
ABSTRACT
Purpose
To report ocular outcome, somatic co-morbidities, genetics, and quality of life in children born with anophthalmia (A) or microphthalmia (M).
Methods
Thirty-five children (19 boys) with A/M underwent ophthalmological examinations and a review of medical records. Parents of 12/22 cases completed the Pediatric Quality of Life Inventory (PedsQL).
Results
Age at examination ranged from 7 months to 18 years (median 2.3 years). Ten cases were totally blind or had light perception. Isolated A/M occurred in 16/35 cases, while somatic, psychomotor, neuroradiological and/or genetic pathology occurred in 19/35 cases both in the bilateral (7/9) and in the unilateral group (12/26). Among 26 unilateral cases, 4/16 with one normal eye had associated problems compared to 9/10 if the contralateral eye was pathological (p < .01). There was an increased risk for heart defects in children with psychomotor delay (p = .04). Pathogenic genetic abnormalities were identified in 10/24 cases. Neuroimaging demonstrated pathology in 14/20 cases with corpus callosum dysgenesis (6/20) being the most common. The median total PedsQL score of parent reports for ages 2–12 was 52.4 (range 22.6–100).
Conclusions
Somatic, psychomotor and/or neuroradiological pathologies were more common in bila-teral than unilateral cases, but the difference was not significant. There was decreased risk in unilateral cases with one normal eye. Genetic defects occurred in both unilateral and bilateral cases. Health-related quality of life was reduced.
Introduction
Clinical anophthalmia (A) is described as the complete absence of the eye globe in the orbit with no ocular structure seen during macroscopic clinical examination. Microphthalmia (M) is defined as an underdeveloped eye, either only a small rudiment or an eyeball of subnormal size, more explicitly a bulb with a total axial length that is at least <2 standard deviations (SD) below the mean for age. The combined prevalence of A and M has been reported to be 0.4–3.2/10, 000 live births (Citation1–7). A/M can occur in isolation, together with other ocular malformations or somatic co-morbidities or as part of a syndrome and demonstrate extensive clinical and genetic complexity and heterogeneity (Citation1).
A/M account for 11% of blind children worldwide (Citation1) and are among the top five causes of severe visual impairment in Sweden (Citation8). The potential for vision is dependent on whether the case is uni- or bilateral, the size of the bulb and/or-associated ocular or cerebral pathology. The most common ocular malformation associated with A/M is “coloboma” which is-an ocular fissure closure defect (OFCD) in the same and/or in the contralateral eye (Citation9). In A/M, there is also a risk of deficient orbital growth with subsequent evolvement of facial asymmetry (Citation9,Citation10).
A/M may be caused by-numerical chromosomal abnormalities like trisomy 18 (Citation11) or trisomy 13 (Citation12) as well as by variants in many single genes, including PAX6, SOX2, OTX2 and CHD7 (Citation13,Citation14).
The mutations can be de novo or inherited. In severe bilateral cases of A/M, a genetic cause has now been identified in approximately 80% of cases, with de novo heterozygous loss-of-function mutations in SOX2 or OTX2 being the most common (Citation15).
In addition to optimising visual outcome, early oculoplastic treatment with socket expansion using-“conformers” is important for eyes with an axial length of <16 mm. The goal is simultaneous expansion of the eyelids, soft tissue and the orbital bones in order to minimise facial asymmetry and to facilitate wearing a prosthesis (Citation16). To promote visual function, clear rather than opaque conformers are fitted-in cases with light perception or positive visual evoked potential (VEP).
The purpose of the study was to evaluate and compare visual outcome, ocular pathology, somatic associated morbidities and genetic causative factors in children born with unilateral or bilateral A/M. Further aims were to evaluate their health-related quality of life (HRQoL)
Material and methods
St. Erik Eye Hospital’s database was searched for patients <18 years of age with the diagnoses of congenital A/M, ICD10 numbers Q11.1 and Q 11.2. After excluding patients who had been treated surgically for congenital cataract, 46 patients with A/M were identified. In total 35 patients eligible for inclusion agreed to participate in this cross-sectional cohort study with prospective longitudinal follow-up and were assessed at our departments of Paediatric Ophthalmology and Oculoplastic and Orbital services between 2018 and April 2021. One patient reached the age of 18 before assessment-but was still included. Eligible cases were followed up 9–27 months after the first notification.
