
ABSTRACT
Background
Variants in RCBTB1 were recently described to cause a retinal dystrophy with only eight families described to date and a predominant phenotype of macular atrophy and peripheral reticular degeneration. Here, we further evaluate the genotypic and phenotypic characteristics of biallelic RCBTB1-associated retinal dystrophy in a North American clinic population.
Methods
A retrospective analysis of genetic and clinical features was performed in individuals with biallelic variants in RCBTB1.
Results
Three unrelated individuals of French-Canadian descent with rare biallelic RCBTB1 variants were identified. All individuals shared a novel p.(Ser342Leu) missense variant; one patient was homozygous whereas the other two each possessed a second unique novel variant p.(Gln120*) and p.(Pro224Leu). All three had macula-predominant disease with symptom onset in the fifth decade of life.
Conclusion
This report adds to the genetic diversity of RCBTB1-associated disease. These cases confirm the later-onset, relative to many other retinal dystrophies, and macular focus of disease described in most cases to-date. They are thus a reminder of considering hereditary disease in the differential for later-onset macular atrophy.
KEYWORDS:
Introduction
Inherited retinal dystrophies (IRDs) are a genetically and phenotypically diverse group of diseases that affect more than 2 million individuals worldwide and result in significant visual disability and blindness (Citation1,Citation2). Mutations in nearly 300 genes are known to cause these conditions (https://sph.uth.edu/retnet/) and while mutations in certain genes account for a significant fraction of disease, the remaining genetic forms are rare. Although many disease-causing genes have roles in retina-specific pathways, an increasing number of genes are being identified with more ubiquitous roles in tissue homeostasis and stress response (https://sph.uth.edu/retnet/).
RCBTB1 is a protein that comprises a regulator of chromosome condensation 1 (RCC1)-like domain (RLD) and two broad complex, tramtrack, and bric-a-brac (BTB) domains (Citation3). It has been proposed to have a function in cell cycle regulation (Citation3), and some evidence points to its potential role as a tumor suppressor gene (Citation4). Expression array data on total RNA samples has shown ubiquitous expression in all human adult tissues (Citation5). Targeted analysis of expression has revealed relatively high expression of RCBTB1 mRNA in human neural retina compared to retinal pigment epithelium (RPE) (Citation5). In the retina specifically, it has been hypothesized that RCBTB1 may act as a substrate adaptor in the ubiquitinylation pathway (Citation6) and possibly modify the localization of oxidative stress-response transcription factors (Citation5,Citation7).
At the time of writing, only eight IRD families have been reported with retinal disease due to mutations in RCBTB1 (OMIM 607867) (Citation5,Citation8–10). The available phenotypic data from reports of retinal disease suggest that although mutations in RCBTB1 can cause typical retinitis pigmentosa (RP), the more common presentation may be a later-onset retinal dystrophy characterized by prominent macular atrophy (Citation5,Citation8,Citation9). Here, we describe three unrelated patients of French-Canadian descent with recessive disease carrying 3 novel pathogenic RCBTB1 variants. This series increases the number of reported patients with non-syndromic disease and describes a novel variant shared by all three patients. All three of these patients have later-onset disease with functional consequences due to macular involvement.
Methods
This retrospective study was conducted at Massachusetts Eye and Ear (MEE) under a protocol approved by the local institutional review board. The study met the tenets of the Declaration of Helsinki. Individuals with two rare, likely biallelic variants in RCBTB1 were identified from the patient population of a clinic specializing in inherited retinal dystrophies.
Genetic analysis
Blood samples were obtained from probands and available family members, and DNA was isolated from peripheral blood lymphocytes by standard procedures. Genetic testing was performed through the MEE Ocular Genomics Institute for two subjects (OGI1042_002062 and OGI3572_005183) and by a commercial diagnostic genetic testing lab for the third (OGI3572_005182). For testing performed at MEE, the Genetic Eye Disease panel and previously described analysis methods were used (Citation11). Guidelines of the American College of Medical Genetics (Citation12) were used for the interpretation of sequence variants. Variants were verified to have an allele frequency of less than 1% in the Genome Aggregation Database (gnomAD) (Citation13). The Combined Annotation Dependent Deletion (CADD) tool including PHRED-scaling was used to score the deleterious of the missense variants with scores of greater than 20 suggesting that a variant is more likely to have a deleterious effect (Citation14). Predictions from in silico modeling including SIFT (Citation15), PolyPhen-2 (Citation16), and MutationTaster (Citation17) were also used to help assess variant pathogenicity. Finally, structural modeling was used to assess the impact of missense mutations on polar bond organization and surrounding structure. Tridimensional structure of the RCBTB1 protein was generated with protein modeling software (PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC) using AlphaFold Database-predicted (Citation18) (https://alphafold.ebi.ac.uk) structure of human RCBTB1 as an input (AlphaFold ID: AF-Q8NDN9-F1).
