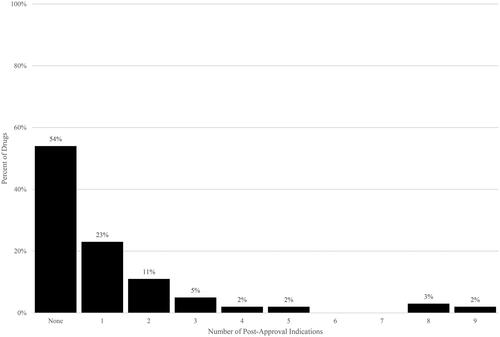
Figure 2. Post-approval industry-funded trial starts per drug. Source: Authors’ calculations of ClinicalTrials.gov data. Notes. Post-approval trials have start dates after US drug approval date.
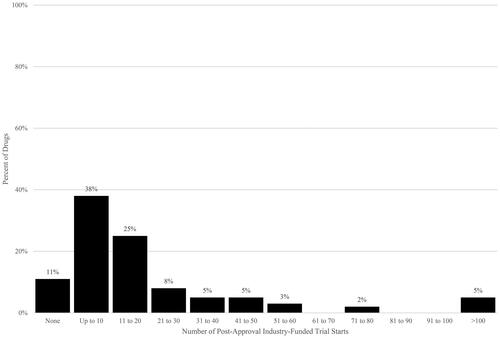
However, because our analysis is a cross-sectional snapshot taken at a point in time, the drugs were observed for varying periods of time, with fewer years of post-approval observation for some drugs and many more for others. As a result, additional trials and indications will be observed over time, particularly for drugs with more recent original approval dates, as reflected in the trends across approval-year cohorts in , which show higher rates of activity for earlier cohorts with more years during which to observe clinical trial activity and indication approvals than for later cohorts.
Our findings that for cardiovascular drugs, roughly half of labeled indication approvals and three-quarters of clinical trial starts occur after initial drug approval align with other estimates. Others have found that for all drugs approved between 1995 and 2019 (across disease areas), post-approval indications represented approximately 40% of all (i.e. total initial plus post-approval) indicationsCitation9. Others found that for 88 new medicines (71 small molecules and 17 biologics) approved between 2010 and 2012, 47 (53%) received at least one post-approval indication, that post-approval indications represented 58% of all indications and that 44% of them were approved seven or more years after the drug’s initial approvalCitation10,Citation11.
Thirty-eight percent of post-approval indications were in new diseases not previously approved for that drug (comparing the indication with all previous approved indications, whenever approved). Others were for treatment within the same disease (for instance, for treatment of other lipid disorders within the broad disease area “dyslipidemia”), for prevention rather than treatment, and/or for different patient populations (such as pediatric approvals). ()
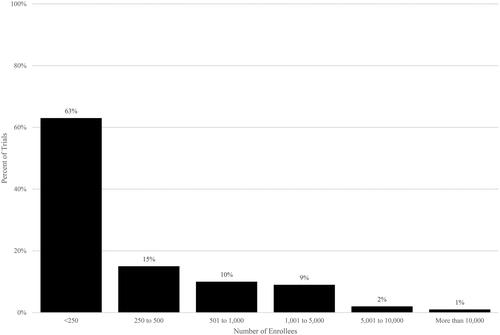
The distribution of enrollment in clinical trials is skewed. The majority of trials had fewer than 250 patients but twelve percent had more than 1,000 patients. Mean enrollment in post-approval trials was 733 (). Post-approval trials reflected all phases (24% Phase 1, 9% Phase 2, 40% Phase 3, 26% Phase 4; data not shown).
Figure 3. Trial enrollment in post-approval industry-funded trials. Source: Authors’ calculations of ClinicalTrials.gov data. Notes. Post-approval trials have start dates after US drug approval date.
