
Abstract
Keratinocyte and fibroblast dysfunctions contribute to delayed healing of diabetic wounds. Small extracellular vesicles (sEV) are key mediators of intercellular communication and are involved in the pathogenesis of several diseases. Recent findings suggest that sEV derived from high-glucose-treated keratinocyte (HaCaT-HG-sEV) can transport LINC01435 to inhibit tube formation and migration of HUVECs, thereby delaying wound healing. This study aimed to elucidate sEV-related communication mechanisms between keratinocytes and fibroblasts during diabetic wound healing. HaCaT-HG-sEV treatment and LINC01435 overexpression significantly decreased fibroblast collagen level and migration ability but significantly increased fibroblast autophagy. However, treatment with an autophagy inhibitor suppressed LINC01435 overexpression-induced decrease in collagen levels in fibroblasts. In diabetic mice, HaCaT-HG-sEV treatment decreased collagen levels and increased the expression of the autophagy-related proteins Beclin-1 and LC3 at the wound site, thereby delaying wound healing. Conclusively, LINC01435 in keratinocyte-derived sEV activates fibroblast autophagy and reduces fibroblast collagen synthesis, leading to impaired diabetic wound healing.
SIGNIFICANCE STATEMENT
Diabetic foot ulcers are a serious complication of diabetes and can lead to amputation and death. Therefore, it is crucial to comprehensively elucidate the mechanisms of delayed diabetic wound healing, with emphasis on the role of keratinocyte-derived small extracellular vesicles. In vivo and in vitro experiments showed that keratinocyte-derived small extracellular vesicles suppressed diabetic wound healing, which is partly attributed to the effects of their content (LINC01435) in fibroblasts. This study suggests that LINC01435 could be targeted to regulate diabetic wound healing.
Introduction
Diabetes is a chronic and complex metabolic disorder that imposes serious health and socioeconomic burden worldwide (Zheng et al. Citation2018). Diabetic foot ulcers (DFUs) are one of the most serious and chronic complications of diabetes and are extremely harmful because of their high disability and mortality rates (American Diabetes Association Citation2020). According to a systematic review and meta-analysis, amputation rate is 10–20 times higher among people with diabetes than among those without diabetes (R. Liu et al. Citation2021). However, the mechanisms underlying refractory DFUs remain unclear. Therefore, further studies are necessary to elucidate the underlying mechanisms and explore potential therapeutic strategies for the prevention and treatment of DFUs.
The skin microenvironment refers to the spatial structure below the skin surface, including the extracellular matrix (ECM), various cells (keratinocytes, fibroblasts, endothelial cells, and immune cells), and cytokines (Schindler Citation2011). During the physiological process of skin wound repair, cells in the skin microenvironment perform specific functions and interact with each other, accompanied by ECM remodelling to facilitate wound healing (Singer and Clark Citation1999). However, the biological functions of various cells, such as proliferation and migration, collagen synthesis, growth factor secretion, and angiogenesis, are suppressed in diabetic wounds (Falanga Citation2005, Galkowska et al. Citation2006, Hao et al. Citation2011, Okizaki et al. Citation2015, Zhu et al. Citation2012). In the skin microenvironment, keratinocytes are the main constituent cells of the epidermis, accounting for more than 80% of epidermal cells, and are responsible for building the skin barrier (Kruse et al. Citation2015, Zhou et al. Citation2019). Additionally, dermal fibroblasts play critical roles inbiological processes in the skin. Specifically, fibroblasts proliferate and migrate, secrete collagen and other ECM, and form granulation tissue with endothelial cells during the proliferation stage, creating conditions for re-epithelialization. Re-epithelialization is characterised by collagen degradation and remodelling, connective tissue shrinkage, and scar formation (Martin Citation1997, Rodrigues et al. Citation2019).
Abnormal interactions between cells in the skin microenvironment play an important role in the regulation of diabetic wound healing. Recent studies suggest that small extracellular vesicles (sEV) constitute an important cell-cell information transmission system (Akbar et al. Citation2019, Mathieu et al. Citation2019, Shah et al. Citation2018). sEV, double-membrane tea saucer-like vesicles with a diameter of 30–150 nm, are widely distributed in various body fluids, transmit signal molecules (e.g. proteins, lipids, DNA, mRNA, lncRNAs, and miRNAs), and participate in the occurrence and development of several diseases (Chaput et al. Citation2006, Stahl and Raposo Citation2019, Xie et al. Citation2019). A preliminary study showed that sEV derived from the wound exudate of patients with DFU significantly inhibited skin wound healing in animal models of diabetes, indicating that sEV play a key role in diabetic wound healing (J. Liu et al. Citation2021). Additionally, lncRNA microarray assay and quantitative reverse transcription polymerase chain reaction (qRT-PCR) showed that keratinocyte-derived sEV carrying LINC01435 enter vascular endothelial cells and inhibit the angiogenic and migration abilities of vascular endothelial cells, thereby influencing the recovery of diabetic wounds (Fu et al. Citation2022). Therefore, it is necessary to investigate whether LINC01435 in keratinocyte-derived sEV can be taken up by fibroblasts and regulate diabetic wound healing.
Autophagy is an essential cellular process that plays dual roles in various physiological processes. Refractory diabetic wounds may be related to dysregulation of autophagy (Yu et al. Citation2015). Autophagy is significantly upregulated in fibroblasts in diabetic skin wounds, leading to decreased migration, increased collagen degradation, and decreased collagen secretion (Sun et al. Citation2016). Additionally, sEV derived from vascular endothelial cells under diabetic conditions can inhibit the expression of the ERK1/2 pathway and trigger fibroblast autophagy, resulting in reduced collagen secretion and delayed healing of diabetic wounds (Zeng et al. Citation2019). However, the regulatory role of LINC01435 in the activities of keratinocytes and fibroblasts during diabetic wound healing remains unknown.
Therefore, this study was aimed at exploring whether LINC01435 in keratinocyte-derived sEV affects the biological function of fibroblasts and participates in the healing of diabetic wounds. This may provide a potential approach for promoting wound healing.
