
ABSTRACT
Background
Rods and cones are photoreceptor neurons in the retina that are required for visual sensation in vertebrates, wherein the perception of vision is initiated when these neurons respond to photons in the light stimuli. The photoreceptor cell is structurally studied as outer segments (OS) and inner segments (IS) where proper protein sorting, localization, and compartmentalization are critical for phototransduction, visual function, and survival. In human retinal diseases, improper protein transport to the OS or mislocalization of proteins to the IS and other cellular compartments could lead to impaired visual responses and photoreceptor cell degeneration that ultimately cause loss of visual function.
Results
Therefore, studying and identifying mechanisms involved in facilitating and maintaining proper protein transport in photoreceptor cells would help our understanding of pathologies involving retinal cell degeneration in inherited retinal dystrophies, age-related macular degeneration, and Usher Syndrome.
Conclusions
Our mini-review will discuss mechanisms of protein transport within photoreceptors and introduce a novel role for an unconventional motor protein, MYO1C, in actin-based motor transport of the visual chromophore Rhodopsin to the OS, in support of phototransduction and visual function.
Introduction
The human retina contains two different types of photoreceptor cells, rods that are responsible for high-sensitivity photon detection under low-light conditions and for peripheral vision, and cones that are responsible for color vision and high central visual acuity (Citation1,Citation2). In the human retina, rods outnumber the cones (Citation3,Citation4). However, zebrafish and ground squirrel have cone-dominant retinas (Citation5), while retinas in birds, humans and primates have rod distribution more prominent in the periphery and cones being restricted to the central retina (Citation1,Citation6). In humans and non-human primates, the fovea (or macula) at the center of retina contains only cones and is responsible for high visual acuity (Citation7,Citation8).
The retinal photoreceptor cell is further distinguished into morphologically distinct compartments that coordinate proteins to optimize each specialized function. Here, rod and cone photoreceptor cells consist of distinct compartments; the outer segment (OS) containing membrane discs housing proteins involved in phototransduction, the inner segments (IS) in which protein biosynthesis occurs, and the synaptic terminal transmits electric excitation that is light-driven to downstream neurons. The site where photons are absorbed and the visual signal is generated through phototransduction is in the most distal photoreceptor compartment, the OS, which is adjacent to the Retinal Pigment Epithelium (RPE) (Citation9,Citation10). The OS is connected to the IS through a single physical bridge called the connecting cilium (CC) (Citation9,Citation10).
The OS of cones differs from that of rods in structural organization and 3D shape. Cones owe their name to the conical shape of their OS consisting of parallel disc-like membranes. The discs in cones are open and contiguous with the plasma membrane, unlike rod OS discs that are separated from the plasma membrane (Citation11). In cones of lower vertebrates, these open discs persist over the full OS length, while mammalian cones may contain discs separated from the plasma membrane like in rods (Citation1). The IS serves as the primary housekeeping compartment of the photoreceptor cell and includes two main sub-compartments, the ellipsoid and myoid. The ellipsoid is located directly below the connecting cilium and is packed with mitochondria that help satisfy the metabolic requirements of photoreceptors. The myoid zone is the portion of the IS of photoreceptors that is between the mitochondria-rich ellipsoid zone and the cell body/nucleus of the photoreceptor IS. This myoid zone contains protein synthesis apparatus (Citation1,Citation6). Rods have thin elongated mitochondria while cones have short wider mitochondria, which allows for an easy distinction between the respective IS in electron micrographs (Citation12). The sequestration of mitochondria to the ellipsoid is a morphological adaptation proposed to bring them closer to the choroidal blood vessels for more efficient supply of oxygen required to produce adenosine triphosphate (ATP) in direct proximity of the energy-demanding OS (Citation13).