Many post-approval clinical trials and indication approvals occur after or near the new IRA DPNP selection and MFP dates
Focusing only on small molecules, which make up the overwhelming majority of our sample (60 of the 65 drugs in the sample), an average of 60% of post-approval indication approvals occurred five years or more after original drug approval (five years after original US drug approval corresponds to two years before potential DPNP selection of small molecules), 46% of indication approvals occurred seven years or more after original US drug approval (i.e. after the timeframe for potential DPNP selection), and 27% of indication approvals occurred nine years or more after original US drug approval (i.e. after the timeframe for MFPs for small molecules to be effective) (). The average time for all original approval-year cohorts from drug approval to post-approval indication approval is 7.0 years.
Table 3. Post-approval indications and industry-funded clinical trials by years from US drug approval (small molecules only).
The corresponding figures for only the earliest approval-year cohort of small molecules (that have the greatest number of years after original approval during which subsequent indication approvals and trial starts can be observed), are: 78% of post-approval indications occurred five years or more post-approval; 57% occurred seven years or more post-approval; and 26% occurred nine years or more post-approval.
For clinical trials, the picture is similar. For small molecules, on average across all approval-year cohorts, 63% of post-approval trials were begun five years or more after original approval, 52% were begun seven years or more after original approval, and 41% were begun at or after the potential MFP effective date for small molecules of nine years after original approval. () The average time for all cohorts from drug approval to clinical trial start was 8.1 years.
Looking only at the earliest approval-year cohort with the greatest number of years after original approval during which to observe subsequent trials, the corresponding figures are: 94% of trials were begun five years or more post-approval; 82% were begun seven years or more post-approval; and 65% were begun nine or more years post-approval.
For later approval-year cohorts, figures are lower, reflecting the fact that there are fewer years during which to observe post-approval trial starts. As a result, the average numbers and percentages of indication approvals and trial starts after a given date is a lower-bound estimate, as the number and percentages of indications approved and clinical trials begun after any given time period (e.g. 5 years, 7 years, 9 years) will increase with time, particularly for more recently-approved drugs earlier in their lifecycle.
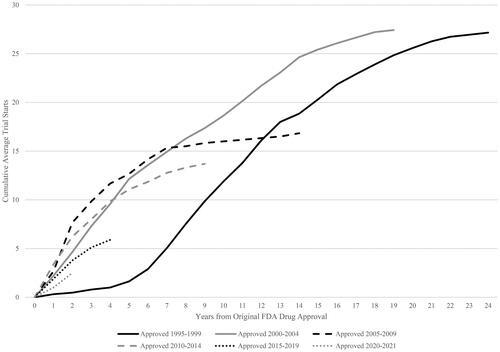
The pattern of the average number of post-approval clinical trial starts over time by five-year original approval-year cohort is shown in . There is some variation by cohort, reflecting the skewed distribution across drugs. However, clinical trial starts continue to increase after five years, seven years and nine years for all relevant cohorts.
Figure 4. Cumulative Average Industry-Funded Post-Approval Trial Starts by Drug Approval Cohort. Source: Authors’ calculations of ClinicalTrials.gov data. Notes. Post-approval trials have start dates after US drug approval date. Cohort is defined by US drug approval year.
Most trials including pediatric and older adult populations occurred post-approval and indications specific to these populations were often approved well after the drug’s original approval
Two populations of special interest, pediatric and older patients, also reflect the prevalence of post-approval indications and trials.
Approximately 8% of all trials in the sample included pediatric populations (8% of post-approval trials and 6% of trials started before approval). Among these relatively less-frequent pediatric trials, the overwhelming majority or 82% were post-approval trials. Pediatric post-approval trials started on average 6.0 years after original approval, and had an average duration of 3.3 years. The percentage of indications for pediatric populations was comparable, with 18 of 146 total indications referencing pediatric populations (12%), and all but two of these being awarded post-approval (89%). The average time after original approval for a pediatric indication approval was 8.9 years, and 10 of the 18 were approved 7 years or more following original approval (the period when small molecules become eligible for DPNP selection).
Whereas pediatric trials are less common, most trials provided for the enrollment of individuals age 65 or older, or roughly three-quarters (77%) of all trials (75% of post-approval trials and 86% of trials started before approval). Among trials that provided for older adults, 75% were post-approval trials. These post-approval trials started on average 7.6 years after original approval and had an average duration of 1.8 years.