Materials and methods
Cell culture and treatment
The human keratinocyte cell line (HaCaT) was purchased from Procell Life Science & Technology, and then cultured in Minimum Essential Medium (MEM; Procell) supplemented with 10% foetal bovine serum (FBS; Gibco). Human primary foreskin fibroblasts were obtained from Sun Yat-sen University and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% FBS. All cells were cultured at 37 °C in a humidified atmosphere containing 95% air and 5% CO2. Cells were regularly tested for mycoplasma contamination. At 50% confluency, HaCaT cells were cultured in a high glucose medium (HG; 33.3 mmol/L(mM); Sigma-Aldrich). Except for normal glucose (NG; 5.55 mM), isotonic mannitol (HM; 5.55 mM of glucose + 27.75 mM of mannitol; Sigma-Aldrich) was used as the osmotic control for high-glucose in MEM containing 10% sEV-free FBS for 48 h, and the cell supernatant was collected for subsequent sEV isolation. sEV-free FBS was prepared by ultracentrifuged at 120,000 g at 4 °C for 16 h to remove sEV.
Based on previous findings, fibroblasts were stimulated with 25 μg/mL of sEV derived from HaCaT pre-treated with high mannitol (HM-sEV), high glucose (HG-sEV), phosphate-buffered saline (PBS; Jet Bio-Filtration).
sEV isolation and identification
sEV were isolated from conditioned cell culture supernatants using the differential centrifugation method (Théry et al. Citation2006). Briefly, the conditioned medium underwent a series of centrifugation steps to remove cell debris (300 × g for 10 min, 2000 × g for 20 min, and 10,000 × g for 30 min). After filtering through 0.22-μm filters (Jet Bio-Filtration), the supernatant was centrifuged at 110,000 × g for 70 min. Finally, pure sEV were resuspended in PBS for subsequent identification and experiments after another centrifugation step (110,000 × g for 70 min) in PBS. All operations were performed at 4 °C. To further characterise sEV, western blotting was performed to examine the expression of the positive markers CD9 and TSG101, and the negative marker Grp94. Transmission electron microscopy (TEM; Hitachi) was employed to confirm sEV morphology, and nanoparticle tracking analysis (NTA; NanoSight 300; Malvern Panalytical) was used to analyse size distribution and concentration.
PKH26-labelled sEV
Purified sEV were labelled with a red fluorescent dye using the PKH26 labelling kit (Sigma–Aldrich), according to the manufacturer’s instructions. Briefly, sEV (isolated from 5 × 106 HaCaT cells) were suspended in 4 μL of PKH26 diluted in 1 ml of Diluent C (1:250), followed by incubation in the dark at room temperature (RT) for 5 min and the addition of 1 ml of sEV-free FBS to terminate the reaction. Subsequently, the labelled sEV were harvested and washed twice in PBS by ultracentrifugation (110,000 × g for 70 min). Finally, the PKH26-labelled sEV were resuspended in DMEM and co-incubated with fibroblasts in an incubator for 24 h. After fixing with 4% paraformaldehyde in PBS for 15 min at RT, the nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI, Solarbio) for 5 min at RT. The uptake of sEV into stained cells was visualised using confocal laser scanning microscopy (LSM; Zeiss).
Western blot analysis
Proteins were extracted from fibroblasts using radioimmunoprecipitation assay (RIPA) lysis buffer (EpiZyme) containing a protease and phosphatase inhibitor cocktail (CWBIO). Total protein concentrations in cells and sEV were quantified using a bicinchoninic acid (BCA) Protein Quantification Kit (EpiZyme). Lysates were diluted with protein loading buffer (EpiZyme) at a ratio of 1:4 and heated with NuPAGE LDS Sample Buffer (Invitrogen, NP0007) at 98 °C for 10 min. Proteins were separated using sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS–PAGE; EpiZyme) and transferred to polyvinylidene fluoride (PVDF) membranes (Merck Millipore). Subsequently, membranes were blocked with 5% non-fat milk (Sangon Biotech) and 0.1% Tween 20 in TBST (TBS, Boster; Tween-20, Solarbio) at RT for 4 h, followed by incubation with corresponding primary antibody overnight at 4 °C. The membranes were incubated with secondary antibodies (Beyotime) at RT for 1 h, followed by the detection of antibody reaction using an ECL reagent (EpiZyme) and a Mini Chemi imaging system (Sage Creation). Band intensity was quantified using ImageJ 5.0 software, and the relative expression levels of the target proteins were normalised to that of β-actin. Primary antibodies against CD9, Tsg101, Grp94, collagen I and III were purchased from Abcam, antibodies against LC3 were obtained from Cell Signalling Technology, antibodies against Beclin-1 were obtained from Proteintech, and β-actin were obtained from ImmunoWay.
Enzyme-linked immunosorbent assay (ELISA)
Collagen type I and III protein levels in the samples were determined using a commercially available ELISA kit (Elabscience), according to the manufacturer’s instructions. The absorbance of each well was measured at 450 nm using a multiscan spectrophotometer (Tecan Spark), and the protein levels in the samples were calculated using a four-parameter logistic (4-PL) standard curve, according to the manufacturer’s instructions.
qRT-PCR
Total RNA was isolated from the cultured cells using sEV RNAiso Plus (Takara) and reverse-transcribed to complementary DNA (cDNA) using the PrimeScript RT Master Mix (TaKaRa). qRT-PCR was performed on a LightCycler 480 II Real-Time PCR instrument (Roche) using TB Green Premix Ex Taq II (TaKaRa). The threshold value upregulated lncRNAs was set at a fold change (FC) of 1.2 and p < 0.05. The relative mRNA expression levels of the target genes were calculated using the 2−ΔΔCt method and normalised to that of ACTB (internal control). Primer sequences are listed in .
Table 1. Primer sequences of mRNA and lncRNA.
Cell proliferation assay
Changes in cell viability in response to the various treatments were assessed using a Cell Counting Kit-8 (CCK-8) assay. Briefly, cells at the log phase of growth were inoculated into 96-well plates at a density of 3 × 103 cells/well. After incubation for 0, 24, 48, 72, and 96 h, a mixture of 10 μL of CCK-8 reagent (BLPBIO) and 90 μL of medium was added to each well. After incubation for 2 h, the optical density (OD) of each well was measured at 450 nm using a Multiscan spectrophotometer (Tecan Spark). DNA proliferation analysis was performed using 5-ethyl-2 deoxyuridine (EdU) Cell Proliferation Kit with Alexa Fluor 555 (Cellorlab), according to the manufacturer’s instructions. Images of EdU-positive cells from five visual fields were obtained using an inverted fluorescence microscope and counted using ImageJ software.