When available, DNA samples were obtained from family members for segeregation analysis.
Clinical evaluation
All clinical assessments were performed in the Massachusetts Eye and Ear (MEE) Inherited Retinal Disorders Service. Visual acuity was assessed using Snellen charts. Kinetic perimetry was evaluated with Goldmann perimetry. Full-field ERGs were conducted with Burian Allen electrodes and a custom ERG system (Citation19). Fundus exam was complemented by fundus photography (Optos 200Tx and California devices, Optos PLC, Dunfermline, Scotland, United Kingdom), widefield fundus autofluorescence imaging (Optos), and spectral-domain optical coherence tomography (SD-OCT: Spectralis, Heidelberg Engineering, Heidelberg, Germany). The region tool within the Heidelberg Spectralis software was used to outline central macular areas of disrupted near-infrared reflectance on high-resolution images for Patient 1 (Heidelberg Engineering Region Finder Module 2.6.1.0).
Results
Genetic analysis
Three unrelated patients (2 males, 1 female) with biallelic rare variants in RCBTB1 were identified (). One of the variants led to a premature stop codon in exon 5 (of 13 exons total): c.358 C > T, p.(Gln120*), likely leading to degradation of the transcript and no protein product. The remaining two RCBTB1 variants found were missense changes that fell within exons encoding the RCC1-like domain (, ). The CADD-PHRED scores for the missense variants were greater than 20 thus suggesting a deleterious effect (p.(Pro224Leu) C-score = 31; p.(Ser342Leu) C-score = 22.9). These scores for the missense variants, as well as combined information from in silico predictions and ACMG classification of each of the three variants (), supported pathogenicity. Structural modeling of the two missense variants found that they did not affect polar bond organization or significantly modify surrounding protein structure, but the analysis did not permit any conclusions regarding whether these variants impaired binding efficiency with other proteins (data not shown).
Table 1. Genetic and clinical data
Table 2. Disease-associated alleles reported to date
Figure 1. Location and features of disease-associated RCBTB1 variants.
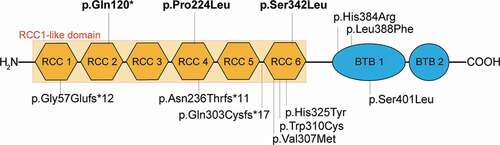
None of the variants have been reported before in association with retinal disease: the missense variants are reported in gnomAD but not in the homozygous state, and the variant with the premature stop is absent in gnomAD. One missense variant (c.1025 C > T, p.(Ser342Leu)) was shared by all three individuals, and it was homozygous in one patient (Patient 3, OGI3572_005183; no testing of family members performed) and in trans with the c.358 C > T, p.(Gln120*) variant in another (Patient 1; OGI1042_002062; confirmed by biparental testing). Family members were not available for testing in Patient 2 (OGI3572_005182), but the gnomAD Variant Co-Occurrence calculator predicts these two variants occur on different haplotypes supporting that these variants are likely in trans (Citation20). All three patients reported French-Canadian ancestry. Two of Patient 2’s four siblings had subjective difficulty with night vision and one carried a possible diagnosis of a retinal degeneration (Supplemental Figure 1). There was no family history of retinal disease or degeneration in the other two patients. Variants in other IRD genes (https://sph.uth.edu/retnet/) were found for two patients (Supplemental Table 1), but none of the variants detected were able to explain the phenotype in the reported cases.
Clinical data
Clinical assessments, with data summarized in , were performed at a single visit for two patients and spanned 3 years for one individual (Patient 1). Patients were between 46 and 62 years of age at initial examination. They described symptoms of decreased central vision, increased glare, and nyctalopia with most of these beginning when they were in their early to mid-40’s. Patient 3 was told that he had retinal abnormalities during a routine exam at age 25 suggesting that anatomic changes predated any functional impact. At the time of initial exam, visual acuity ranged from 20/20 to “count fingers” with an accompanying range of macular disruption apparent on fundus exam and with retinal imaging. The oldest patient (Patient 3) had the most impaired visual acuity at the initial exam.