Discussion
Today, manufacturers consider many factors in making decisions to invest in post-approval activities, such as the value to patients, the expected length of time to earn a return on such investments before generic drug entry occurs and branded drug share plummets, and the cost, duration and expected probabilities of success for the trials involved. While all trials may yield valuable clinical information to influence clinical practice, for many reasons not all will ultimately result in additional approved labeled indications. Investments in clinical trials that begin after original US approval have a limited economic window during which they can be completed and contribute to new labeled indications that could yield economic benefits and a return on these investments. Investments that are expected to yield their benefits largely after the end of the US market exclusivity period are less likely to be justified economically.
The IRA’s DPNP introduces an additional set of considerations to this economic calculus. Some post-approval development programs that were previously economically attractive will no longer be. While the calculus will differ for each drug, all else equal, the DPNP will create disincentives for certain types of drug development activities: disincentives to invest in small molecules versus biologics (because MFPs are effective four years earlier for small molecules); disincentives to invest in drugs that disproportionately serve older and disabled individuals (because they have a higher proportion of Medicare spend and MFP price reduction exposure); disincentives for post-approval indication development and clinical trials generally (due to the reduced likelihood of earning a return); and disincentives to invest in post-approval trials that are particularly lengthy or expensive (for instance, because they require very large numbers of patients and/or follow-up for long periods of time). Pediatric trials and indication development, subject to special legislative incentives and requirements to encourage them, may also experience disincentives as the value of the six-month pediatric exclusivity provision under the Best Pharmaceuticals for Children Act (BPCA) would be undermined for some drugs as a result of the DPNP.Footnoteviii
Cardiovascular drugs will be subject to a number of these potential disincentives:
Most CV drugs are small molecules and are therefore subject to earlier DPNP selection. 60 of the 65 molecules are small molecules and will therefore be subject to the earlier small molecule timelines, i.e. possible DPNP selection at seven years and MFP effective dates at nine years after US approval (in comparison to the eleven and thirteen year MFP selection and effective dates for biologics).
Post-approval indications and trials represent a substantial share of all indications and trials for cardiovascular drugs, and many of them occur years after original approval. Among small molecule drugs, 78% of all trials are begun and 50% of all indications are awarded post-approval. On average across all approval-year cohorts, 63% of small molecule post-approval trials were begun five years or more after original approval, 52% were begun seven years or more after original approval (i.e. after potential DPNP selection), and 41% were begun after the potential MFP effective date of nine years after original US approval for small molecules. For indication approvals, the pattern is similar – on average for all approval-year cohorts, 60% of all post-approval indications were awarded five years or more after original approval, 46% seven years or more after original approval, and 27% nine years or more after original approval.
Post-approval CV drug trials can be lengthy. When trial endpoints may involve relatively infrequent outcomes such as death, a second myocardial infarction, or a major cardiovascular event, such trials must be powered with long durations, large study populations, or both. 27% of the post-approval trials in our sample were longer than two years and 4% were longer than five years. (Data not shown)
The combination of trial duration and trial starts means a substantial share of all post-approval trials are completed after or near the DPNP dates. When a trial begins relative to drug approval and its duration both contribute to the time between original drug approval and trial end, when results can be used to support new indication approval and additional information to clinicians. Either factor or both may result in the trial’s results being available well after the drug’s original approval, and potentially being reconsidered in light of DPNP timelines, potential MFP exposure, and reduced economic returns. Adding the two elements together (i.e. time from drug approval to trial start plus trial duration), for the small molecule drugs 76% of post-approval trials have primary trial end dates five years or more after original approval (i.e. no more than two years before potential DPNP selection), 65% have primary end dates seven years or more after original approval (i.e. after potential DPNP selection), and 53% have primary end dates nine years after original approval (i.e. after potential MFP implementation). ()
