
ABSTRACT
Background
Congenital cataracts are the most common cause of visual impairment worldwide. Inherited cataract is a clinically and genetically heterogeneous disease. Here we report disease-causing variants in a novel gene, CYP21A2, causing autosomal dominant posterior polar cataract. Variants in this gene are known to cause autosomal recessive congenital adrenal hyperplasia (CAH).
Methods
Using whole-exome sequencing (WES), we have identified disease-causing sequence variants in two families of British and Irish origin, and in two isolated cases of Asian-Indian and British origin. Bioinformatics analysis confirmed these variants as rare with damaging pathogenicity scores. Segregation was tested within the families using direct Sanger sequencing.
Results
A nonsense variant NM_000500.9 c.955 C > T; p.Q319* was identified in CYP21A2 in two families with posterior polar cataract and in an isolated case with unspecified congenital cataract phenotype. This is the same variant previously linked to CAH and identified as Q318* in the literature. We have also identified a rare missense variant NM_000500.9 c.770 T > C; p.M257T in an isolated case with unspecified congenital cataract phenotype.
Conclusion
This is the first report of separate sequence variants in CYP21A2 associated with congenital cataract. Our findings extend the genetic basis for congenital cataract and add to the phenotypic spectrum of CYP21A2 variants and particularly the CAH associated Q318* variant. CYP21A2 has a significant role in mineralo- and gluco-corticoid biosynthesis. These findings suggest that CYP21A2 may be important for extra-adrenal biosynthesis of aldosterone and cortisol in the eye lens.
Introduction
Cataract, the opacification of the eye lens, is the most common, but treatable cause of blindness in the world (https://www.who.int/publications-detail/world-report-on-vision). Congenital cataracts are detected at birth or during the first decade of life. Hereditary cataract can be isolated or be a part of other ocular defects like anterior segment mesenchymal dysgenesis, glaucoma, microcornea, or aniridia; and systemic disorders such as heart disease, diabetes, deafness, and Wolframin disease.
Congenital cataract is usually autosomal dominant, followed by recessive and X-linked inheritance. It is a clinically and genetically heterogeneous disease, displaying various phenotypes (Citation1). Cholesterol biosynthetic pathways are linked to cataract as evidenced by lanosterol synthase gene (LSS). The full spectrum of congenital cataract causing genes can be found here (https://cat-map.wustl.edu/) (Citation2,Citation3). In this study, we have identified two heterozygous variants Q319X, and M257T in the CYP21A2 gene (MIM 201910). The variant identified as Q319X (once a legacy N-terminal sequencing error is corrected), is equivalent to the Q318X variant linked to congenital adrenal hyperplasia (CAH) (Citation4). As previously reported, Q319X is predicted to be pathogenic at both a bioinformatic and a structural levels; while M257T, a novel but rare variant, has been predicted as a variant of uncertain significance at bioinformatic level.
CYP21A2 is a member of the cytochrome P450 (CYP450) family of enzymes, and it is also known as steroid 21-hydroxylase, important in the biosynthesis of mineralocorticoids (aldosterone) and glucocorticoids (cortisol). Like other steroid hydroxyases, it requires a heme cofactor to function as a monooxygenase and catalyse the oxidization and hydroxylation of a variety of steroid substrates at position 21 (Citation5), such as progesterone and 17α-hydroxyprogesterone, precursors in the synthesis of aldosterone and cortisol, respectively. For this reason, CYP21A2 is associated with CAH (Citation6), an autosomal recessive endocrine disorder (Citation7,Citation8) that leads to aldosterone deficiency and in its severe form, the pathological loss of sodium ions (Citation9). Here, for the first time, we have identified pathogenic variants in CYP21A2 gene causing an isolated autosomal dominant posterior polar congenital cataract.
Materials and methods
Phenotyping
Patients were identified via the proband attending the Genetic Service at Moorfields Eye Hospital, London, UK. The study protocol adhered to the Tenets of the Declaration of Helsinki and was approved by UCL research ethics committee (project ID—4817/001). All family members participating in this study gave written informed consent and underwent full ophthalmic examination, including slit lamp examination. All affected individuals from two families and two isolated cases were diagnosed as having an isolated congenital cataract.