Protein localization and trafficking in photoreceptor cells
All proteins destined for the photoreceptor OS are synthesized in the IS compartment, so they must be properly trafficked to reach their final functional destination. Therefore, proper trafficking and compartmentalization of proteins to specific cellular compartments of the photoreceptor cell are critical for effective phototransduction and visual function (Citation14). Specific sorting of proteins within the photoreceptor cell requires trafficking and plasma membrane targeting/localization sequences that serve as “biochemical zip codes,” making the photoreceptors among the most interesting cells to study intracellular protein sorting, trafficking, and localization in retinal health and disease conditions (Citation15). It is therefore well established that the development, maintenance, and function of photoreceptors require the trafficking and transport of both cilia and OS specific proteins from the IS to their appropriate cellular compartments. The photoreceptor OS has predominantly transmembrane and lipid-modified proteins that coordinate the protein movement through ciliary transport. The OS compartment is a modified primary cilium therefore mechanisms identified in primary and other sensory cilia can apply to the photoreceptor OS, thereby stimulating studies in both fields. It is known that protein transport occurs throughout the life of the photoreceptor cell as the OS are continuously renewed. Studies demonstrated that in mouse rod OS renewal occurs every 10 days (Citation16). This phenomenon has been proposed to serve as a protective mechanism to minimize the potential accumulation of any damaged molecular components in the phototransduction cascade, which might arise due to the constant photo-oxidative stress associated with light absorption in the photoreceptor OS compartment. Rod OS renewal occurs in an orderly fashion, which was first revealed by pulse/chase studies in rats, mice, and frogs (Citation16). In adult frogs, the disc membranes at the base of rod OS were progressively displaced by newer discs (Citation17). Similar studies also showed cone-shedding as well in squirrel retinas and human fovea, both of which are cone-dominant (Citation18,Citation19). This rapid OS renewal requires a high rate of protein synthesis and efficient protein trafficking. The ability of the photoreceptors to renew their OS and their transmission of signals through polarized neurons therefore makes them an ideal cell type to study protein trafficking and localization. Thus, understanding and identifying new mechanisms regulating and maintaining accurate protein transport and compartmentalization is vital to the function and survival of photoreceptor cells.
Signaling sequences facilitating the transport of Rhodopsin from the Photoreceptor IS to OS
Due to their characteristic shape and large biosynthetic requirement, photoreceptors are considered model cells to study the dynamics of protein trafficking. A central question in this regard is understanding the mechanisms facilitating the post-biosynthesis transport of plasma membrane-associated proteins and the mechanisms that target these proteins to the OS for disc assembly and/or their participation in the phototransduction cascade. Therefore, a mode of rapid and efficient transport and proper localization of proteins from the endoplasmic reticulum (ER) to the proximal OS is essential for photoreceptor cell function and survival. The G-protein coupled receptor (GPCR) Type II Opsin, Rhodopsin, constitutes the majority (approximately 80%) of the OS resident proteins, and is the major player in vertebrate phototransduction (Citation20–23). Rhodopsin is synthesized in the rough endoplasmic reticulum (ER) of the IS of photoreceptors and subsequently undergoes posttranslational modifications in the Golgi before becoming functional, where it is sorted into vesicles destined for the OS. Rhodopsin contains an intracellular OS targeting signal located at its C-terminal, which includes the last four amino acids comprising the ciliary targeting VXPX motif (Citation24–26). Deletion or mutations of these amino acids result in mislocalization of Rhodopsin from the OS (Citation14,Citation27). Furthermore, these mutations or deletions in humans lead to the most severe forms of retinal degeneration (Citation28). Rhodopsin also contains a predicted FR (a Phe-Arg amino acid doublet) targeting signal within its intracellular H-8 α-helix. This FR signal was originally characterized for the ciliary receptor, smoothened, and was shown to play a critical role in Rhodopsin targeting to the primary cilium of cultured cells (Citation29), but it remains to be tested whether mutations of this signal lead to Rhodopsin mis-targeting in photoreceptors. Many signaling proteins located in photoreceptor OS are tightly associated with disc membranes via lipid modifications reviewed in (Citation2,Citation30).
Retinal degenerative disorders
Genetic mutations in rod- or cone-specific genes can lead to inherited retinal degeneration (Citation28,Citation30–35), primarily affecting rods (e.g. retinitis pigmentosa and rod-cone dystrophy) and cones (e.g. cone-rod dystrophy). In IRD genetics is a key contributor, which is associated with irreversible loss of visual function that affects an estimated 1:2500 individuals. It is estimated that about 50% of all IRDs belong to a class of pigmented retinopathies, called Retinitis Pigmentosa (Citation35), which owes its name because ophthalmoscopic examination of such patients shows an ischemic appearance of the retina, together with a pale cole optic nerve head and brown/black pigmented “bone spicules” in the periphery (Citation31,Citation32).