Post-approval CV drug trials may have high enrollment. While mean enrollment in post-approval trials was 733, certain trials had enrollment that was much higher, for instance those that included the term “mortality” in the listed outcome measures had average enrollment that was over 3,500 (the largest with enrollment of over 27,000). For example, the ODYSSEY Outcomes trial of patients who had a previous acute coronary syndrome and who received high-intensity statin therapy enrolled over 18,000 patients and had a trial duration of over five years. Although outside the scope of our analysis, recent trials of GLP-1 receptor agonist drugs originally approved for non-cardiovascular indications like glycemic control are now generating cardiovascular study results that required thousands of patients and many years. For example, the SELECT trial involving over 17,000 enrollees and nearly five years of follow-up recently released results showing semaglutide reduced the overall rate of a combined endpoint of heart attacks, stroke and CV-related death by 20%.Citation12
The more of these factors apply, the more likely it is that post-approval investments may be subject to additional economic headwinds. For instance, a lengthy follow-up trial with a large study population that begins several years after original approval that targets a condition most common among older patients could be subject to particularly strong challenges. Relative to other therapeutic areas, others have found that cardiovascular trials also face disadvantages with respect to total development duration times to original approval, with cardiovascular drugs facing the second-longest time from Phase 1 to launch (11.5 years, compared with 10.5 years for all indications) and the second-lowest overall probability of success measured from Phase 1 to approval (4.8%, compared to an average of 7.9% for all indications).Citation13
Limitations
Our study has several limitations related to the underlying sources of data. With regard to the clinical trial information, ClinicalTrials.gov data is entered by the trial sponsor, is subject to change over time, and may be incomplete (i.e. certain fields may be blank at the point in time of our data download), although manufacturers are required to submit registration information within 21 days after the first human subject is enrolled, and false or misleading information can lead to monetary penalties.Footnoteix Under-reporting would lead to a downward bias in our calculations of the numbers of trials per drug. In addition, the values recorded for study enrollment and durations (defined as the difference between trial start and primary completion dates) may change over time, if trial sponsors later update these entries. Such changes, if any, could lead to shorter or longer trial durations, so the direction of the impact on our estimates is unknown.
There are also limitations associated with the selection criteria we used to identify relevant clinical trials among all industry-funded clinical trials by all companies and for all products. We would have under-estimated the number of clinical trials for a given drug if the sponsor-level screens we applied were overly strict. Conversely, we would have over-estimated the number of clinical trials for a given drug if the sponsor-level screens were insufficiently strict. Those criteria are described in greater detail in the Online Supplement.
With regard to the labeled indication information, we determine the total number of distinct indications for a given drug from its current FDA-approved label, and the approval date for each of the indications reflected there by working backwards to find its first appearance on a previously-approved label. In some cases, manual resolution of similarly phrased versions of the current indication is required. We use a keyword search approach to assign certain characteristics to each indication (i.e. treatment and/or prevention usage approval, pediatric approval), confirmed by manual review. To the degree that our indication matching and keyword approach are subject to error, they may result in totals that are too high or too low.
As noted, by limiting our sample to drugs first approved for cardiovascular indications, our analysis excludes post-approval cardiovascular indications for other drugs, such as GLP-1 receptor agonist drugs originally approved for glycemic control but that have important cardiovascular benefits and use. In addition, results for drug classes beyond cardiovascular medications may differ, reflecting their unique dynamics, and they also merit investigation.
Importantly, our results reflect cross-sectional data captured as of a point in time and therefore are subject to downward truncation bias. As a result, we may have underestimated the number of trials and approved indications and not fully captured the dynamics of post-approval activity over the long-term. Additional approved indications and trials for a given drug may be reflected over time. This downward effect is expected to be most severe for most-recently approved drugs, where we observe only the first several years of their lifetimes. While this downward bias can be counteracted by referring to totals associated with the oldest approval-year cohort drugs for which more post-approval years are available, this will result in fewer observations, and these drugs may differ from later-approved drugs in important ways.
Conclusions
Post-approval indications and industry-funded clinical trials represent a substantial share of all indications and industry-funded trials for cardiovascular drugs, and a substantial share of these indications occur years after initial approval.