Whole-exome sequencing (WES) and bioinformatics analysis
Whole-exome sequencing and bioinformatic analysis were performed using the Phenopolis bioinformatics platform as before (Citation10).
Sanger sequencing
Sanger sequencing was performed to validate the variant identified by WES. Genomic DNA was amplified by PCR using GoTaq 2X master mix (AB gene; Thermo Scientific, Epsom, UK) and CYP21A2 specific primers i) Q319*- Forward primer: acaagcaaaaggctccttcc; Reverse primer: tttcctcactcatccccaac, ii) M257T- Forward primer: AGAGGGATCACATCGTGGAG; Reverse primer: ggaaggagccttttgcttgt designed with http://bioinfo.ut.ee/primer3-0.4.0/. After validating the variant, segregation was performed in all the available family members.
The protein structure of cytochrome P450 (CYP21A2) for wt- CYP21A2, mut-CYP21A2 G319* and mut M257T was analysed using SWISSMODEL (https://swissmodel.expasy.org/interactive) (–d).
Results
Cataract families with bioinformatic analysis
In this study, we have investigated two families, A and B along with two isolated individuals C and D with autosomal dominant congenital cataract.
Family A consists of a three-generation British pedigree comprising 7 affected and 1 unaffected individual presenting with bilateral congenital posterior polar cataract (). There was no reported family or personal history of non-ocular disease. One affected individual (III:5) was sequenced by whole-exome sequencing (WES), followed by phenopolis genetic variant analysis pipeline, resulting in 356 out of a total of 2930 rare coding variants being filtered by allele frequency (Kaviar and GnomAD) and CADD > 15. The top scoring variant for CADD was a rare heterozygous variant c.955 C > T; p.Q319* in exon 8 of CYP21A2, with a score of 43.
Family B is a small Irish pedigree with posterior polar cataract, comprising 2 affected and 1 unaffected family members (). No reported family or personal history of non-ocular disease was found. One affected individual (II:1) was sequenced by WES. Variant annotation and tiered filtering yielded 376 out of a total of 3087 rare coding variants. Furthermore, a rare heterozygous variant c.955 C > T; p.Q319* in exon 8 of CYP21A2, with a highest CADD score of 43 was found.
Individuals C and D, both affected by congenital cataract (specific phenotype not present in clinical notes), with no personal history of non-ocular disease, underwent WES. Following this, variant analysis and tiered filtering yielded 491/3425 rare coding variants and further one rare variant c.955 C > T; p.Q319* in exon 8 of CYP21A2 (C) with CADD score of 43 and one rare variant c.770 T > C; p.M257T in exon 7 of CYP21A2 (D) with CADD score of 23.8.
Direct Sanger sequencing
Figure 1. (a) Family A: Abridged British pedigree with posterior polar cataract; (b) Family B: Abridged Irish pedigree with posterior polar cataract. The diagonal line indicates a deceased family member. Squares and circles symbolize males and females, respectively. Open and filled symbols indicate unaffected and affected individuals, respectively. The arrow indicates the family members who participated in the WES analysis. All the available members in the family were sequenced to show the segregation.
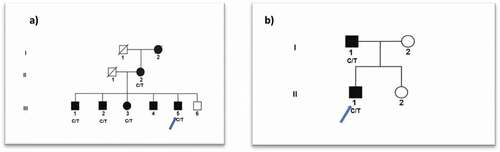
Table 1. Pathogenicity scores of variants in CYP21A2 gene in ADCC patients
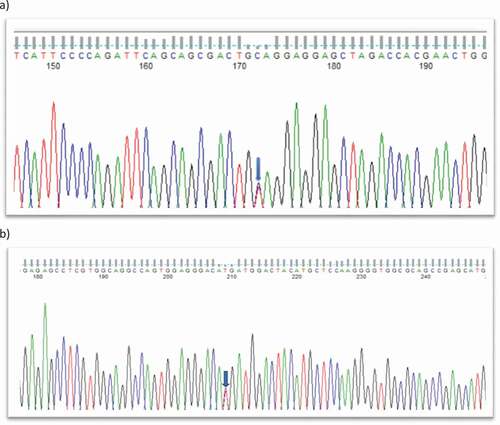
Figure 2. (a) Sequence analysis of CYP21A2: nonsense variant c.955 C > T in all affected member of family A, family B with autosomal dominant posterior polar cataract and an isolated individual C with congenital cataract; (b): missense variant c.770 T > C in individual D with congenital cataract.