The clinical ophthalmological examinations included, where possible, best corrected visual acuity (BCVA) with acuity cards (Teller acuity cards, Precision Vision, Illinois, USA) or-optotypes (LEA-Test Ltd, Helsinki, Finland or Konstantin Motakis Ortho KM, Lund, Sweden), microscopy and fundus evaluation. Facial photography and scrutiny of ophthalmological, paediatric and genetic medical records were performed.
The ocular diagnosis of A or M was based on a clinical evaluation, which showed a non-existent eye or a small eye with coexisting microcornea. The total axial length was measured using magnetic resonance imaging (MRI), computed tomography (CT) scans or ocular ultrasound in 21/35 cases and was defined as the length from the cornea to the central area of the retina. Microphthalmia was confirmed if the axial length was <2 SD below mean for age. Testing with visual evoked potential (VEP) had been performed or was initiated in cases where the visual potential was unclear.
The patients born with A/M were categorised according to four different subgroups using a modified version of Skalicky (Citation17) and Groot (Citation18). 1) M with a smaller but otherwise normal bulb, or an ocular remnant or anophthalmia (ORA), 2) M with OFCD, 3) M with anterior segment dysgenesis (ASDG) or persistent foetal vitreous (PFV) or 4) M with combined defects in the anterior or posterior segment (COMB). When the A/M was bilateral, it was classified according to pathology leading to visual impairment in the best eye.
The paediatric resident evaluated information from medical records regarding family history, pregnancy, labour, and somatic associated morbidities such as heart defects, psychomotor delay and abnormal cerebral findings on MRI. The genetic testing was performed as part of clinical analysis and comprised of microarray testing, investigation with A/M gene panels and/or whole exome or genome sequencing. The results from clinical genetic testing were collected and reviewed by the clinical genetic resident.
A logistic regression analysis was performed including age, gender, bilaterality and subgroups as predictors for estimating the associated risk of heart defects, other severe somatic comorbidities, psychomotor delay, genetic pathology, and abnormalities found using-neuroradiological scanning.
The Pediatric Quality of Life Inventory (PedsQL), a validated tool that covers physical, emotional, social and school functioning, was used for measuring HRQoL. Self-reports (children aged 5–18 years) and parent reports (children aged 2–12 years) were used in the current study. The scores were calculated according to instructions from the developers (Citation19).
The study was performed according to the Helsinki Declaration, with ethical approval from the local committee. Informed consent was obtained from the parents/guardians and from adolescents above the age of 15.
Results
Demographics and presentation
Among the 35 cases, unilateral A/M occurred in 26/35 (74%) and bilateral in 9/35 (26%). There were 19 boys (54%) and age at examination ranged from 7 months to 18 years (median 2.3 years). Three children were prematurely born, nine were delivered by emergency or elective caesarean section, three children had consanguineous parents, and two children were born after in vitro fertilisation. Maternal age ranged from 21 to 48 years (median 32 years).
The majority of cases, 28/35, presented during the first week of life. In 19 children, the parents noticed something odd about the eye or eyes in the first days after birth. Two children, including one with congenital toxoplasmosis, were diagnosed prenatally on ultrasound ().
Table 1. Demographic data on children with microphthalmia and anophthalmia including age at examination, completed gestational age and birth weight, mode of delivery, diagnosis, unilaterality or bilaterality and microphthalmia subgroup
Visual characteristics including visual evoked potential (VEP)
The best-corrected visual acuity (BCVA) ranged from amaurosis to 1.3. Eight cases with bilateral A/M and two cases with unilateral A/M were blind with amaurosis or at best light perception (WHO categories 4 and 5 visual impairment).
VEP, performed in 7/35 cases, demonstrated distinct cortical response in two children (one had total amaurosis and the other only reacted to light). VEP showed low or uncertain responses in four patients.
Bilateral or unilateral disease
The majority of the bilateral cases, 8/9, were from subgroup ORA ().