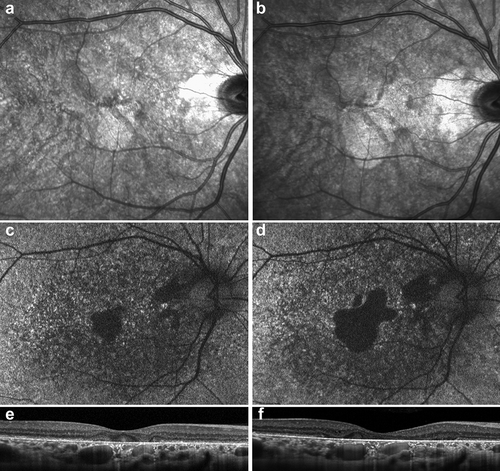
Fundus exam in all individuals showed macular atrophy of varying severity and characterized by nummular areas of atrophy that could be either fovea-sparing or fovea-threatening even in earlier stages of macular disease and later becoming confluent (). Abnormalities of the peripheral retina varied in extent and included subtle pigment mottling (Patient 1), fine pigment deposits (Patient 2, 3), and chorioretinal atrophy (Patient 2). At least one patient had a reticular pattern of pigment deposition in the peripheral retina. Fundus autofluorescence () emphasized the distribution of clinically apparent atrophy but was also more diffusely peripherally disrupted in two patients (Patients 2, 3). In addition to delineating areas of clear atrophy, OCT showed reduced definition of the RPE and ellipsoid zone even in seemingly intact areas of retina (). Numerous small cystoid spaces were present in the inner and outer retina overlying areas of atrophy in Patient 2. Retinal imaging over an approximately three-year interval in Patient 1 illustrated significant progression of macular involvement that corresponded to visual acuity decreasing from 20/20 in the right eye and 20/25 in the left eye to 20/30 and 20/600, respectively (). The area of abnormal reflectance on near-infrared imaging corresponding to the atrophy expanded by 1.1 mm2/year in the right eye and 1.2 mm2/year in the left eye (Starting values: 2.18 mm2 OD, 3.24 mm2 OS; final values 3.24 mm2 OD, 6.96 mm2 OS).
Figure 2. Fundus imaging of patients with RCBTB1-associated retinal dystrophy.
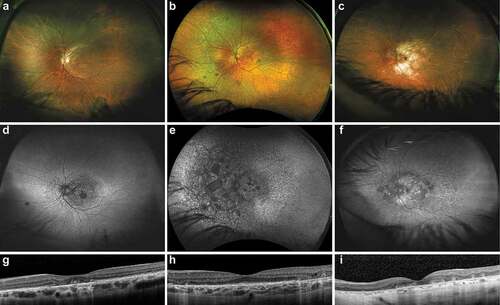
Figure 3. Evolution of macular findings in a single patient with RCBTB1-associated retinal dystrophy.
Goldmann perimetry demonstrated relative central scotomas but full peripheral fields. Full-field ERG demonstrated normal to mildly abnormal rod- and cone-isolated responses, but even when abnormal, responses remained robust ().
Patient 2 had additional medical diagnoses of tremor, anxiety, and nephrolithiasis; Patients 1 and 3 were in good health.
Discussion
In this cohort of patients with RCBTB1-associated retinal dystrophy, we thoroughly characterize the clinical phenotype and identify three novel disease-causing variants including a suspected founder mutation in the French-Canadian population. The functional (perimetry, ERG) and anatomic (widefield color, autofluorescence, OCT) descriptions of this cohort complement prior reports but also provide new information on the clinical spectrum of disease. Our description of prominent macular atrophy and peripheral pigmentary abnormalities corresponds to the findings described in five of six families in the original report of RCBTB1-associated disease (Citation5) as well as in available data from two additional individuals (Citation9,Citation21). Features of more typical retinitis pigmentosa were present in one family (Citation5) but appear to be a phenotypic outlier in RCBTB1-associated disease. Full-field ERG recordings in the current cohort demonstrated the potential for varying degrees of rod and cone dysfunction consistent with the range of changes previously reported (Citation21). Even in the patient with the most appreciably impacted ERGs (Patient 1), however, the response amplitudes were better preserved than those seen in typical RP. Indeed, the impact on visual acuity and central perimetry was consistent with the macular focus of degeneration. The progression over a three-year interval from 20/20 to counting fingers acuity in Patient 1 demonstrates the rapidity with which fovea-threatening disease can impact functional vision. The rate of atrophy expansion was similar to what has been described previously (Citation21).