Further we have also found 6 polymorphisms (rs61732108, rs61732108, rs61732108, rs61732108, rs147821751, and rs6455) in CYP21A2 in our ADCC panel of 60 patients.
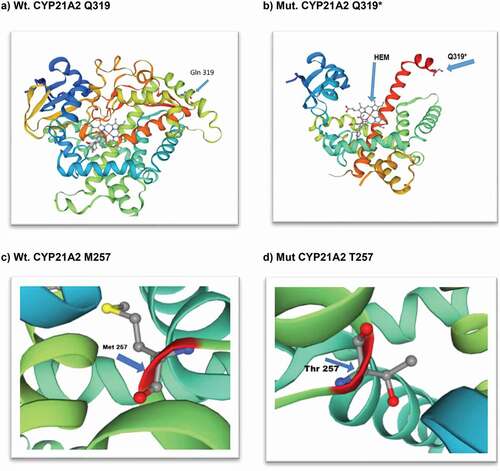
We have demonstrated the crystal structure of human wild type CYP21A2 (30–485) amino acids and truncated protein at position Q319*, losing 166 amino acids, where several residues are crucial for various important functions of the protein and for missense variant Met257Thr ().
Figure 3. Structural view of human cytochrome P450 21A2 hydroxyprogesterone complex (CYP21A2): X-ray diffraction, 3.31 monomer 1x HEM (https://swissmodel.expasy.org/interactive); (a) wild-type CYP21A2 (30–485 amino acids); (b) mutant stop codon amino acid at 319 (Glutamine); (c) wild-type amino acid Methionine at position 257; (d) mutant missense amino acid Threonine at position 257.
Discussion
Cytochrome (CYP) P450 is a superfamily of 18 members encoding 57 genes in the human genome (Citation11). It has a wide spectrum of functions such as synthesis of steroid hormones (e.g., oestrogen and testosterone), fatty acids, and sterols (cholesterol and bile acids). CYP enzymes are crucial for the synthesis and breakdown of non-protein ligands which bind to receptors or activate G protein-mediated pathways regulating growth, differentiation, apoptosis, homeostasis, and other neuroendocrine functions (Citation12).
CYP21A2 gene consists of 10 exons, and it is a part of the RCCX module (RP2-C4B-CYP21A2-TNXB) located on chromosome 6p21.3 (Citation13). CYP21A2 shares 98% sequence homology with a non-functional pseudogene (CYP21A1P) that is arranged in tandem at the same site (Citation14). Where truncations occur, such as R316X (Citation15) and Q318X, the genotype-phenotype correlation can be complicated by the gene duplication despite enzyme inactivation, due to the removal of the heme binding domain by these truncations (Citation9,Citation16). The severe salt wasting phenotype requires all alleles to be affected (Citation9). Within the spectrum of observed phenotypes associated with CAH, there is a report of keratoconus being linked to nonclassical CAH (Citation17).
The expression of CYP450 family members has also been identified in ocular tissues; CYP46A1 (24-hydroxycholesterol) has been identified in the brain (Citation18) and retina (Citation19); while CYP1B1 variants are responsible for primary congenital glaucoma (Citation20) and CYP4V2 variants cause Bietti corneo-retinal crystalline dystrophy and retinitis pigmentosa (Citation21,Citation22).
CYP21A2 is expressed in lens cells (Citation4) suggesting that congenital variants in CYP21A2 can present with, albeit rare, eye phenotypes as observed for multiple other genes associated with both syndromic and non-syndromic (isolated ocular) disorders. The lens is an eye tissue that actively synthesizes sterols (Citation23,Citation24) and steroid medication is a well-established cause of cataract, presenting characteristically as a posterior subcapsular cataract (Citation25,Citation26).
CYP21A2 is a key player in the biosynthesis of aldosterone and cortisol. Aldosterone is expressed in the lens (Citation27). This mineralocorticoid is important in the modulation and regulation of ion channels (Citation28) and could therefore contribute to volume regulation and ion transport in the lens. Cholesterol-dependent transmembrane channel protein, TRPV4, acts as a mechanosensor and is highly expressed in lens epithelial cells and differentiating fiber cells, which are also responsible for volume regulation and ion exchange (Citation29,Citation30). Based on this, we hypothesise that CYP21A2 is likely important to the renin–angiotensin–aldosterone system (RAAS) (Citation31) in the lens.