Retinitis Pigmentosa
Retinitis Pigmentosa (Citation31,Citation35) is a group of inherited retinal disorders characterized by early symptoms that includes night blindness and a progressive loss of peripheral vision. Varying modes of RP inheritance include 50–60% autosomal recessive, 30–40% autosomal dominant, and 5–15% X-linked (Citation31,Citation35). The majority of genetic defects identified in RP are related to rod photoreceptor-specific proteins that include components of the rod photo-transduction cascade, proteins involved in maintaining the structural integrity of ROS, and intracellular protein trafficking (Citation34). In the early stages of RP, rod photoreceptors are more severely affected than cones. As the rod cells degenerate, patients experience night blindness and a progressive loss of peripheral vision. As RP progresses, clinical symptoms include loss of central vision and visual acuity (Citation35).
Non-Syndromic Retinitis Pigmentosa
Non-syndromic RP has no extra-ocular manifestations and can be inherited as autosomal dominant, autosomal recessive, or X-linked trait (Citation31,Citation36). Majority of the patients with RP usually are non-syndromic, meaning the symptoms are related to vision loss. Some clinical studies involving ERG in patients have reported a difference in cone function in syndromic and non-syndromic RP (Citation31,Citation37). More than 10% of adRP cases reported are due to mutations in Rhodopsin.
Syndromic retinitis pigmentosa
Syndromic retinitis pigmentosa (Citation31,Citation35) is a pathological condition with both ocular and extra-ocular manifestations. There are many syndromic forms of RP, among the most common are Usher syndrome and Bardet-Biedl syndrome (BBS). Typical symptoms for Usher syndrome include hearing loss and vision loss from RP and in some cases vestibular dysfunction. In contrast, phenotypes in BBS include visual loss, obesity, reduced kidney function, intellectual impairment polydactyly, and hypogenitalism. In RP visual loss usually begins with night blindness (nyctalopia), followed by progressive loss of central visual function. It is estimated that mutations in more than 80 different genes contribute to syndromic and non-syndromic RP. These mutations manifest in different forms of syndromic RP characterized by different pathologies like ciliopathies (Usher syndrome and Bardet-Biedl syndrome), neurodegenerative disorders like hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa and pallidal degeneration (HARP syndrome), polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, cataract (PHARC syndrome), Batten disease, and mitochondrial disorders like neuropathy, ataxia, and retinitis pigmentosa (NARP syndrome) (Citation36,Citation37).
Association of mutant Rhodopsin mislocalization with retinitis pigmentosa
Over 100 mutations in the rod opsin (Rhodopsin) have been associated with RP (Citation38,Citation39). Of these, nearly all RP-related Rhodopsin mutations are autosomal dominant (adRP) and account for 30% of adRP cases (Citation35,Citation39). Mechanistically, mutations in the Rhodopsin gene causes opsin mislocalization, altered activity and inability to bind to its ligand, 11-cis retinal, endoplasmic retention and stress, and apoptosis (Citation40,Citation41). Mutations in Rhodopsin have been identified across the cytoplasmic, transmembrane as well as the intradiscal domain. Most of these mutations are reported to result in misfolding of opsin proteins, while some class I Rhodopsin mutations have been reported to cause mislocalization of proteins to the IS, ONL, and cell bodies (Citation27,Citation42). There are a number of classes of Rhodopsin mutations with those such as class I and II resulting in defective trafficking of Rhodopsin to the OS, due to mistrafficking or misfolding (for a complete review refer to (Citation35)). For example, these mutations mainly occur in the plasma membrane targeting motif of Rhodopsin, the VXPX motif. The Ter349Glu mutation extends the C-terminal of Rhodopsin protein and causes a severe form of adRP. Other mutations in the C-terminal like the Gln344Ter and Ser334Ter result in truncated forms of Rhodopsin, while the P347L/P347S and V345M mutations cause amino acid substitutions (Citation27,Citation39). These class I mutations have been reported to affects OS transport and photoreceptor survival (Citation35,Citation43–45). The class I mutants function like wild-type opsin and have been shown to localize to the cytoplasmic membrane and bind vitamin A/11-cis retinaldehyde (11-cis RAL) to form spectrally active Rhodopsin (Citation46). This suggests the existence of an ER mechanism, which prevents further trafficking of misfolded Rhodopsin to the photoreceptor OS (Citation35,Citation38). Opsin class II mutants have been shown in vitro to accumulate abnormally in the rough ER membrane and are not trafficked efficiently to the cytoplasmic membrane in contrast to the WT opsin and fail to bind 11-cis RAL to form a spectrally active photopigment. Therefore, class II mutants are proposed to be defective in protein folding and/or stability (Citation34,Citation38,Citation39). The P23H mutation, one of the earliest identified mutations in RP causes misfolding of Rhodopsin and its retention in the ER, leading to activation of the UPR complex and in ER stress (Citation35).