The typical RCBTB1-associated cases in our cohort and others are characterized by central vision symptoms appearing and progressing in a patient’s 40s or 50s, with subsequent onset of nyctalopia. In this respect, the disease is similar to other later-onset disorders that can have significant impact on central vision through macular involvement, such as a C1QTNF5-associated late-onset retinal dystrophy (LORD) (Citation22,Citation23) or autosomal dominant RPE65-associated retinal degeneration (Citation24). In conjunction with reticular peripheral retinal pigmentation, the potential overlap with nonexudative age-related macular degeneration is also apparent; this diagnosis, and inherited retinal dystrophies more generally, should be considered in younger patients with macular atrophy but no drusen. However, as previously described, there are several examples of genes (e.g., PRPH2 (Citation25,Citation26), ABCA4 (Citation27)) in which mutations can cause dystrophies that vary from macular-dominant disease to features consistent with typical RP. Our cases highlight the importance of including evaluation of RCBTB1 in genetic analysis of patients with apparently non-syndromic later-onset retinal dystrophy. Finally, unlike a previous report (Citation5) which detailed 7 cases with associated systemic findings, our small cohort also did not have features suggestive of syndromic disease; they thus add to 5 of the 12 previously reported cases of RCBTB1-associated retinal dystrophy with non-syndromic disease (Citation5,Citation8,Citation9).
The three novel RCBTB1 variants found in this cohort expand the list of disease-associated variants. All three patients, from unrelated pedigrees, shared the c.1025 C > T, p.(Ser342Leu) variant. Even though all three patients self-reported French-Canadian ethnicity, this variant is predominantly present in Finnish individuals in gnomAD and therefore, at present, it is unclear if the c.1025 C > T p.(Ser342Leu) change is a founder allele or a mutational hot-spot. The p.(Ser342Leu) change affects the sixth RCC repeat of the RCC1-like domain, which harbors three additional missense variants associated with disease (, ), and the surrounding amino acids are highly conserved throughout evolution (Citation5). Patient 3 who was homozygous for the c.1025 C > T p.(Ser342Leu) mutation had the earliest onset and most anatomically advanced phenotype of the three cases, which may also be related to the fact that he was more than a decade older than the other cases. Of the remaining two patients, one carried a null allele in exon 5 (c.358 C > T, p.(Gln120*)) and the other carried a variant affecting the fourth RCC repeat (c.671 C > T, p.(Pro224Leu)). Given the limited number of cases in the literature, additional cases will be needed to determine whether there is any genotype–phenotype correlation in age-of-onset or disease severity.
Initial insights regarding the molecular function of RCBTB1 came from oncology with a focus on cell cycle regulation. The identification of RCBTB1 in 2001 was facilitated by the commonly encountered deletion of chromosome 13q14, the region within which it falls, in B-cell chronic lymphocytic leukemia (Citation3). The RCBTB1 protein was proposed to have a function in cell cycle regulation based upon the two protein domains encoded within its sequence (regulator of chromosome condensation-like (RCC1); broad complex, tramtrack, and bric-a-brac (BTB)) (Citation3). Indeed, the function of RCBTB1 as a tumor suppressor gene has been supported by findings including decreased RCBTB1 transcript levels in multiple cancer cell lines and the activation of DNA repair pathways following its expression (Citation4). RCBTB1 expression levels influence resistance to chemotherapy-induced apoptosis in cell lines with a clinical correlate of an association between deletion and metastatic progression in patients with sarcomas, particularly in individuals with prior chemotherapy (Citation4,Citation28). Increasing RCBTB1 levels by inhibiting the microRNA miR-26a-2 increases susceptibility to chemotherapy-induced apoptosis in a liposarcoma cell line and thus offers one potential method of modifying expression levels of this gene (Citation28).