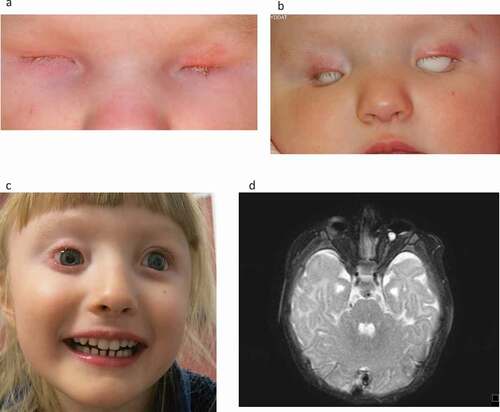
Figure 1. (a–d) Girl born with total blindness (amaurosis) due to right sided anophthalmia and left sided microphthalmia. There was no bulb on the right side and a very small, rudimentary bulb 5 mm on the left side. (a) New-born child, one month of age. (b) Treatment with expanding conformers, here 10 months of age. (c) Six years of age with bilateral prostheses. (d) MRI of the brain and orbits showing anophthalmia right eye and microphthalmia left eye (Published with permission from parents) .
Of the unilateral cases, 9/26 had OFCD, 5/26 had ORA () and 5/26 had PFV. The other eye was normal in 16/26 unilateral cases, 4/16 had associated co-morbidities, while 9/10 with a pathological contralateral eye had associated co-morbidities (p < .01).
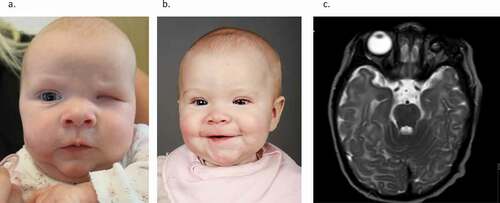
Figure 2. (a–c) Child born with small microphthalmic left eye. (a) Two months of age. (b) Treated with expanding conformers-at eight months of age she is wearing a-left eye prosthesis. Right eye normal. (c) Magnetic resonance tomography (T-2 weighted) of the brain and orbits. Note the microphthalmic left eye. (Published with permission from the parents).
Ocular anomalies in contralateral eyes included cases of papillochorioretinal coloboma (4), retinal degeneration or scars (3) and single cases of optic nerve aplasia (1), foveal hypoplasia (1) or nystagmus (1). For examples of prostheses please see .
Figure 3. Prostheses from one child with bilateral anophthalmia.

Refraction
Refraction, which could be-measured in 23 cases, was median +2 Dioptres (D) (range +6.5 D—12 D) in the contralateral eyes of unilateral cases. Two patients could not be measured. In the eyes of most of the patients’ (8/9) with bilateral AM, refractions could be not measured apart from a single eye where the refraction was +0.5 D.
ORA subgroup
All eight cases with bilateral ORA were blind according to WHO categories 4 or 5. Case 9 with chromosome 18q-deletion-syndrome had a severe heart defect and CNS malformations, and died during the study period. Case 32 had a de novo chromosome 13q31.3-q34 duplication. A SOX2 mutation was identified in case 10 who had psychomotor delay and a hypoplastic chiasm. In four cases, no genetic abnormalities were identified, and one child is still under investigation.
Among the five unilateral cases with ORA, three children had subnormal visual acuity with severe myopia, retinal degeneration, nystagmus and/or macular hypoplasia in the contralateral eye. The child with macular hypoplasia, case 22, had a micro-syndrome with BCOR-mutation. He had subnormal hearing and speech problems and a second-degree relative with A. He is described in a previous cohort (Citation20). The child with retinal degeneration, case 13, had a chromosome 17p13.1p13.2-deletion, microcephaly, epilepsy, a congenital heart defect and died during the study period. The third child had normal genetic findings but presented with unilateral cleft lip and jaw, intellectual impairment and vertebral anomalies ().
Table 2. Somatic comorbidities in children with anophthalmia and microphthalmia including subgroup, isolated or non-isolated A/M, genetic findings, magnetic resonance imaging of the bulb and CNS, and disorders in different organ systems
Table 3. Ocular characteristics including diagnosis, visual functions, VEP, intraocular pressure, treatment with expander (E) and/or prosthesis (P)
OFCD subgroup
In total, 10 children/adolescents had OFCD. None of them were totally blind. Case 12 with bilateral OFCD had a RARB-mutation and MCOPS12-syndrome with neuromuscular disease and psychomotor delay.
Among the nine unilateral cases with OFCD, genetic testing revealed mutations in CHD7 in two children, verifying a clinical diagnosis of CHARGE syndrome and a WDR37 mutation—neurooculocardiogenitourinary syndrome—in one child.
One child showed psychomotor delay, absent corpus callosum and lacunes in the ocular fundus and was suspected to have Aicardi syndrome. Another case had undergone surgery because of aortic coarctation and also had hypospadias. Genetic testing was normal in both children ().