The timing of additional post-approval clinical trial starts, primary completions and indication approvals often occurs after or near IRA DPNP drug selection and implementation dates, suggesting that the new DPNP economic incentives may have an impact on decisions about future cardiovascular drug post-approval investment. As most approved cardiovascular drugs are small molecules that will face earlier potential DPNP selection and MFP implementation dates, these effects could be heightened relative to sectors with a greater share of biologics. Baseline analyses of additional therapeutic areas, reflecting other dynamics, also would be valuable, together with later empirical analyses comparing historical patterns to post-IRA innovation activity in cardiovascular and other therapeutic areas. Our historical baseline analysis is necessarily retrospective in nature and firms may be expected to make strategic adjustments in the face of the IRA’s DPNP.
Particular types of cardiovascular indications may be subject to greater disincentives for future investment, namely those associated with small populations, higher than average shares of older patients, smaller revenue opportunities, or those that face higher risk in achieving successful approval (and thus lower risk-adjusted expected net revenues). Likewise, particular types of post-approval cardiovascular clinical trials may be subject to greater potential disincentives, namely those that require especially large populations, are of particularly long duration, or start later after initial drug approval. These reduced incentives are due to the DPNP provisions that reduce the value of post-approval investments by introducing price caps for some sales after a certain number of years.
While many uncertainties remain, including what actual MFPs will be announced and implemented, and how extensive and rapid the spillover effect will be from government purchases to prices in the commercial market, the level and timing of post-approval indication and clinical trial activity for cardiovascular drugs suggests there could be adverse effects on manufacturer post-approval investment behavior as a result of the DPNP’s changes to the economic incentives for post-approval investment. This is an important issue for policymakers and researchers to monitor going forward.
Transparency
Declaration of financial/other relationships
HG has been a consultant to several government agencies and business organizations and has served as an expert witness in pharmaceutical patent-related litigation on behalf of both plaintiffs and defendants. GL is an employee of Analysis Group, Inc., a consulting company that has provided services to brand-name and generic biopharmaceutical manufacturers and government agencies. The analysis presented was designed and executed entirely by the authors, and therefore, they are responsible for any errors or misstatements.
Author contributions
HG and GL contributed to the conception and design of the study, the analysis and interpretation of the data, the drafting and revising of the manuscript, and gave final approval of the manuscript to be published. All authors agree to be accountable for the aspects of the work as described above.
Reviewer disclosures
Peer reviewers on this manuscript have received an honorarium from JME for their review work but have no other relevant financial relationships to disclose.
Supplemental Material
Download PDF (225 KB)Acknowledgements
The authors thank Jacob Klimek and Dody Eid of Analysis Group, Inc. for valuable data preparation assistance.
Additional information
Funding
Notes
i Social Security Act, §1192.
ii Specifically, the price ceiling for a given drug is the lower of either the Part D average net price or Part B average sales price, as relevant, or a percentage of the average non-federal average manufacturer price (“non-FAMP”, the price paid to manufacturers by wholesalers and distributors in the commercial market after wholesaler discounts and chargebacks), that is 75%, 65%, or 40%, depending on whether the drug is a small molecule 9 to 12 years after US approval (75%), a small molecule or biologic 12 to 16 years after approval (65%), or a small molecule or biologic more than 16 years since approval (40%).
iii The only exception being a transitional temporary price floor for small biotech drugs in 2029 and 2030, following expiration of the small biotech exclusion in place for 2026 through 2028. SSA, §1194(d).
iv Social Security Act, §1194.