Overview of Rhodopsin trafficking from the photoreceptor ER to OS
Biosynthesis and folding of Rhodopsin in the endoplasmic reticulum (ER) is followed by its transport to Golgi apparatus by chaperone proteins like BiP (Citation40–49). In addition, Rhodopsin also undergoes glycosylation and palmitoylation which appears to have an indirect role in the trafficking and stability of the opsin molecule (Citation45,Citation50,Citation51). From the ER, Rhodopsin is carried to the Golgi apparatus where it is sorted and bound to other scaffolding proteins like Arf4, Rab11, Rab8 and transported to the connecting cilium. It has previously been shown that Arf4, a GTP binding protein, interacts with the C-terminus region of Rhodopsin, that is known to contain the VXPX motif (Citation24,Citation45,Citation52). Rab8 is a GTPase, that forms complex with Rabin8 and has been reported to mediate Rhodopsin trafficking from the Golgi apparatus to connecting cilium. Arf-ASAP scaffolding complex helps Rab8-Rab11 mediated ciliary targeting of Rhodopsin. However, in addition to VXPX motif, this interaction also involves the FR motif of Rhodopsin (Citation29).
Interflagellar transport (IFT) has been studied for its role in transport of Rhodopsin through the connecting cilium and the detailed mechanism of this mode of transport has been discussed previously (Citation53). Most of these IFT proteins are localized at the ciliary portion of photoreceptors (Citation54). The IFT transport involves a large polypeptide complex of 16 proteins that are involved in anterograde and retrograde transport of through the ciliary bodies. These complexes are organized as IFT-A and IFT-B which are known for their role in protein trafficking in photoreceptors. Defects in IFT-B proteins like IFT52 and IFT57 causes OS defects (Citation55), whereas IFT-A proteins like IFT40 and IFT20 are required for proper localization of Rhodopsin. Deletion of IFT20 causes Rhodopsin accumulation near the Golgi complex, while deletion of IFT40 causes its accumulation near plasma membrane of the photoreceptor IS (Citation56). The role of kinesins and dynesins in microtubule mediated Rhodopsin trafficking has been discussed below (Citation57–60).
Chaperone mediated Rhodopsin transport to the photoreceptor OS
Tubby-like protein 1 (TULP1) is a photoreceptor-specific protein localized to the IS, CC, and synaptic terminal known to be involved in protein trafficking. TULP1 is highly expressed in human retina and mutations in TULP1 have been associated with retinal degeneration in Retinitis Pigmentosa (Citation31,Citation35,Citation57–60). TULP1 plays a critical role in the transport and localization of proteins from IS to OS of the retina. Previous studies of Tulp1−/− photoreceptors have revealed that Rhodopsin, cone opsins, and several other phototransduction proteins destined for the OS are mislocalized within photoreceptor compartments, including the IS, cell bodies, and ONL, prior to photoreceptor OS cell degeneration and death. In Tulp1−/− mice retinas, chaperone proteins and arrestins were mislocalized, however transducins were not affected (Citation59). Thus, opsins appear to be a cargo for TULP1, as studies have reported Rhodopsin accumulation in photoreceptor cells in Tulp1−/− mutant mice (Citation60). Thus, it can be concluded that Tulp1 is required for the correct transport of specific integral membrane proteins and their respective binding partners. Other classes of OS resident proteins do not appear to be affected in this mutant mouse model. These differences support the hypothesis that TULP1 plays a specific and critical role in photoreceptor OS protein transport pathways. Recently, it was shown that mutant TULP1 results in its retention to the ER and subsequently the activation of the unfolded protein response (UPR) complex, causing ER stress and cellular apoptosis (Citation61,Citation62). Thus, it can be hypothesized that the retinal degenerative phenotypes observed in humans with these TULP1 mutations could be due to ER stress.