ASDG and PFV subgroups
Four cases had unilateral ASDG and five cases had unilateral PFV. The contralateral eyes had normal visual behaviour for age, but five children were too young to be using symbols. No other birth defects or anomalies were found in the ASDG-group. One child with PFV had unilateral cryptorchidism ().
Combined subgroup
Three children were diagnosed with having combined defects in the anterior as well as the posterior segment. One child had a cataract and retinal detachment with amaurosis in the microphthalmic eye. The other eye was of normal size but amaurotic due to optic nerve aplasia. Genetic evaluation was normal. Case 16 had sclerocornea and a rudimentary lens in the severe microphthalmic left eye and optic nerve aplasia, and retinal atrophy in the amaurotic right eye. Genetic evaluation revealed an OTX2 mutation. The third child had unilateral M due to congenital toxoplasmosis and inactive chorioretinal scars in both eyes ()
Somatic comorbidities, neurodevelopmental disorders and neuroradiological findings
Isolated A/M affecting visual function and craniofacial appearance in otherwise healthy children, occurred in 16/35 cases (46%). A syndromic, non-isolated form of A/M with somatic comorbidities, neurodevelopmental disorders, and/or abnormal neuroradiological findings was found in 19/35 cases (54%), and was more common in 7/9 bilateral than in unilateral cases (12/26) but the difference was not significant.
There was no difference in age between the isolated and non-isolated groups, with median ages of 2.04 (range 0.6–18.0 years) and 2.33 years (range 0.5–16.5 years), respectively.
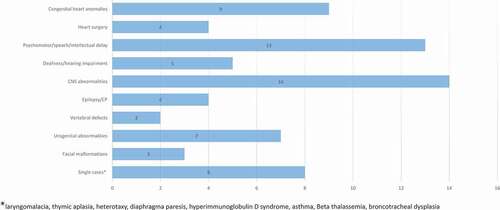
Echocardiography was performed in 15 patients. Congenital heart defects were present in nine patients and consisted of Tetralogy of Fallot, transposition of the great arteries, truncus arteriosus, aortic coarctation, atrial and ventricular septal defects, aortic valve insufficiency, patent ductus arteriosus and pulmonary atresia. Four patients had undergone cardiac surgery.
Neurodevelopmental disorders were present in 13 cases including psychomotor or speech delay and intellectual disability, but several of the children were too young for proper evaluation during the study period.
Logistic regression analysis showed a significantly increased risk for congenital heart defects in children with delayed psychomotor development when controlled for age, sex, subgroup and ocular diagnosis, with an odds ratio (OR) of 16.8 (p = .04). Furthermore, children with congenital heart defects showed an increased risk of delayed psychomotor development when controlled for age, sex, subgroup, and ocular diagnosis, OR 17.1 (p = .03).
Three patients had epilepsy, five were either deaf or had hearing impairment, one had cerebral palsy and one had both ADHD and autism (). Congenital toxoplasmosis was diagnosed in one patient prenatally. A TORCH panel test was checked in three other patients and was found to be negative, whereas four additional patients had negative cytomegalovirus serology tests. MRI or CT of the brain were performed in 19 cases and 1 case, respectively. Pathology was demonstrated in 14/20 with combinations of corpus callosum dysgenesis (6), microcephaly (3), vermis hypoplasia (2), affected pituitary gland (1), aquaeductus stenosis and calcifications (2), ventricular dilation (2) and polymicrogyria (1). Brain ultrasound was performed in four additional cases and were normal in all.
Figure 4. Somatic co-morbidities in patients with anophthalmia or microphthalmia.
Both the patient with the SOX2-mutation and the patient with the OTX2-mutation had hypoplastic chiasmas and the latter also had corpus callosum hypoplasia. Hormonal screening was normal in both patients.
Genetic pathology
Genetic testing according to clinical practice had been performed in 24/35 cases and pathological findings were found in 10/24 (42%). Genetic testing was normal in 14/24 of the tested cases and had not been performed/had not been considered clinically necessary in 10 cases. One child is still under investigation.
Structural variants occurred in three patients including a chromosome 18q21.1-qter deletion, a chromosome 17p13.1-p13.2-microdeletion and a chromosome 13q31.3-q34 duplication. Monogenic variants were identified in seven patients with the most common being CHD7- mutations that occurred in two patients with CHARGE syndrome.