v Manufacturers who reject CMS’s MFP “final offer” must either be subject to an excise tax that would increase from 186% and reach 1,900% of total US revenues from all purchasers for the drug after nine months, withdraw all drugs from coverage by Medicare and Medicaid (not just the DPNP MFP drug), or, according to recent CMS Guidance, transfer their interest in the drug to another entity. Congressional Research Service, Tax Provisions in the Inflation Reduction Act of 2022 (HR5376). [cited 2024 Feb 1] Available from: https://crsreports.congress.gov/product/pdf/R/R47202. As a result of the magnitude of these impacts, the Congressional Budget Office (CBO) assumed that no manufacturer would opt to reject the MFP and withdraw from the DPNP. Congressional Budget Office. How CBO Estimated the Budgetary Impact of Key Prescription Drug Provisions in the 2022 Reconciliation Act, February 2023. [cited 2024 Feb 1] Available from: https://www.cbo.gov/system/files/2023-02/58850-IRA-Drug-Provs.pdf). Similarly, the Joint Committee on Taxation assumed no revenues would be generated by such an excise tax provision, as no manufacturer would elect to be subject to it. Congressional Budget Office. Re: Effects of Drug Price Negotiation Stemming From Title 1 of H.R. 3, the Lower Drug Costs Now Act of 2019, on Spending and Revenues Related to Part D of Medicare, October 11, 2019. [cited 2024 Feb 1] Available from: https://www.cbo.gov/system/files/2019-10/hr3ltr.pdf.
vi As confirmed by Citeline’s Pharmaprojects database. (Citeline, Inc., New York City).
vii As defined in the Citeline Pharmaprojects database. (Citeline, Inc., New York City).
viii 21 U.S.C. 355a.
ix 42 CFR Part 11.
References
- Tsao CW, Aday AW, Almarzooq ZI, American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee., et al. Heart disease and stroke statistics-2023 update: a report from the American Heart Association. Circulation. 2023;147(8):e93–e621. doi: 10.1161/CIR.0000000000001123.
- Ward C, Ewald E, Koenig K, et al. Prevalence and health care expenditures among Medicare beneficiaries aged 65 years and over with heart conditions. Medicare Current Beneficiary Survey, data highlight. 2017. https://www.cms.gov/Research-Statistics-Data-and-Systems/Research/MCBS/Downloads/HeartConditions_DataBrief_2017.pdf
- Mills KT, Stefanescu A, He J. The global epidemiology of hypertension. Nat Rev Nephrol. 2020;16(4):223–237. doi: 10.1038/s41581-019-0244-2.
- Avalere Health. Updated reconciliation package changes drugs eligible for negotiation. 2022. https://avalere.com/insights/updated-reconciliation-package-changes-drugs-eligible-for-negotiation
- Congressional Budget Office. How CBO Estimated the Budgetary Impact of Key Prescription Drug Provisions in the 2022 Reconciliation Act. 2023. https://www.cbo.gov/system/files/2023-02/58850-IRA-Drug-Provs.pdf
- Grabowski H, Long G, Mortimer R, et al. Continuing trends in U.S. brand-name and generic drug competition. J Med Econ. 2021;24(1):908–917. doi: 10.1080/13696998.2021.1952795.
- Grabowski H, DiMasi JA, Long G. Post-Approval innovation for oncology drugs and the Inflation Reduction Act. 2024. Under review; available from the authors.
- ClinicalTrials.gov. Protocol registration data element definitions for interventional and observational studies. 2024. https://clinicaltrials.gov/policy/protocol-definitions
- Berger B, et al. Regulatory approval and expanded market size. NBER Working Paper No. 28889, 2021. https://www.nber.org/system/files/working_papers/w28889/w28889.pdf
- Longo N. Partnership for Health Analytic Research. Implications of the Inflation Reduction Act price setting provisions on post-approval indications for small molecule medicines, 2023. https://www.pharllc.com/wp-content/uploads/2023/05/Implications-of-the-IRA-on-Post-Approval-Small-Molecules-2006-2012_Final.pdf.
- Long N. New government price setting policy threatens post-approval research. (Blog post) November 10, 2022. https://catalyst.phrma.org/new-government-price-setting-policy-threatens-post-approval-research
- Lincoff AM, Brown-Frandsen K, Colhoun HM, SELECT Trial Investigators., et al. Semaglutide and cardiovascular outcomes in obesity without diabetes. N Engl J Med. 2023;389(24):2221–2232. doi: 10.1056/NEJMoa2307563.
- Quantitative Life Sciences. Clinical development success rates and contributing factors, 2011-2020. 2021. https://www.bio.org/clinical-development-success-rates-and-contributing-factors-2011-2020