Nearly, 200 variants have been found in the CYP21A2 gene causing autosomal recessive CAH; from severe classic CAH to non-classic CAH (NCCAH) with a milder phenotype (Citation32), reflecting that other factors may be responsible for the genotype-phenotype variability in patients with CAH and NCCAH. Here, we have found pathogenic variants, p.Q319X, and one rare novel variant p. M257T in CYP21A2 causing isolated congenital posterior polar cataract and congenital cataract with undefined phenotype, respectively. The p.Q319X variant has been found previously in different ethnic groups in various parts of the world, displaying a wide spectrum of clinical features from severe to mild phenotypes (Citation9). This variant p.Q319X is frequently found in the Iranian (Citation16), Italian (Citation33), and Brazilian (Citation34) populations, both with CAH and NCCAH, harbouring either homozygous or compound heterozygous forms; and reported to coexist with the wildtype allele on the same chromosome, showing mild symptoms in patients (Citation9).
In the families reported herein, the p.Q319X and p.M257T variants cause isolated congenital autosomal dominant congenital cataracts (ADCC) in European and Indian Asian ethnicities, further extending the genetic basis of congenital cataracts. It is possible that haploinsufficiency accounts for the autosomal dominant nature of the cataract caused by these variants—although other genetic modifying factors and/or environmental influences may be involved. It is commonplace in the inherited retinal disease group of disorders that specific alleles can be associated with a huge range of differing phenotypes both within and between families, as well as causing both syndromic and non-syndromic manifestations. Moreover, the p.Q319X variant association with both syndromic recessive disease and isolated ADCC is in keeping with the phenotypes reported for certain WFS1 variants (Citation35).
Conclusions
Based on the data presented, we speculate that mineralocorticoids (aldosterone) and glucocorticoids (cortisol) play important roles in the physiopathology of cataract, and identify a novel cause of ADCC. As CYP21A2 is critical to their biosynthesis, this suggests the lens also actively contributes to the renin–angiotensin–aldosterone system (RAAS) of the eye. This report also alerts clinicians to the importance of a broader examination by endocrinologists, urologists and ophthalmologists for suspected CAH cases.
Author’s Contribution
V.B. conceived, analyzed the data, wrote, and provided critical revision of the manuscript. A.I. provided clinical information. A.K., R.Q., and M.M. provided critical revision of the manuscript. N.P. contributed to WES data analysis.
Acknowledgments
We thank the family members for their cooperation in this study. For this work, VB and MM were supported by grants from the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology (BRC-D-CON 546795), Moorfields Eye Hospital Special Trustees.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Ionides A, Francis P, Berry V, Mackay D, Bhattacharya S, Shiels A, Moore A. Clinical and genetic heterogeneity in autosomal dominant cataract. Br J Ophthalmol. 1999;83(7):802–08. doi:https://doi.org/10.1136/bjo.83.7.802.
- Shiels A, Hejtmancik JF. Biology of inherited cataracts and opportunities for treatment. Annu Rev Vis Sci. 2019;5:123–49. doi:https://doi.org/10.1146/annurev-vision-091517-034346.
- Berry V, Georgiou M, Fujinami K, Quinlan R, Moore A, Michaelides M. Inherited cataracts: molecular genetics, clinical features, disease mechanisms and novel therapeutic approaches. Br J Ophthalmol. 2020;104(10):1331–37. doi:https://doi.org/10.1136/bjophthalmol-2019-315282.
- Zhang Y, Yao K, Yu Y, Ni S, Zhang L, Wang W, Lai K. Effects of 1.8 GHz radiofrequency radiation on protein expression in human lens epithelial cells. Hum Exp Toxicol. 2013;32(8):797–806. doi:https://doi.org/10.1177/0960327112472353.