One patient had neurooculocardiogenitourinary syndrome due to WDR37 mutation, another had MCOPS3 due to a SOX2 mutation, another had MCOPS12-syndrome due to a RARB mutation, another had a MCOPS5-syndrome due to an OTX2-mutation, and another had Lenz microphthalmia syndrome due to a maternally inherited hemizygous BCOR-mutation occurred in single patients respectively. The BCOR-mutation occurred in the patient with an uncle with A. No mutations in PAX6 were found ().
Biometric data, socket expansion treatment and surgery
The axial length of the bulbs was re-measured in 20 patients using available MRI and CT scans and in one case, ocular ultrasound (). The use of expanders or prostheses for each child is specified in .
In all 22/35, children were referred to an ocularist for socket expansion with custom-made conformers to promote eyelid and orbital growth. Clear conformers were fitted in three cases where the child reacted to light or showed a VEP response.
Thirteen children did not need any expansion of the socket and had no prostheses due to mild microphthalmus often with preserved vision (11/13) or general poor health, where expansion/prosthesis was not initiated or wearing one was rejected-(2/13).
Four children had not started expansion at the time of this investigation. Two children did not need expansion but wore prostheses.
Health related quality of life—PedsQL
Parents of 14/22 children completed the PedsQL questionnaire. Five children and two adolescent filled in the questionnaire themselves. Thirteen children were younger than 2 years of age and thus were not eligible. Two children died. Additionally, one family only committed to medical records.
The median total score of parental-reports for-children aged 2–12 was 52.4 (range 22.6–100). The median physical health summary score was 57.8 (range 0–100). The median psychosocial summary score was 57.3 (range 35–100). Nine out of fourteen parents except for two reported lower physical summary scores than psychosocial scores.
Five children and two adolescents aged 5–18 completed the PedsQL self-reports, which resulted in a median total score of 80.4 (range 35.9–96.7). The median physical health summary score was 90.1 (range 25–100) and the median psychosocial summary score was 70 (range 41.7–95). Four of the children had corresponding parental reports. Two of these children reported higher psychosocial and physical summary scores than the parents reported, while for two other children, the parents estimated a greater physical quality of life than the children did ().
Table 4. Health-related quality of life reported by parents and children according to age
Discussion
The purpose of the study was to evaluate and compare visual outcome, ocular pathology, somatic associated morbidities and genetic causative factors in children born with unilateral or bilateral A/M. Further aims were to evaluate the HRQoL in children with A/M.
The main results from the current study show high variability in both the unilateral and bilateral groups of children and adolescents with A/M. Almost one-third (29%) of the cases had total blindness or only light perception. Isolated A/M occurred in 46%, while more than half of the cases (54%) had associated somatic comorbidities, neuroradiological abnormalities and/or neurodevelopmental delay. These comorbidities were more common in the bilateral than in the unilateral group, but the difference was not significant. Among the unilateral cases, those with a normal contralateral eye had a reduced risk of associated problems (p < .01). There was an increased risk of heart defects in children with psychomotor delay when controlled for age, sex, subgroup and ocular diagnosis, odds ratio (OR) of 16.8 (p = .04). Pathogenic genetic abnormalities were identified in 43%. According to parental reports, QoL seemed reduced in children with A/M.
Demographics
In the current study, 9% (3/35) of the children were prematurely born in comparison with 6% (1/18) in patients in another recent Swedish study (Citation20). There was a slight over-representation (26%) of emergency and elective caesarean sections in our study compared to the rate of caesarean sections registered in the Swedish Birth Register 2000–2018 (14.8–17.7%) (Citation21).
There was also a slight predominance for males in our cohort (19 vs 16) although not statistically significant, which accords with the literature (Citation2,Citation20).
Bilateral A/M occurred in 26% of our patients, which is in accordance with the study from the Netherlands where 24% were bilateral (Citation18) but lower than in Hawaii (96 cases) where 45% of the cases were bilateral (Citation4). This may be due to different selection criteria. Taha Najim et al., who included 18 cases, had no bilateral cases (Citation20). Only three patients (9%) in the current study had consanguineous parents as opposed to 47% in another study (Citation22).
Visual function including VEP
Severe visual impairment with amaurosis or light perception was present in 29% of children (10/35). The other, contralateral eye, was normal in 62% of the 26 unilateral cases in this study, compared to the 50% reported by Taha Najim (Citation20).