Cell migration assay
A scratch assay was used to assess the horizontal migration ability of fibroblasts. Briefly, cells were seeded into a 6-well plate, and the monolayer was scratched with a sterile 200-μL pipette tip when the cells had attached. After washing with PBS to remove the floating cells, the medium was replaced with RPMI-1640 culture medium containing 1% FBS. Wounded areas were photographed at 0 and 24 h and calculated using ImageJ software. Cell migration rate was calculated using the following equation: Migration area (%) = (A0 − An)/A0 × 100, where A0 represents the blank area at 0 h and An represents the remaining area at the measuring point.
Additionally, migration assay was performed in 24-well transwell chambers (Corning) with 8.0 μm pore size to assess the vertical migration abilities of fibroblasts. Briefly, cells (6 × 104 per well) in 300 μL of DMEM medium containing 2.5% FBS were incubated in the upper chamber for 16 h, and lower chamber was filled with 700 μL of medium with 15% FBS. After incubated at 37 °C for 16 h, migrating cells were fixed with 0.4% paraformaldehyde and stained with 0.1% crystal violet (Beyotime). Images of five randomly selected fields were taken, and the number of migrating cells was calculated using ImageJ 5.0 software.
Flow cytometric analysis
To assess cell apoptosis, fibroblasts from different treatment groups were collected and resuspended in 100 μL of binding buffer. Subsequently, the cells were treated with 5 μL of Annexin V-FITC (Elabscience) and 10 μL of propidium iodine (PI) reagent and then incubated at RT in the dark for 15 min. For cell cycle assay, cells were collected and fixed with pre-cold 70% ethanol at 4 °C overnight, following by resuspension in 100 μL of RNaseA at 37 °C for 30 min and incubated with 400 μL of PI at RT for 30 min in the dark. Finally, fluorescent cells were quantified using fluorescence-activated cell sorting (FACS, Becton Dickinson). The experiment was repeated three times with three pairs of wells in each round.
Transmission electron microscopy (TEM)
Briefly, cells were fixed with 2.5% glutaraldehyde in the dark at 4 °C for 2 h and collected by centrifugation (500 × g for 3 min). Subsequently, cell pellets were embedded in 1% agarose, followed by post-fixing in 1% osmium tetroxide at RT for 2 h. The pellets were dehydrated using graded concentrations of ethanol (50, 70, 80, 90, 95, 100, and 100%) and acetone (100 and 100%), and embedded in epoxy resin agent (SPI) at 37 °C overnight and at 60 °C for 48 h. The samples were cut into 60–80 nm ultra-thin sections and stained with 2% uranium-acetate saturated alcohol solution and lead citrate for 15 min, respectively. Finally, the morphology of the autophagic structures was examined using TEM (Hitachi, Ltd.).
Autophagic flux
After transfection with tandem mRFP-GFP-LC3 (Hanbio), the fibroblasts were treated with sEV or transfected with plasmids, followed by fixation with 4% paraformaldehyde for 15 min and incubation with DAPI for 5 min to counterstain the nuclei. Finally, images were obtained using a confocal laser-scanning microscope (Zeiss), and the numbers of autophagosomes and autolysosomes were counted using ImageJ software.
Cell transfection
The LINC01435 overexpression plasmid (pcDNA3.1) was obtained by inserting the coding sequence (CDSs) of LINC01435 into pcDNA3.1. Cells in the control group were transfected with pcDNA3.1-empty vector. Additionally, LINC01435 expression in fibroblast cells was silenced. Plasmids were transfected using the INVI DNA RNA Transfection Reagent (Invitrogen), according to the manufacturer’s instructions.
Animal experiment
Seven-week-old male BKS-Leprem2Cd479/Gpt (db/db) mice were purchased from the Guangzhou Yaokang Animal Centre and raised at the Laboratory Animal Centre of Sun Yat-sen University. After moulting the backs of the mice, two circular full-thickness skin wounds (diameter: 6 mm) were cut using a skin biopsy punch. The db/db mice were randomly divided into three groups, and HM-sEV or HG-sEV (200 μg dissolved in 100 μL of PBS) or PBS (100 μL) were subcutaneously injected once around the wounds at four injection sites (25 μL per site). Images of the wounds were digitally photographed at 0, 6, and 11 days after wound modelling, and the wound areas were measured using the ImageJ software. Finally, the mice were sacrificed 11 days after the operation, and tissue specimens were harvested for subsequent experiments. All animal experiments were approved by the Animal Research Committee of the Sun Yat-sen University (approval number: SYSU-IACUC-2020-B0129).
Haematoxylin and Eosin (H&E), Masson’s, Immunohistochemical (IHC), and Immunofluorescence (IF) staining
Briefly, skin tissues were fixed with 4% paraformaldehyde, paraffin-embedded, cut into sections and baked at 65 °C for 2 h before deparaffinization. Subsequently, the sections were stained with H&E (Servicebio) and Masson’s trichrome (Servicebio).
For IHC and IF assays, tissue sections were deparaffinized, subjected to antigen retrieval with EDTA (pH 8.0; ZSGB-BIO), permeabilized with Triton X-100 (Solarbio), and blocked with a blocking buffer (Beyotime). Subsequently, the sections were incubated with primary antibodies at 4 °C overnight, followed by incubation with the respective secondary antibodies at 37 °C for 1 h. Finally, the sections were stained with DAPI (Solarbio) in the dark for 5 min for IF assay. Images were obtained using a Nikon Ni-U camera (for HE, Masson’s, and IHC staining) or a Zeiss confocal laser-scanning microscope (for IF staining), and five random fields were observed in each section. Epidermal thickness and the average score of the selected areas were calculated using the ImageJ software.