The precise retinal functions of RCBTB1 remain under investigation. A more specific role for RCBTB1 within the ubiquitination pathway is emerging that has implications for understanding the mechanisms of RCBTB1-associated retinal disease. Protein tagging with ubiquitin, through the action of ubiquitinylation enzymes (E1, E2, E3), targets the complex to the proteasome for degradation or other cellular processes. RCBTB1 interacts with this pathway in two ways: it is proposed to be a substrate adaptor for a cullin3 (CUL3) E3 ligase, and it also binds to the mouse homolog of UBE2E3, an E2 ubiquitin-conjugating enzyme, through an interaction facilitated by the BTB domain (Citation6). UBE2E3, like RCBTB1, is expressed in the retina and is important for mediating the localization and activity of the stress-response transcription factor NRF2 (Citation7). Loss of NRF2 is known to play a role in retinal vulnerability to oxidative stress (Citation29,Citation30). Findings by Coppieters et al. suggest how RCBTB1-associated retinal disease might impact pathways important for regulating oxidative stress and suggest points for intervention. Individuals with RCBTB1-associated retinal dystrophy showed lower levels of NRF2 expression than control individuals based on mRNA expression in peripheral blood mononuclear cells (Citation5). As previously noted (Citation5), however, the pathologic accumulation of other proteins destined for degradation in the proteasome cannot be excluded as a disease mechanisms in RCBTB1-associated retinopathy (Citation31,Citation32). RPE cells derived from induced pluripotent stem cells (iPSCs) in individuals with RCBTB1-associated retinal disease are also beginning to provide insights into aberrations at a retina-specific level (Citation33).
If the mechanism of RCBTB1-associated disease is one of accumulation of oxidative damage, then multiple therapeutic strategies might be envisioned. Animal studies in which NRF2 was overexpressed using an AAV-vector in mouse models of RP showed prolonged cone survival (Citation34). Oral neuroprotective strategies based on preventing oxidative damage may also have therapeutic potential. For example, N-acetyl cysteine was previously shown to reduce oxidative damage and increased cone function and survival in animal models of RP (Citation35), and a recent report of a Phase I clinical trial for oral N-acetylcysteine suggested that it was safe, well-tolerated and may improve the function of macular cones in patients with RP (Citation36). It remains to be seen whether these strategies based on reducing oxidative damage may have a differential genotype-specific effect based on a specific pathogenic mechanism for particular IRDs. Recent work in RPE derived from iPSCs from a patient with RCBTB1-associated retinopathy also suggests the potential benefits of AAV-mediated RCBTB1 gene augmentation (Citation33).
Conclusion
Our report provides a comprehensive description of the phenotype of three cases of retinal dystrophy of varying severity associated with biallelic RCBTB1 mutations and describes three novel disease-causing variants including a potential founder mutation. This case series increases the number of reported families with nonsyndromic disease. The macular atrophy common to all three indivdiuals emphasizes the importance of considering retinal dystrophies in the differential of later-onset retinal disease. Further work is needed to elucidate the role of RCBTB1 in the retina, both to understand retinal pathobiology of disease and to identify potential treatments that may intervene at the level of the gene or downstream effectors.
Supplemental Material
Download PDF (19.5 KB)Supplemental Material
Download PDF (110.1 KB)Acknowledgments
The authors would like to thank the patients and their family members for their participation in this study and the Ocular Genomics Institute Genomics Core members for their experimental assistance. The authors would like to thank the Exome Aggregation Consortium, the Genome Aggregation Database (gnomAD), and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about and http://gnomad.broadinstitute.org/about.
Disclosure statement
The authors do not have a proprietary interest in the contents of this manuscript or financial disclosures relevant to its content. Naomi E. Wagner is an employee and stockholder of Invitae, Inc. (September 2020–present). Jason I. Comander is a consultant for AGTC, Beam Therapeutics, Biogen, Gensight, Vedere, and Wave Life Sciences. Rachel Huckfeldt is a consultant for AGTC, ProQR Therapeutics, and Vida Ventures.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- Berger W, Kloeckener-Gruissem B, Neidhardt J.The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29(5):335–75.doi:https://doi.org/10.1016/j.preteyeres.2010.03.004.
- O’Neal TB, Luther EE. Retinitis pigmentosa. Treasure Island (FL): StatPearls; 2020.
- Mabuchi H, Fujii H, Calin G, Alder H, Negrini M, Rassenti L, Kipps TJ, Bullrich F, Croce CM.Cloning and characterization of CLLD6, CLLD7, and CLLD8, novel candidate genes for leukemogenesis at chromosome 13q14, a region commonly deleted in B-cell chronic lymphocytic leukemia. Cancer Res. 2001;61(7):2870–77.