There were significant mismatches when comparing VEP results with clinical manifestations of the children. For example, two clinically blind children had normal VEP responses. Although the number of performed investigations is very small, this indicates that the VEP method may be unreliable in young children and that the results should be interpreted with caution, especially when discussing visual prognosis with parents.
Somatic comorbidities, neurodevelopmental disorders and neuroradiological findings
A/M occurred as an isolated finding in 46% of the cases. Genetic testing was normal, had not been performed, or had not been considered clinically necessary in these children. It can be discussed whether repeated future genetic analysis-could reveal new mutations in these cases. Neurodevelopmental problems can also become evident with increasing age.
The majority of the cases, 54%, were non-isolated, and the children suffered from systemic extraocular malformations, functional impairments and/or cerebral pathology. This is within the previously described range of 32.1–61.1% (Citation5,Citation18,Citation20). However, systemic involvement was more common in bilateral (78%) than in unilateral cases (50%), but the difference was not significant unlike a previous Swedish study in which bilateral cases had more frequent systemic involvement (Citation2).
The most common somatic anomalies in our study were cardiac defects, which occurred in 26% of all cases. The prevalence of hearing impairment was 14%, and the prevalence of psychomotor or speech delay or intellectual disabilities was 37%. All numbers are in line with the study by Taha Najim et al., who reported cardiac defects in 25%, hearing impairment in 12% and psychomotor delay in 31% of cases. This is somewhat surprising as their study material-consisted of only unilateral cases. Brain pathologies were found in 40% of our cases after imaging with ultrasound, MRI or CT, while Taha Najim et al. reported 25% (Citation20).
Genetic pathology
Pathogenic genetic abnormalities occurred in 42% of the tested cases, which is in the same range as previous studies (20–61%) (Citation4,Citation7,Citation20,Citation23) and higher than in the study by Shah et al. (Citation9). Genetic testing was normal or had not been considered clinically necessary in the other cases. It is open to discussion as to whether in these cases, repeated future genetic analysis could reveal new mutations.
Two patients in our study had genetically confirmed CHARGE syndrome (Citation14), while one patient had a SOX2 mutation. This differs from Groot and co-workers who reported that four bilateral cases (out of 58) had a SOX2 mutation (Citation18).
One limitation of this study could be that only 24/35 patients who underwent genetic screening as referrals were only sent when genetic investigation was part of clinical practise. Also, the type of genetic investigation differed between different patients. If more extensive genetic investigations with WGS screening were conducted for all patients, it is possible that a genetic cause of A/M would be found in a greater number. Identification of a genetic cause is important information for patients with A/M and their parents, as it may alter the prognosis and management. It would also permit genetic counselling and prenatal testing for parents who are planning for another child.
One patient had congenital toxoplasmosis. However, TORCH panels were only performed in a few children. Environmental factors such as congenital infections with toxoplasma gondii and cytomegalovirus, for example, but as well as herpes simplex, influenza, coxsackie and Zika viruses and parvovirus 19 (Citation24) can be causative to A/M, as can maternal diabetes (Citation7) or vitamin A deficiency, exposure to X-rays, solvent misuse and thalidomide exposure (Citation1).
Health-related quality of life PedsQL
In this study, both parental reports and self-reports resulted in variable findings about HR QoL, but the majority of the cases indicated a significant reduction in HR QoL. We were unable to make further comparisons because there were only four corresponding self and parental reports.
Previous studies also had similar results, with reduced HR QoL and a greater impact on the psychosocial domain than on the physical well-being reported by the children compared to the parental reports (Citation25–27). This will be further investigated in deep interviews in future studies.
Conclusion
In conclusion, children born with bilateral and unilateral anophthalmia or microphthalmia were at risk of not only visual impairment but also somatic, neurodevelopmental and/or genetic comorbidities. As associated morbidities and delayed psychomotor development may be difficult to assess in small children, a multidisciplinary approach and regular follow-ups throughout childhood and adolescence are important.
Since genetic defects seem to be of great importance not only in bilateral A/M but also in unilateral A/M, we suggest that all patients should undergo genetic evaluation.
Clinical guidelines for the diagnosis and treatment of patients with anophthalmia or microphthalmia adapted from Ragge et al. (Citation16)
Suspicion of microphthalmia if the horizontal corneal diameter is <9 mm in neonates
Diagnosis of microphthalmia, if the axial length of the bulb is <15.8 mm on ultrasound or MRI in a neonate
Refer to a paediatric ophthalmologist
Refer to oculoplastics and prosthetics unit if bulb length is <16 mm neonatally.