Statistical analysis
Data are presented as the mean ± standard deviation (SD) or mean ± standard error of mean (SEM) derived from at least three independent experiments. All statistical analyses were performed using GraphPad Prism Software (GraphPad Software, San Diego, CA, USA). Student’s t-tests were used for two-group comparisons, while one-way analysis of variance (ANOVA) or one-way multivariate analysis of variance (MANOVA) was used for multiple group comparisons, followed by Fisher’s least significant difference (LSD) post-hoc test. Statistical significance was set at p < 0.05. Significance levels were indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001; ns: not significant.
Results
Characterisation of HaCaT-Derived sEV
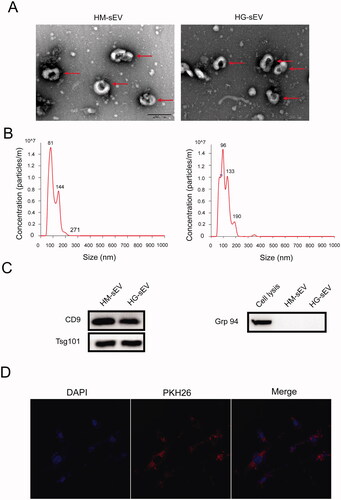
To investigate the role of HaCaT cell-derived sEV in the biological functions of human skin fibroblasts and diabetic wound healing, sEV were isolated and characterised using TEM, NTA, and western blotting. TEM showed that the purified sEV exhibited the typical cup- or sphere-shaped characteristics of membrane-bound vesicles (). NTA using Nanosight showed that the average diameters of the sEV ranged from 30–200 nm (). Moreover, western blotting confirmed the presence of the sEV surface markers CD9 and TSG101, and the absence of the negative marker Grp94 (). Furthermore, fibroblasts were incubated with PKH26-labelled sEV for 24 h to assess sEV uptake by the fibroblast. Laser scanning confocal microscopy showed the presence of PKH26 in fibroblasts treated with HaCaT-sEV, indicating that sEV can transmit between cells ().
Figure 1. Characterisation of the HaCaT-derived small extracellular vesicles (sEV). (A) The ultrastructure of the two types of sEV was observed using transmission electron microscopy (TEM). Scale bar: 200 nm. Red arrow indicates the representative morphology of the sEV. (B) The size distribution of the sEV was measured using NanoSight analysis. (C) sEV-related markers and negative markers were detected using western blotting. (D) Laser scanning confocal microscopy was used to detect the presence of HaCaT-derived sEV (pre-labelled with PKH26) in fibroblasts.
HaCaT-HG-sEV inhibits the biological function of fibroblasts
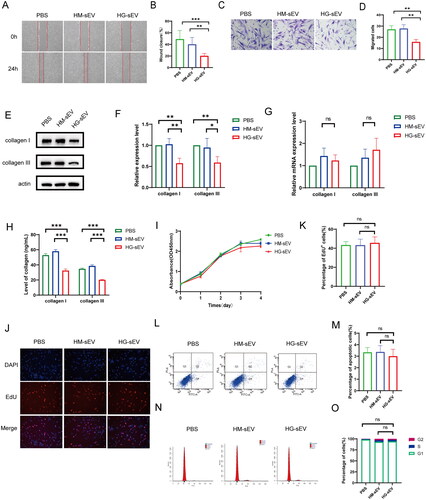
Several biological function assays were performed to investigate the functional role of HaCaT cell-derived sEV in fibroblast and wound healing HaCaT cells. Scratch and transwell assays indicated that HG-sEV treatment significantly decreased the horizontal and vertical migration of fibroblasts compared with HM-sEV treatment (). Compared with HM-sEV treatment, HG-sEV treatment significantly reduced collagen I and III protein levels in fibroblasts () and suppressed collagen secretion (). However, there was no significant difference in the mRNA levels of collagen I and III in fibroblasts between the groups (). Additionally, CCK-8 and EdU assays showed that HG-sEV treatment did not affect fibroblast proliferation (). Similarly, flow cytometry showed that there were no significant differences in cell apoptosis and cell cycle between the HG-sEV and PBS- or HM-sEV groups ().
Figure 2. HaCaT-HG-sEV inhibits the biological function of fibroblasts. (A–D) The migration ability of fibroblasts treated with PBS, HM-sEV, or HG-sEV was examined using scratch and transwell assays. (E, F) The protein expression levels of collagen types I and III in fibroblasts treated with PBS, HM-sEV, or HG-sEV were examined using western blotting. (G) The mRNA expression of collagen I and III was validated using quantitative real-time polymerase chain reaction (qRT-PCR). (H) The levels of collagen I and III were examined using enzyme-linked immunosorbent assay (ELISA) kit. (I–K) Cell proliferation level was measured using CCK-8 and Edu staining assay. (L–O) Cell apoptosis and cell cycle were analysed using flow cytometry. *p < 0.05, **p < 0.01, ***p < 0.001.
sEV-packaged LINC01435 suppresses biological function in human skin fibroblasts
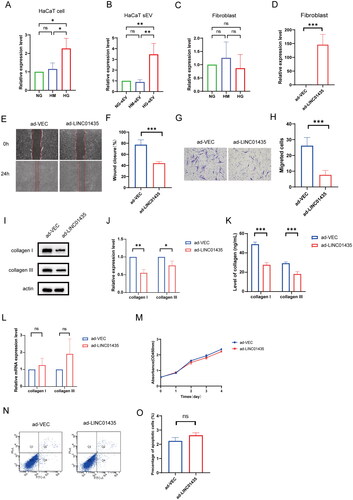
LncRNAs can be selectively packed into sEV and ingested by target cells to modulate various physiological and pathological processes. In a previous study, we identified differentially expressed lncRNAs in sEV derived from HaCaT pre-treated with or without advanced glycation end products (AGEs) (Fu et al. Citation2022). In this study, we further confirmed the expression of LINC01435 in HaCaT cells and human skin fibroblasts after treatment with HM or HG using qRT-PCR. Among the lncRNAs, LINC01435 was highly enriched in HaCaT cells pre-treated with high glucose versus NG or HM, and highly expressed in HG-sEV than in NG-sEV or HM-sEV derived from HaCaT cells (). However, there was no significant difference in LINC01435 expression between NG-, HM- and HG-treated fibroblasts, further supporting our hypothesis (). To elucidate the relationship between LINC01435 and fibroblast migration, fibroblasts were transfected with vector overexpressing LINC01435 (). Compared with that in the pcDNA3.1-vector group (ad-VECs), LINC01435 overexpression suppressed the migration ability of fibroblasts (). Notably, transfection with pcDNA3.1-LINC01435 (ad-LINC01435) significantly decreased the protein expression or secretion of collagen in fibroblasts (). However, there were no significant differences in collagen mRNA levels and fibroblast apoptosis and proliferation between the ad-VECs and ad-LINC01435 groups (). Overall, these results suggest that LINC01435 packaged in HG-sEV derived from HaCaT cells can inhibit collagen level and fibroblast migration.