- Pallan PS, Wang C, Lei L, Yoshimoto FK, Auchus RJ, Waterman MR, Guengerich FP, Egli M. Human cytochrome P450 21A2, the major steroid 21-hydroxylase: structure of the enzyme\textperiodcentered progesterone substrate complex and rate-limiting c–h bond cleavage. J Biol Chem. 2015;290(21):13128–43. doi:https://doi.org/10.1074/jbc.M115.646307.
- Parsa AA, New MI. Steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. 2017;165(Pt A):2–11. doi:https://doi.org/10.1016/j.jsbmb.2016.06.015.
- El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet. 2017;390(10108):2194–210. doi:https://doi.org/10.1016/S0140-6736(17)31431-9.
- Carvalho B, Marques CJ, Santos-Silva R, Fontoura M, Carvalho D, and Carvalho F. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: an update on genetic analysis of CYP21A2 gene. Exp Clin Endocrinol Diabetes. 2020;129(7): 477–481 . doi:https://doi.org/10.1055/a-1108-1419.
- New MI, Abraham M, Gonzalez B, Dumic, M, Razzaghy-Azar, M, Chitayat, D, Sun, L. Genotype–phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci USA. 2013;110(7): 2611–6 . https://www.pnas.org/content/110/7/2611.short
- Berry V, Ionides A, Pontikos N, Georgiou M, Yu J, Ocaka LA, Moore AT, Quinlan RA, Michaelides M. The genetic landscape of crystallins in congenital cataract. Orphanet J Rare Dis. 2020;15(1):333. doi:https://doi.org/10.1186/s13023-020-01613-3.
- Nebert DW, Gonzalez FJ. P450 genes: structure, evolution, and regulation. Annu Rev Biochem. 1987;56:945–93. doi:https://doi.org/10.1146/annurev.bi.56.070187.004501.
- Nebert DW. 1991. MINIREVIEW: proposed role of drug-metabolizing enzymes: regulation of steady state levels of the ligands that effect growth, homeostasis, differentiation, and neuroendocrine functions. Mol Endocrinol. 5(9):1203–14. doi:https://doi.org/10.1210/mend-5-9-1203
- White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci U S A. 1986;83(14):5111–15. doi:https://doi.org/10.1073/pnas.83.14.5111.
- Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. 1986. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proc Nat Acad Sci. 83(9):2841–45. doi:https://doi.org/10.1073/pnas.83.9.2841
- Lee HH, Chao HT, Lee YJ, Shu SG, Chao MC, Kuo JM, Chung BC. Identification of four novel mutations in the CYP21 gene in congenital adrenal hyperplasia in the Chinese. Hum Genet. 1998;103(3):304–10. doi:https://doi.org/10.1007/s004390050821.
- Rabbani B, Mahdieh N, Ashtiani MTH, Larijani B, Akbari MT, New M, Parsa A, Schouten JP, Rabbani A. 2012. Mutation analysis of theCYP21A2Gene in the Iranian population. Genet Test Mol Biomarkers. 16(2):82–90. doi:https://doi.org/10.1089/gtmb.2011.0099
- Incorvaia C, Parmeggiani F, Costagliola C, Perri P, Tittoni M, Sebastiani A. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency associated with bilateral keratoconus. Am J Ophthalmol. 2003;135(4):557–59. doi:https://doi.org/10.1016/S0002-9394(02)01979-7.
- Björkhem I, Lütjohann D, Diczfalusy U, Ståhle L, Ahlborg G, Wahren J. 1998. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res. 39(8):1594–600. doi:https://doi.org/10.1016/s0022-2275(20)32188-x
- Bretillon L, Diczfalusy U, Björkhem I, Maire MA, Martine L, Joffre C, Acar N, Bron A, Creuzot-Garcher C. Cholesterol-24S-hydroxylase (CYP46A1) is specifically expressed in neurons of the neural retina. Curr Eye Res. 2007;32(4):361–66. doi:https://doi.org/10.1080/02713680701231857.
- Suri F, Yazdani S, Narooie-Nejhad M, Zargar SJ, Paylakhi SH, Zeinali S, Pakravan M, Elahi E. Variable expressivity and high penetrance of CYP1B1 mutations associated with primary congenital glaucoma. Ophthalmology. 2009;116(11):2101–09. doi:https://doi.org/10.1016/j.ophtha.2009.04.045.