Role of dyneins and kinesins in protein transport within photoreceptor cells
Cytoplasmic dynein protein is an end-directed motor complex that has been studied as a candidate for transport of Rhodopsin within the PR cells. It has previously been shown that dynein light chain (DLC) Tctex-1 acts as a cargo adaptor molecule in the interaction between Rhodopsin and dynein (Citation63). In zebrafish, DLC was shown to be required for post-golgi vesicular trafficking of Rhodopsin protein. Mutation in dynein showed mislocalization of Rhodopsin (Citation64). Interestingly in zebrafish lacking dynein-2, neither opsins nor arrestins were mislocalized, but the loss of dynein-2 function caused a significant reduction in a-wave and b-wave in the ERGs (Citation65). Dynein-1 is involved in the localization of organelles like golgi, mitochondria and endoplasmic reticulum. Subsequently in Dync1l1−/− mice, the ER in the photoreceptors is found in the OPL in addition to IS and ONL of the retina normally seen (Citation66). In these mice, the loss of this dynein subunit caused accumulation of OS proteins and partial blocking of vesicle export from the endoplasmic reticulum (Citation66). Recently, dynein was also found to transport melanosomes along the microtubules in RPE cells (Citation67). In retinal PR cells, dynein2 is involved in retrograde inter-flagellar transport (IFT) of proteins through the connecting cilium (Citation68), whereas dynein 1 is involved in the material transport of PR cells.
While dyneins are involved in retrograde inter flagellar transport, kinesins like kinesin-1 are involved in anterograde axonal transport. Kinesin-1 is activated by the presence of cargo molecules, which interact with the heavy chain and light chains of the motor protein simultaneously (Citation69). In the conditional knockout model of KIF3a, a subunit of kinesin-2 with Kif3aflox/flox expressing Rho cre, Rhodopsin mislocalization was observed in the photoreceptor IS. However, the photoreceptor growth was normal and devoid of any structural defects (Citation70). Complete loss of KIF3a in mice also caused accumulation of arrestins and opsins within the IS of the photoreceptors at 1 month of age. Loss of KIF3a also resulted in the subsequent death of photoreceptor cells (Citation71). In another study, deletion of Rho prevented the cell death in KIF3a depleted rod photoreceptor cells. This study also showed that the translocation of arrestin was blocked in KIF3a depleted rod photoreceptor cells (Citation72). In zebrafish, mutations in KIF3a caused rapid rod degeneration due to Rhodopsin accumulation (Citation73). However, recent studies showed that KIF3a is required for the transport of proteins in the cones but not rod photoreceptor (PR) cells in mice with KIF3a depleted PR cells (Citation74).
Unconventional motor proteins in cell physiology
Myosin-I molecular motors have been proposed to have various cellular roles related to membrane dynamics and protein trafficking. Higher vertebrates have been shown to express eight different Myosin-I genes MYO1A-MYO1H, with the corresponding proteins named MYO1A–MYO1H (Citation75). Myosin-I motors are classified into short-tailed myosins (MYO1A, MYO1B, MYO1C, MYO1D, MYO1G and MYO1H) or long-tailed myosins (MYO1E, MYO1F) based on the presence of additional glycine-rich (TH2) and SH3 (TH3) domains in the long-tailed isoforms (). In general, the cellular localization of Myosin-I isoforms is dependent on both the preference of their ATP-motor domains for different actin filament populations and for specific anionic phospholipids that are found on different cellular membranes (Citation76).
Figure 1. Myosin Phylogenetic Tree. The Myosin family protein sequences were aligned in MEGA-X software version 10.2.6 and the phylogenetic tree was generated. The closest resembling member from each of the seven groups were used to represent the domains and regions from the UniProt database. Phylogenetic and molecular evolutionary analyses were conducted using MEGA version X (Citation77).