Figure 3. sEV-packaged LINC01435 suppresses biological function in human skin fibroblasts. (A–C) qRT-PCR was used to detect the differential expression of LINC01435 in HaCaT cells, HaCaT-sEV, and fibroblasts stimulated using high glucose. (D) qRT-PCR was used to confirm successful transfection of plasmid carrying LINC01435 into fibroblasts. (E–H) The migration ability of LINC01435-overexpressing fibroblasts was examined using scratch and transwell assays. (I, J) The protein expression of collagen I and III in LINC01435-overexpressing fibroblasts was examined using western blotting. (K) The levels of collagen I and III were examined using ELISA kits. (L) mRNA expression of collagen I and III was validated using qRT-PCR. (M) Cell proliferation level was measured using a CCK-8 assay. (N, O) Cell apoptosis was analysed using flow cytometry. *p < 0.05, **p < 0.01, ***p < 0.001.
LINC01435 suppresses the activities of fibroblasts by triggering autophagy
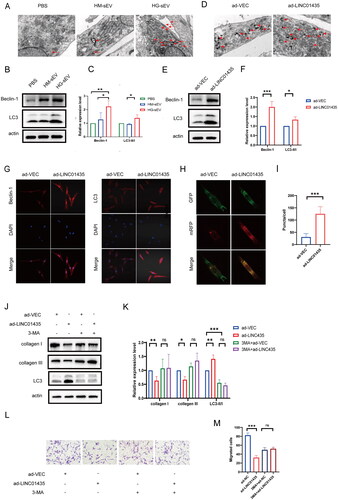
As autophagy is an important regulatory mechanism for fibroblast migration and collagen synthesis (Qiang et al. Citation2021, Zhao et al. Citation2019), we examined the role of LINC01435 on fibroblast autophagy. TEM showed an increased autophagosomes in sEV-treated fibroblasts compared with that in PBS-treated cells (). During autophagy, Beclin-1 and the lipid kinase Vps34 form a complex to promote autophagic membrane remodelling and LC3-I transforms to LC3-II, indicating that LC3-II/LC3-I ratio (LC3-II/I) and Beclin-1 expression could be used to assess the degree of autophagy. Western blot analysis showed an increase in Beclin-1 expression and LC3-II/I ratio in fibroblasts treated with HG-sEV (). Similarly, TEM showed that LINC01435 overexpression increased autophagy (), Beclin-1 expression, and LC3-II/I ratio in fibroblasts (). IF staining confirmed an increase in Beclin-1 and LC3 levels in LINC01435-overexpressing fibroblasts compared with that in the vector (control) group (). Additionally, the number of autophagosomes increased in LINC01435-overexpressing fibroblasts, suggesting an increase in autophagic flux in the fibroblasts (). To further investigate whether autophagy is involved in the inhibitory effect of LINC01435, LINC01435-overexpressing fibroblasts were treated with 3-methyladenine (3-MA; an inhibitor of PI3K), followed by the detection of fibroblast migration. 3-MA treatment suppressed LINC01435 overexpression-induced increase in autophagy in fibroblasts (). However, there was no significant difference in collagen expression and the migration ability of fibroblasts between the ad-VEC and ad-LINC01435 groups after 3-MA treatment (). Collectively, these results suggest that LINC01435 can reduce collagen protein levels in fibroblasts, probably by triggering fibroblast autophagy.
Figure 4. LINC01435 suppresses the biological function of fibroblasts by triggering autophagy. (A) TEM analysis of fibroblasts treated with PBS, HM-sEV, or HG-sEV. Arrow heads indicate autophagosomes. (B, C) The expression of the autophagy-related proteins LC3 and Beclin-1 in fibroblasts treated with PBS, HM-sEV, or HG-sEV was examined using western blotting. (D) TEM analysis of LINC01435-overexpressing fibroblasts. Arrow heads indicate autophagosomes. (E, F) The expression of the autophagy-related proteins LC3 and Beclin-1 in LINC01435-overexpressing fibroblasts was detected using western blotting. (G) The expression of the autophagy-related proteins LC3 and Beclin-1 in LINC01435-overexpressing fibroblasts was detected using immunofluorescence assay. (H, I) Autophagy flux was observed in LINC01435-overexpressing fibroblasts infected with MRFP-GFP-LC3 using laser confocal microscopy, and semi-quantitative analysis of autophagosomes and autolysosomes (including the yellow and red fluorescent spots in merged images) was conducted. (J–M) Treatment with 3-MA restored collagen protein expression and the migration ability in LINC01435-overexpressing fibroblasts. *p < 0.05, **p < 0.01, ***p < 0.001.