MRI of the brain and orbits to detect pathology and measure axial length
VEP in selected cases, but the results should be interpreted with caution
Multidisciplinary approaches including paediatricians and ear, nose and throat specialists.
Long-term follow-up is needed as associated comorbidities in the other eye or other organs and psychomotor delay or behavioural problems may not be evident in the neonatal period
Genetic evaluation is strongly recommended both in children born with unilateral or bilateral microphthalmia/anophthalmia
Acknowledgments
We would like to thank ocularists Eva Johansson, Anna Povse, Niklas Schesny and Benjamin Rosenberg for evaluating the questionnaires as well as Associate Professor Britt Marie Anderlid for reading the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Verma AS, and FitzPatrick DR. Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007;20(4): 345–350 .
- Kallen B, Tornqvist K. The epidemiology of anophthalmia and microphthalmia in Sweden. Eur J Epidemiol. 2005;20(4):345–50.doi:https://doi.org/10.1007/s10654-004-6880-1.
- Dolk H, Busby A, Armstrong BG, Walls PH, Cuzick J, et al. Geographical variation in anophthalmia and microphthalmia in England, 1988-94. Bmj. 1998;317(7163):905–09. discussion 910. doi:https://doi.org/10.1136/bmj.317.7163.905.
- Forrester MB, Merz RD. Descriptive epidemiology of anophthalmia and microphthalmia, Hawaii, 1986-2001. Birth Defects Res A Clin Mol Teratol. 2006;76(3):187–92.doi:https://doi.org/10.1002/bdra.20237.
- Roos L, Jensen H, Grønskov K, Holst R, Tümer Z, et al. Congenital microphthalmia, anophthalmia and coloboma among live births in Denmark. Ophthalmic Epidemiol. 2016;23(5):324–30.doi:https://doi.org/10.1080/09286586.2016.1213859.
- Morrison D, Hanson D, Williamson I, van Heyningen K, Fleck V, Jones B, Chalmers I, Campbell J . National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet. 2002;39(1):16–22.doi:https://doi.org/10.1136/jmg.39.1.16.
- Chambers TM, Agopian AJ, Lewis RA, Langlois PH, Danysh HE, Weber KA, Shaw GM, Mitchell LE, Lupo PJ . Epidemiology of anophthalmia and microphthalmia: prevalence and patterns in Texas, 1999-2009. Am J Med Genet A. 2018;176(9):1810–18. doi:https://doi.org/10.1002/ajmg.a.40352.
- de Verdier K, Ulla E, Löfgren S, Fernell E, et al. Children with blindness - major causes, developmental outcomes and implications for habilitation and educational support: a two-decade, Swedish population-based study. Acta Ophthalmol. 2018;96(3):295–300.doi:https://doi.org/10.1111/aos.13631.
- Shah SP, Taylor AE, Sowden JC, Ragge N, Russell-Eggitt I, Rahi JS, Gilbert CE, et al. Anophthalmos, microphthalmos, and Coloboma in the United Kingdom: clinical features, results of investigations, and early management. Ophthalmology. 2012;119(2):362–68.doi:https://doi.org/10.1016/j.ophtha.2011.07.039.
- Krastinova D, Kelly MB, Mihaylova M. Surgical management of the anophthalmic orbit, part 1: congenital. Plast Reconstr Surg. 2001;108(4):817–26.doi:https://doi.org/10.1097/00006534-200109150-00001.
- Acar DE, Acar U, Ozdemir O, Ozen ZT, Cakar ES, et al. Microphthalmia in a case of Edward syndrome. Semin Ophthalmol. 2014;29(2):114–17.doi:https://doi.org/10.3109/08820538.2013.874486.
- Petry P, Polli JB, Mattos VF, Rosa RCM, Zen PRG, Graziadio C, Paskulin GA, Rosa RFM, et al. Clinical features and prognosis of a sample of patients with trisomy 13 (Patau syndrome) from Brazil. Am J Med Genet A. 2013;161a(6):1278–83.
- Bardakjian TM, Schneider A. The genetics of anophthalmia and microphthalmia. Curr Opin Ophthalmol. 2011;22(5):309–13.doi:https://doi.org/10.1097/ICU.0b013e328349b004.