- Li A, Jiao X, Munier FL, Schorderet DF, Yao W, Iwata F, Hayakawa M, Kanai A, Chen MS, Lewis RA, et al. Bietti crystalline corneoretinal dystrophy is caused by mutations in the novel gene CYP4V2. Am J Hum Genet. 2004;74(5):817–26. doi:https://doi.org/10.1086/383228.
- Wang Y, Guo L, Cai S-P, Dai M, Yang Q, Yu W, Yan N, Zhou X, Fu J, Guo X, et al. Exome sequencing identifies compound heterozygous mutations in CYP4V2 in a pedigree with retinitis pigmentosa. PLoS One. 2012;7(5):e33673. doi:https://doi.org/10.1371/journal.pone.0033673.
- Cenedella RJ. Sterol synthesis by the ocular lens of the rat during postnatal development. J Lipid Res. 1982;23(4):619–26. doi:https://doi.org/10.1016/S0022-2275(20)38126-8.
- Hitchener WR, Cenedella RJ. Absolute rates of sterol synthesis estimated from [3H]water for bovine lens epithelial cells in culture. J Lipid Res. 1985;26(12):1455–63. doi:https://doi.org/10.1016/S0022-2275(20)34251-6.
- James ER. The etiology of steroid cataract. J Ocul Pharmacol Ther. 2007;23(5):403–20. doi:https://doi.org/10.1089/jop.2006.0067.
- Gupta V, Wagner BJ. Search for a functional glucocorticoid receptor in the mammalian lens. Exp Eye Res. 2009;88(2):248–56. doi:https://doi.org/10.1016/j.exer.2008.04.003.
- Hampl R, Stárka L, Obenberger J, Rozsíval P. Content and binding of aldosterone in human ocular lens. Endocrinol Exp. 1984;18(1):59–64.
- Valinsky WC, Touyz RM, Shrier A. Aldosterone and ion channels. Vitam Horm. 2019;109:105–31.
- Kumari S, Kumar A, Sardar P, Yadav M, Majhi RK, Kumar A, Goswami C. 2015. Influence of membrane cholesterol in the molecular evolution and functional regulation of TRPV4. Biochem Biophys Res Commun. 456(1):312–19. doi:https://doi.org/10.1016/j.bbrc.2014.11.077
- Mamenko M, Zaika O, Jin M, O’Neil RG, Pochynyuk O. Purinergic activation of Ca2+-permeable TRPV4 channels is essential for mechano-sensitivity in the aldosterone-sensitive distal nephron. PLoS One. 2011;6(8):e22824. doi:https://doi.org/10.1371/journal.pone.0022824.
- Holappa M, Vapaatalo H, Vaajanen A. Local ocular renin–angiotensin–aldosterone system: any connection with intraocular pressure? A comprehensive review. Ann Med. 2020;52(5):191–206. doi:https://doi.org/10.1080/07853890.2020.1758341.
- Hannah-Shmouni F, Morissette R, Sinaii N, Elman M, Prezant TR, Chen W, Pulver A, Merke DP. 2017. Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US Ashkenazi Jews and Caucasians. Genet Med. 19(11):1276–79. doi:https://doi.org/10.1038/gim.2017.46
- Carrera P, Bordone L, Azzani T, Brunelli V, Garancini MP, Chiumello G, Ferrari M. Point mutations in Italian patients with classic, non-classic, and cryptic forms of steroid 21-hydroxylase deficiency. Hum Genet. 1996;98(6):662–65. doi:https://doi.org/10.1007/s004390050280.
- de Carvalho DF, Miranda MC, Gomes LG, Madureira G, Marcondes JAM, Billerbeck AEC, Rodrigues AS, Presti PF, Kuperman H, Damiani D, et al. Molecular CYP21A2 diagnosis in 480 Brazilian patients with congenital adrenal hyperplasia before newborn screening introduction. Eur J Endocrinol. 2016;175(2):107–16. doi:https://doi.org/10.1530/EJE-16-0171.
- Berry V, Gregory-Evans C, Emmett W, Waseem N, Raby J, Prescott D, Moore AT, Bhattacharya SS. 2013. Wolfram gene (WFS1) mutation causes autosomal dominant congenital nuclear cataract in humans. Eur J Hum Genet. 21(12):1356–60. doi:https://doi.org/10.1038/ejhg.2013.52