- Zhou X, Munger K.Clld7, a candidate tumor suppressor on chromosome 13q14, regulates pathways of DNA damage/repair and apoptosis. Cancer Res. 2010;70(22):9434–43.doi:https://doi.org/10.1158/0008-5472.CAN-10-1960.
- Coppieters F, Ascari G, Dannhausen K, Nikopoulos K, Peelman F, Karlstetter M, Xu M, Brachet C, Meunier I, Tsilimbaris MK, et al. Isolated and syndromic retinal dystrophy caused by biallelic mutations in RCBTB1, a gene implicated in ubiquitination. Am J Hum Genet. 2016;99(2):470–80.doi:https://doi.org/10.1016/j.ajhg.2016.06.017.
- Plafker KS, Singer JD, Plafker SM.The ubiquitin conjugating enzyme, UbcM2, engages in novel interactions with components of cullin-3 based E3 ligases. Biochemistry. 2009;48(15):3527–37.doi:https://doi.org/10.1021/bi801971m.
- Plafker KS, Plafker SM.The ubiquitin-conjugating enzyme UBE2E3 and its import receptor importin-11 regulate the localization and activity of the antioxidant transcription factor NRF2. Mol Biol Cell. 2015;26(2):327–38.doi:https://doi.org/10.1091/mbc.E14-06-1057.
- Huang Z, Zhang D, Chen SC, Thompson JA, McLaren T, Lamey T, De Roach JN, McLenachan S, Chen FK.Generation of three induced pluripotent stem cell lines from an isolated inherited retinal dystrophy patient with RCBTB1 frameshifting mutations. Stem Cell Res. 2019;40:101549. doi:https://doi.org/10.1016/j.scr.2019.101549.
- Yang J, Xiao X, Sun W, Li S, Jia X, Zhang Q.Variants in RCBTB1 are associated with autosomal recessive retinitis pigmentosa but not autosomal dominant FEVR. Curr Eye Res. 2021 Jun; 46(6) ;839–844.doi:https://doi.org/10.1080/02713683.2020.1842457.
- Wu JH, Liu JH, Ko YC, Wang CT, Chung YC, Chu KC, Liu TT, Chao HM, Jiang YJ, Chen SJ, et al. Haploinsufficiency of RCBTB1 is associated with Coats disease and familial exudative vitreoretinopathy. Hum Mol Genet. 2016;25(8):1637–47.doi:https://doi.org/10.1093/hmg/ddw041.
- Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, Wang DY, Au ED, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med. 2015;17(4):253–61.doi:https://doi.org/10.1038/gim.2014.172.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–24.doi:https://doi.org/10.1038/gim.2015.30.
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.doi:https://doi.org/10.1038/s41586-020-2308-7.
- Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M.CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D94.doi:https://doi.org/10.1093/nar/gky1016.
- Kumar P, Henikoff S, Ng PC.Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.doi:https://doi.org/10.1038/nprot.2009.86.
- Adzhubei I, Jordan DM, Sunyaev SR.Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;76. Chapter 7:Unit7 20. doi:https://doi.org/10.1002/0471142905.hg0720s76.
- Schwarz JM, Cooper DN, Schuelke M, Seelow D.MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–62.doi:https://doi.org/10.1038/nmeth.2890.
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–89.doi:https://doi.org/10.1038/s41586-021-03819-2.
- Reichel E, Bruce AM, Sandberg Ma, Berson EL.An electroretinographic and molecular genetic study of X-linked cone degeneration. Am J Ophthalmol. 1989;108(5):540–47.doi:https://doi.org/10.1016/0002-9394(89)90431-5.
- gnomAD. Variant co-occurence. [accessed Oct 27 2021]. https://gnomad.broadinstitute.org/variant-cooccurrence?dataset=gnomad_r2_1&variant=13-50126354-G-A&variant=13-50123614-G-A.
- Huang Z, Zhang D, Thompson JA, Jamuar SS, Roshandel D, Jennings L, Mellough C, Charng J, Chen SC, McLaren TL, et al. Deep clinical phenotyping and gene expression analysis in a patient with RCBTB1-associated retinopathy. Ophthalmic Genet. 2021;1–10. doi:https://doi.org/10.1080/13816810.2021.1966053.