HG-sEV suppresses cutaneous wound healing in db/db mice In Vivo
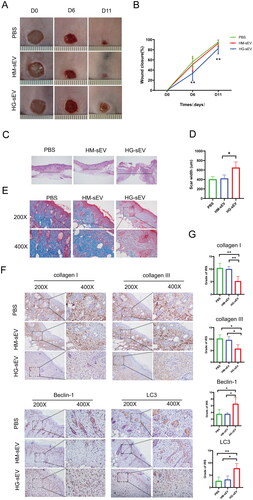
To confirm the in vitro effects and mechanisms of HG-sEV in vivo, the site of diabetic wounds in db/db mice was injected with HG-sEV. BKS-Leprem2Cd479/Gpt (db/db) mice (mouse model of diabetes) were used to simulate diabetic wound healing. Wound closure rates were observed after treatment with PBS, HM-sEV, and HG-sEV. Wound closure rate was significantly lower (p < 0.05) in the HG-sEV group than in the PBS and HM-sEV groups at 6 and 11 days post-wounding (). Wound tissues were collected at 11 days post-wounding for H&E, Masson’s, and IHC staining. H&E staining showed a decrease in epithelial regeneration in HG-sEV-treated wounds compared with that in PBS- and HM-sEV-treated wounds (), further confirming the negative effect of HG-sEV in diabetic wound healing. Additionally, Masson’s staining revealed decreased collagen deposition in HG-sEV-treated wounds (). Furthermore, collagen- and autophagy-related proteins were detected in the skin tissues using IHC staining. Compared with that in the PBS and HM-sEV groups, there was decrease in the expression of collagen I and III proteins in the dermis of HG-sEV-treated mice, and an increase Beclin-1 and LC3 expression (). In addition, compared to the PBS group and HM-sEV group, there was a noticeable reduction in the number of hair follicles in the wound sites of the HG-sEV group, suggesting a slower wound healing process in the HG-sEV group. Collectively, these data indicate that HaCaT-HG-sEV treatment induces autophagy and decreases collagen deposition during diabetic wound healing process in mice.
Figure 5. HG-sEV suppress cutaneous wound healing in db/db mice in vivo. (A, B) Representative images of cutaneous wounds treated with PBS, HM-sEV, or HG-sEV at 0, 6, and 11 days post-wounding. (C) HE staining of wound sections after different treatments at 11 days post-wounding. The double-headed arrows represent the edge of the scar. (D) The extent of scar widths of three groups was measured at 11 days post-wounding. (E) Transmitted light images of Masson’s staining and Sirius red staining of three groups to evaluate collagen maturity. (F, G) Immunohistochemical staining to determine collagen I and III, Beclin-1, and LC3B protein expression in skin tissues and related quantitative analyses were performed according to the IRS. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
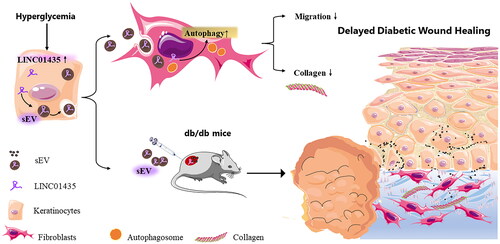
Cellular interactions govern the functions of multiple cells during cutaneous wound healing. Under diabetic conditions, the biological functions of keratinocytes and fibroblasts are impaired (Y. Wang et al. Citation2023). However, the specific mechanism underlying delayed diabetic wound healing has not been clarified. In the present study, LINC01435-containing HG-sEV derived from keratinocytes were taken up by fibroblasts during diabetic healing, which enhanced autophagy and impaired fibroblast collagen synthesis and migration (). To the best of our knowledge, this is the first evidence of the direct involvement of LINC01435 signalling in keratinocyte–fibroblast crosstalk in diabetes-related dermopathy. Moreover, the results indicate the clinical value and potential of targeting non-coding RNA as a treatment strategy for diabetic ulcers.
Figure 6. Schematic of the potential mechanism by which HaCaT-sEV regulates the biological functions of fibroblasts in diabetic wound healing. In this study, LINC01435-containing HG-sEV were transferred to skin fibroblasts, triggering autophagy and decreasing collagen synthesis in fibroblasts, thereby delaying wound healing in vivo.
Keratinocytes and fibroblasts are crucial repair cells, and the interaction between them plays an indispensable role in the wound healing process (Martin and Nunan Citation2015). Our study revealed a novel crosstalk of the skin epidermal-dermal axis under diabetic conditions during wound healing. LINC01435 in keratinocyte-derived sEV was taken up by fibroblasts, demonstrating a direct sEV exchange between epidermal and dermal cells. Additionally, treatment with keratinocyte-derived sEV decreased fibroblast migration and collagen synthesis. sEV can participate in pathophysiological processes by working locally or secreting molecules into the extracellular space to modulate the function of skin cells during wound healing (Bang et al. Citation2014, Fong et al. Citation2015, Wang et al. Citation2019). Therefore, we locally administered HG-sEV derived from keratinocytes to the sites of diabetic wounds and observed a delay in wound closure. Collectively, LINC01435 in keratinocyte-derived sEV serves as a vital regulator of the communication between keratinocytes and fibroblasts.
sEV are lipid bilayer vesicles containing various cellular components, including nucleic acids (DNA, mRNA, miRNA, circRNA, and lncRNA), lipids, and proteins (enzymes, heat shock proteins, and various tissue-specific proteins) (Hong et al. Citation2019). Notably, HaCaT-HG-sEV enriched with LINC01435 can enter endothelial cells and increase the expression of histone deacetylases (HDAC8) by inducing the transport of Yin Yang 1 (YY1) into nucleus, thereby inhibiting the tube formation and migration ability of endothelial cells and affecting angiogenesis (Fu et al. Citation2022). Given that sEV serve as vital messengers for information transmission and material communication between cells (Kim et al. Citation2019, Li et al. Citation2019, Narauskaitė et al. Citation2021), we investigated whether HaCaT-HG-sEV can mediate fibroblasts function by transmitting LINC01435. LINC01435 overexpression decreased the collagen level and migration ability of fibroblasts. Overall, these results indicate that HaCaT-derived sEV can carry internalised lncRNAs to different target cells to regulate different biological processes, thereby mediating wound healing in diabetes.