- Hsu P, Ma A, Wilson M, Williams G, Curotta J, Munns CF, Mehr S, et al. CHARGE syndrome: a review. J Paediatr Child Health. 2014;50(7):504–11.doi:https://doi.org/10.1111/jpc.12497.
- Williamson KA, FitzPatrick DR. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur J Med Genet. 2014;57(8):369–80.doi:https://doi.org/10.1016/j.ejmg.2014.05.002.
- Ragge NK, Subak-Sharpe ID, Collin JR. A practical guide to the management of anophthalmia and microphthalmia. Eye (Lond). 2007;21(10):1290–300.doi:https://doi.org/10.1038/sj.eye.6702858.
- Skalicky SE, White AJR, Grigg JR, Martin F, Smith J, Jones M, Donaldson C, Smith JEH, Flaherty M, Jamieson RV, et al. Microphthalmia, anophthalmia, and coloboma and associated ocular and systemic features: understanding the spectrum. JAMA Ophthalmol. 2013;131(12):1517–24.doi:https://doi.org/10.1001/jamaophthalmol.2013.5305.
- Groot ALW, Kuijten MMP, Remmers J, Gilani A, Mourits DL, Kraal‐Biezen E, Graaf P, Zwijnenburg PJ, Moll AC, Tan S, et al. Classification for treatment urgency for the microphthalmia/anophthalmia spectrum using clinical and biometrical characteristics. Acta Ophthalmol. 2020;98(5):514–20.doi:https://doi.org/10.1111/aos.14364.
- Varni JW, Seid M, Kurtin PS. PedsQL 4.0: reliability and validity of the pediatric quality of life inventory version 4.0 generic core scales in healthy and patient populations. Med Care. 2001;39(8):800–12.doi:https://doi.org/10.1097/00005650-200108000-00006.
- Taha Najim R, Topa A, Jugård Y, Casslén B, Odersjö M, Andersson Grönlund M, et al. Children and young adults with anophthalmia and microphthalmia: diagnosis and Management. Acta Ophthalmol. 2020;98(8):848–58.doi:https://doi.org/10.1111/aos.14427.
- Socialstyrelsen. Statistikdatabas för graviditeter, förlossningar och nyfödda; 2021 [cited 2021 7 January]. Available from: https://www.socialstyrelsen.se/statistik-och-data/statistik/statistikdatabasen/.
- Galindo-Ferreiro A, Elkhamary SM, Alhammad F, AlGhafri L, AlWehaib M, Alessa D, Aldossari S, Akaishi P, Khadekar R, AlShaikh O, et al. Characteristics and management of congenital anophthalmos and microphthalmos at a tertiary eye hospital. Orbit. 2019;38(3):192–98.doi:https://doi.org/10.1080/01676830.2018.1521843.
- Slavotinek A. Genetics of anophthalmia and microphthalmia. Part 2: syndromes associated with anophthalmia-microphthalmia. Hum Genet. 2019;138(8–9):831–46.doi:https://doi.org/10.1007/s00439-018-1949-1.
- Marques VM, Santiago CS, Marques IG, Nunes Brasil SM, Lima MDG, Costa TT . Neurological complications of congenital zika virus infection. Pediatr Neurol. 2019;91:3–10.
- Dahlmann-Noor A, Tailor V, Abou-Rayyah Y, Adams G, Brookes J, Khaw PT, Bunce C, Papadopoulos M, et al. Functional vision and quality of life in children with microphthalmia/anophthalmia/coloboma-a cross-sectional study. J Aapos. 2018;22(4):281–285.e1.doi:https://doi.org/10.1016/j.jaapos.2018.01.015.
- Casslén B, Jugård Y, Taha Najim R, Odersjö M, Topa A, Andersson Grönlund M, et al. Visual function and quality of life in children and adolescents with anophthalmia and microphthalmia treated with ocular prosthesis. Acta Ophthalmol. 2020;98(7):662–70.doi:https://doi.org/10.1111/aos.14424.
- Castañeda YS, Cheng-Patel CS, Leske DA, Wernimont SM, Hatt SR, Liebermann L, Birch EE, Holmes JM, et al. Quality of life and functional vision concerns of children with cataracts and their parents. Eye (Lond). 2016;30(9):1251–59.doi:https://doi.org/10.1038/eye.2016.134.