- Soumplis V, Sergouniotis PI, Robson AG, Michaelides M, Moore AT, Holder GE, Webster AR.Phenotypic findings in C1QTNF5 retinopathy (late-onset retinal degeneration). Acta Ophthalmol. 2013;91(3):e191–5.doi:https://doi.org/10.1111/aos.12010.
- Vincent A, Munier FL, Vandenhoven CC, Wright T, Westall CA, Heon E.The characterization of retinal phenotype in a family with C1QTNF5-related late-onset retinal degeneration. Retina. 2012;32(8):1643–51.doi:https://doi.org/10.1097/IAE.0b013e318240a574.
- Jauregui R, Cho A, Oh JK, Tanaka AJ, Sparrow JR, Tsang SH.Phenotypic expansion of autosomal dominant retinitis pigmentosa associated with the D477G mutation in RPE65. Cold Spring Harb Mol Case Stud. 2020;6(1):a004952.doi:https://doi.org/10.1101/mcs.a004952.
- Wells J, Wroblewski J, Keen J, Inglehearn C, Jubb C, Eckstein A, Jay M, Arden G, Bhattacharya S, Fitzke F, et al. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat Genet. 1993;3(3):213–18.doi:https://doi.org/10.1038/ng0393-213.
- Renner AB, Fiebig BS, Weber BH, Wissinger B, Andreasson S, Gal A, Cropp E, Kohl S, Kellner U.Phenotypic variability and long-term follow-up of patients with known and novel PRPH2/RDS gene mutations. Am J Ophthalmol. 2009;147(3):518–30 e1.doi:https://doi.org/10.1016/j.ajo.2008.09.007.
- Cremers FPM, Lee W, Collin RWJ, Allikmets R.Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog Retin Eye Res. 2020;79:100861. doi:https://doi.org/10.1016/j.preteyeres.2020.100861.
- Lee DH, Amanat S, Goff C, Weiss LM, Said JW, Doan NB, Sato-Otsubo A, Ogawa S, Forscher C, Koeffler HP.Overexpression of miR-26a-2 in human liposarcoma is correlated with poor patient survival. Oncogenesis. 2013;2:e47. doi:https://doi.org/10.1038/oncsis.2013.10.
- Himori N, Yamamoto K, Maruyama K, Ryu M, Taguchi K, Yamamoto M, Nakazawa T.Critical role of Nrf2 in oxidative stress-induced retinal ganglion cell death. J Neurochem. 2013;127(5):669–80.doi:https://doi.org/10.1111/jnc.12325.
- Sachdeva MM, Cano M, Handa JT.Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp Eye Res. 2014;119:111–14. doi:https://doi.org/10.1016/j.exer.2013.10.024.
- Vasireddy V, Jablonski MM, Khan NW, Wang XF, Sahu P, Sparrow JR, Ayyagari R.Elovl4 5-bp deletion knock-in mouse model for Stargardt-like macular degeneration demonstrates accumulation of ELOVL4 and lipofuscin. Exp Eye Res. 2009;89(6):905–12.doi:https://doi.org/10.1016/j.exer.2009.07.021.
- Illing ME, Rajan RS, Bence NF, Kopito RR.A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem. 2002;277(37):34150–60.doi:https://doi.org/10.1074/jbc.M204955200.
- Huang Z, Zhang D, Chen SC, Jennings L, Carvalho LS, Fletcher S, Chen FK, McLenachan S.Gene replacement therapy restores RCBTB1 expression and cilium length in patient-derived retinal pigment epithelium. J Cell Mol Med. 2021;25(21):10020–27.doi:https://doi.org/10.1111/jcmm.16911.
- Xiong W, Garfinkel AE M, Li Y, Benowitz LI, Cepko CL.NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J Clin Invest. 2015;125(4):1433–45.doi:https://doi.org/10.1172/JCI79735.
- Lee SY, Usui S, Zafar AB, Oveson BC, Jo YJ, Lu L, Masoudi S, Campochiaro PA.N-Acetylcysteine promotes long-term survival of cones in a model of retinitis pigmentosa. J Cell Physiol. 2011;226(7):1843–49.doi:https://doi.org/10.1002/jcp.22508.
- Campochiaro PA, Iftikhar M, Hafiz G, Akhlaq A, Tsai G, Wehling D, Lu L, Wall GM, Singh MS, Kong X.Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. J Clin Invest. 2020;130(3):1527–41.doi:https://doi.org/10.1172/JCI132990.