Autophagy participates in regulating various cellular functions and plays an essential role in both the proliferation and remodelling of wound healing by mediating the survival and maintenance of fibroblasts (Cao et al. Citation2018, Nakatogawa et al. Citation2009). Therefore, we examined the role of autophagy in the crosstalk of the skin epidermal-dermal axis. In a previous study, autophagy was significantly activated and upregulated, which decreased the migration ability of fibroblasts and collagen synthesis and secretion, resulting in refractory diabetic skin wounds. Similarly, autophagy was enhanced in fibroblasts treated with HaCaT-HG-sEV or overexpressing LINC01435. However, 3-MA treatment inhibited fibroblast autophagy and rescued the collagen protein levels, which was consistent with the previous findings (Dai et al. Citation2019, Shi et al. Citation2021). Specifically, endothelial cell-derived sEV can influence cutaneous wound healing by triggering autophagy and decreasing collagen synthesis in fibroblasts (Zeng et al. Citation2019). Downregulation of autophagy in fibroblasts can induce apoptosis, thereby degrading excessive fibrin in cells, improving the excessive deposition of ECM, and inhibiting fibrosis (Cao et al. Citation2018). Collectively, these findings suggest that autophagy plays a dual role in the regulation of wound healing, and further research is needed to accurately regulate autophagy in cells to facilitate wound healing.
To enhance the therapeutic potential of sEV for wound healing and promote their translation into clinical practice, manipulating source cells and modifying sEV has become an important issue. Several studies have examined the modification of sEV by preconditioning, genetic manipulation, biomaterial incorporation, and nanoparticle loading (Al-Masawa et al. Citation2022, Luo et al. Citation2021). For example, biological scaffolds comprising ADSC-sEV, collagen-1, and platelet-rich plasma exhibit excellent proangiogenic and anti-inflammatory effects to accelerate wound healing (Y. Wang et al. Citation2023). Polydopamine nanoparticles and mesenchymal stem cell-secreted sEV have been loaded into multifunctional hydrogels for the treatment of radiation-induced skin wounds (Fang et al. Citation2023). However, further studies are necessary to develop strategies for modifying LINC01435 in sEV derived from keratinocytes in diabetic ulcers.
Conclusively, the present study demonstrates a novel mechanism of impaired diabetic wound healing. Mechanistically, LINC01435 in keratinocyte-derived sEV trigger fibroblast autophagy, thereby decreasing collagen synthesis in fibroblasts. These findings provide potential regulatory and modification targets for sEV as a therapeutic strategy for the treatment of diabetic ulcers.
Authors contributions
XSH, LQC and LLL: Performed most experiments and analyses and wrote the manuscript. DHZ, JHL, ZXL, and WF: Performed the experiments. DFL, TTZ, KS, WW, and SFC: Contributed to data interpretation and discussion. MR and LY: Contributed to the study conception and design. All authors approved the version to be published. All authors reviewed and approved the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available on request from the corresponding author (LY).
Additional information
Funding
References
- Akbar, N., et al., 2019. Extracellular vesicles in metabolic disease. Diabetologia, 62 (12), 2179–2187. doi: 10.1007/s00125-019-05014-5.
- Al-Masawa, M.E., et al., 2022. Efficacy and safety of small extracellular vesicle interventions in wound healing and skin regeneration: A systematic review and meta-analysis of animal studies. Theranostics, 12 (15), 6455–6508. doi: 10.7150/thno.73436.
- American Diabetes Association, 2020. Microvascular complications and foot care: Standards of medical care in diabetes-2020. Diabetes care, 43 (Supplement_1), S7–S13. doi: 10.2337/dc20-S011.
- Bang, C., et al., 2014. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. The journal of clinical investigation, 124 (5), 2136–2146. doi: 10.1172/jci70577.
- Cao, C., et al., 2018. Inactivation of Beclin-1-dependent autophagy promotes ursolic acid-induced apoptosis in hypertrophic scar fibroblasts. Experimental dermatology, 27 (1), 58–63. doi: 10.1111/exd.13410.
- Chaput, N., et al., 2006. Dendritic cell derived-exosomes: biology and clinical implementations. Journal of leukocyte biology, 80 (3), 471–478. doi: 10.1189/jlb.0206094.
- Dai, J., et al., 2019. Negative regulation of PI3K/AKT/mTOR axis regulates fibroblast proliferation, apoptosis and autophagy play a vital role in triptolide-induced epidural fibrosis reduction. European journal of pharmacology, 864, 172724. doi: 10.1016/j.ejphar.2019.172724.
- Falanga, V., 2005. Wound healing and its impairment in the diabetic foot. Lancet (London, England), 366 (9498), 1736–1743. doi: 10.1016/s0140-6736(05)67700-8.
- Fang, Z., et al., 2023. A multifunctional hydrogel loaded with two nanoagents improves the pathological microenvironment associated with radiation combined with skin wounds. Acta biomaterialia, 159, 111–127. doi: 10.1016/j.actbio.2023.01.052.
- Fong, M.Y., et al., 2015. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nature cell biology, 17 (2), 183–194. doi: 10.1038/ncb3094.
- Fu, W., et al., 2022. Long noncoding RNA LINC01435 impedes diabetic wound healing by facilitating YY1-mediated HDAC8 expression. iScience, 25 (4), 104006. doi: 10.1016/j.isci.2022.104006.
- Galkowska, H., et al., 2006. Chemokines, cytokines, and growth factors in keratinocytes and dermal endothelial cells in the margin of chronic diabetic foot ulcers. Wound repair and regeneration: official publication of the wound healing society [and] the European tissue repair society, 14 (5), 558–565. doi: 10.1111/j.1743-6109.2006.00155.x.
- Hao, S.Y., et al., 2011. Activation of skin renin-angiotensin system in diabetic rats. Endocrine, 39 (3), 242–250. doi: 10.1007/s12020-010-9428-z.
- Hong, P., et al., 2019. The functions and clinical application potential of exosomes derived from adipose mesenchymal stem cells: a comprehensive review. Stem cell research & therapy, 10 (1), 242. doi: 10.1186/s13287-019-1358-y.
- Kim, H., et al., 2019. Exosome-guided phenotypic switch of M1 to M2 macrophages for cutaneous wound healing. Advanced science (Weinheim, Baden-Wurttemberg, Germany), 6 (20), 1900513. doi: 10.1002/advs.201900513.
- Kruse, C.R., et al., 2015. The external microenvironment of healing skin wounds. Wound repair and regeneration: official publication of the wound healing society [and] the European tissue repair society, 23 (4), 456–464. doi: 10.1111/wrr.12303.
- Li, M., et al., 2019. Macrophage-derived exosomes accelerate wound healing through their anti-inflammation effects in a diabetic rat model. Artificial cells, nanomedicine, and biotechnology, 47 (1), 3793–3803. doi: 10.1080/21691401.2019.1669617.
- Liu, J., et al., 2021. MiR-195-5p and miR-205-5p in extracellular vesicles isolated from diabetic foot ulcer wound fluid decrease angiogenesis by inhibiting VEGFA expression. Aging, 13 (15), 19805–19821. doi: 10.18632/aging.203393.
- Liu, R., et al., 2021. Lower extremity reamputation in people with diabetes: a systematic review and meta-analysis. BMJ open diabetes research & care, 9 (1), e002325. doi: 10.1136/bmjdrc-2021-002325.
- Luo, L., et al., 2021. Regulating the production and biological function of small extracellular vesicles: current strategies, applications and prospects. Journal of nanobiotechnology, 19 (1), 422. doi: 10.1186/s12951-021-01171-1.
- Martin, P., 1997. Wound healing–aiming for perfect skin regeneration. Science (New York, NY), 276 (5309), 75–81. doi: 10.1126/science.276.5309.75.
- Martin, P., and Nunan, R., 2015. Cellular and molecular mechanisms of repair in acute and chronic wound healing. The British journal of dermatology, 173 (2), 370–378. doi: 10.1111/bjd.13954.
- Mathieu, M., et al., 2019. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nature cell biology, 21 (1), 9–17. doi: 10.1038/s41556-018-0250-9.
- Nakatogawa, H., et al., 2009. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nature Reviews. Molecular cell biology, 10 (7), 458–467. doi: 10.1038/nrm2708.
- Narauskaitė, D., et al., 2021. Extracellular vesicles in skin wound healing. Pharmaceuticals (Basel, Switzerland), 14 (8), 811. doi: 10.3390/ph14080811.
- Okizaki, S., et al., 2015. Suppressed recruitment of alternatively activated macrophages reduces TGF-β1 and impairs wound healing in streptozotocin-induced diabetic mice. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie, 70, 317–325. doi: 10.1016/j.biopha.2014.10.020.
- Qiang, L., et al., 2021. Keratinocyte autophagy enables the activation of keratinocytes and fibroblastsand facilitates wound healing. Autophagy, 17 (9), 2128–2143. doi: 10.1080/15548627.2020.1816342.
- Rodrigues, M., et al., 2019. Wound healing: A cellular perspective. Physiological reviews, 99 (1), 665–706. doi: 10.1152/physrev.00067.2017.
- Schindler, O.S., 2011. Current concepts of articular cartilage repair. Acta orthopaedica belgica, 77 (6), 709–726.
- Shah, R., et al., 2018. Circulating extracellular vesicles in human disease. The New England journal of medicine, 379 (10), 958–966. doi: 10.1056/NEJMra1704286.
- Shi, W., et al., 2021. p75NTR silencing inhibits proliferation, migration, and extracellular matrix deposition of hypertrophic scar fibroblasts by activating autophagy through inhibiting the PI3K/Akt/mTOR pathway. Canadian journal of physiology and pharmacology, 99 (4), 349–359. doi: 10.1139/cjpp-2020-0219.
- Singer, A.J., and Clark, R.A., 1999. Cutaneous wound healing. The New England journal of medicine, 341 (10), 738–746. doi: 10.1056/nejm199909023411006.
- Stahl, P.D., and Raposo, G., 2019. Extracellular vesicles: Exosomes and microvesicles, integrators of homeostasis. Physiology (Bethesda, MD), 34 (3), 169–177. doi: 10.1152/physiol.00045.2018.
- Sun, K., et al., 2016. AGEs trigger autophagy in diabetic skin tissues and fibroblasts. Biochemical and biophysical research communications, 471 (3), 355–360. doi: 10.1016/j.bbrc.2016.02.020.
- Théry, C., et al., 2006. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current protocols in cell biology, Chapter 3 (1), Unit 3.22. doi: 10.1002/0471143030.cb0322s30.
- Wang, W.M., et al., 2019. Exosomes in chronic inflammatory skin diseases and skin tumors. Experimental dermatology, 28 (3), 213–218. doi: 10.1111/exd.13857.
- Wang, Y., et al., 2023. Adipose mesenchymal stem cell derived exosomes promote keratinocytes and fibroblasts embedded in collagen/platelet-rich plasma scaffold and accelerate wound healing. Advanced materials (Deerfield Beach, FL), 35 (40), e2303642. doi: 10.1002/adma.202303642.
- Xie, F., et al., 2019. Extracellular vesicles in cancer immune microenvironment and cancer immunotherapy. Advanced science (Weinheim, Baden-Wurttemberg, Germany), 6 (24), 1901779. doi: 10.1002/advs.201901779.
- Yu, T., et al., 2015. Targeting autophagy in skin diseases. Journal of molecular medicine (Berlin, Germany), 93 (1), 31–38. doi: 10.1007/s00109-014-1225-3.
- Zeng, T., et al., 2019. Endothelial cell-derived small extracellular vesicles suppress cutaneous wound healing through regulating fibroblasts autophagy. Clinical science (London, England: 1979), 133 (9), CS20190008. doi: 10.1042/cs20190008.
- Zhao, Y., et al., 2019. LncRNA NR_003923 promotes cell proliferation, migration, fibrosis, and autophagy via the miR-760/miR-215-3p/IL22RA1 axis in human Tenon’s capsule fibroblasts. Cell death & disease, 10 (8), 594. doi: 10.1038/s41419-019-1829-1.
- Zheng, Y., et al., 2018. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nature reviews. Endocrinology, 14 (2), 88–98. doi: 10.1038/nrendo.2017.151.
- Zhou, L., et al., 2019. TET2-interacting long noncoding RNA promotes active DNA demethylation of the MMP-9 promoter in diabetic wound healing. Cell death & disease, 10 (11), 813. doi: 10.1038/s41419-019-2047-6.
- Zhu, P., et al., 2012. Involvement of RAGE, MAPK and NF-κB pathways in AGEs-induced MMP-9 activation in HaCaT keratinocytes. Experimental dermatology, 21 (2), 123–129. doi: 10.1111/j.1600-0625.2011.01408.x.