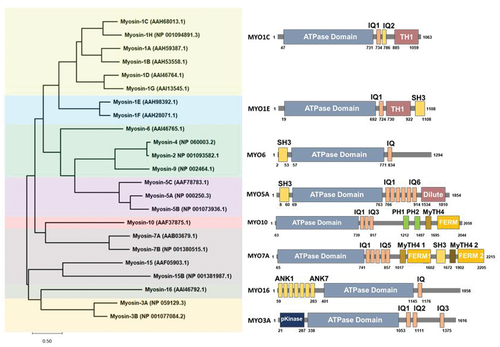
MYO1 protein member family and disease phenotypes
MYO1 protein family member isoforms show varying expression levels across different tissues and thus likely have unique functions in the cell types they are expressed in (). MYO1B, MYO1C, and MYO1E proteins are widely expressed and found in most cell types (Citation78,Citation79). The loss of MYO1C or MYO1E has been associated with kidney disease (Citation78,Citation79), in addition to deafness associated with MYO1C loss (Citation62). Mutations in MYO1A have been associated with hearing loss (Citation100,Citation101), although MYO1A protein is highly expressed in the small intestine and colon where it is also associated with colon cancer (Citation102). MYO1D protein is highly expressed in the central and peripheral nervous system (Citation81,Citation93), liver and small intestine (Citation93), and has been linked to autism spectrum disorders (Citation13). MYO1F and MYO1G proteins are found predominantly expressed in hematopoietic cells, where MYO1E is also found (Citation103,Citation104). MYO1F is the third myosin-I isoform that is associated with deafness (Citation95,Citation96) and found to be causative in acute monocytic leukemia (Citation103). MYO1H is found highly expressed in the testis, with lower expression in adipocytes and heart tissue (Citation105) and has been implicated as a marker associated with the mandibular prognathism phenotype (Citation99). Although loss of Myosin-1 motors is associated with many disorders, there is evidence that partial rescue and overlapping functions by closely related Myosin-1 isoforms minimizes the observed cellular and whole-animal knockdown phenotypes (Citation77, Citation106–108), consistent with well-documented examples in lower eukaryotes.
Table 1. Tissue expression patterns and proposed function of various Myosin 1 isoforms in vertebrates
Myosin motor proteins in actin mediated protein transport
In most vertebrate cells, it is proposed that force production for cell shape change is initiated by actin filaments or microtubules, or both. Being highly asymmetric, both rod and cone photoreceptor cells have highly polarized and paraxially aligned actin and microtubule cytoskeletons. From the distal end of the IS, microvillus-like calyceal processes extend up to and form structures around the base of the OS. Paraxial bundles of actin filaments have been shown to form the cores of the calyceal processes, which then continue through the peripheral cytoplasm of the ellipsoid, and then splay out into a longitudinally oriented, actin filament array in the photoreceptor myoid. The calyceal, ellipsoid, and myoid actin filaments are polarized and uniformly oriented with their fast-growing (plus) ends toward the OS, since each type of myosin motor can walk in only one direction along an actin filament and the polarity of actin filaments determines the vector of force production for actin-based motility. Thus, based on these observations, it is proposed that a plus-end-directed (+) myosin motor protein would walk along a photoreceptor IS actin bundle toward the OS, while a minus-end-directed (-) myosin motor protein would walk toward the nucleus.
Actin and microtubule cytoskeletons together with their associated motors and other accessory proteins, provide the cell with structure and organization. These tracks, along with molecular motors travel, are polarized, with so-called (+) ends and (−) ends, and the (+) ends of both types of tracks are directed toward the plasma membrane (Citation106). Myosin motor proteins have been proposed to regulate the dynamic organization of the plasma membrane by creating and sustaining an actin-dependent force on the cell surface and additionally by transporting and fusing membrane vesicles with it. Myosins are a large superfamily of motor proteins with more than 20 classes identified that primarily use the energy derived from ATP hydrolysis to interact and translocate along actin filaments (Citation77,Citation107). They commonly comprise three domains: (1) a conserved head responsible for actin binding, ATPase activity, and generation of movement, (2) a short neck that usually interacts with myosin light chains, and (3) a variable tail that commonly binds the motor “cargo” and determines the functional specificity of the motor (). It serves several purposes such as mediating the association of myosins with each other, anchoring myosins for movement relative to actin filaments, and specifying binding to different cargoes (Citation107). Although cells can harness the polymerization of actin to generate some forms of movement, many cellular movements depend on interactions between actin filaments and myosin, an ATPase that moves along actin filaments by coupling the hydrolysis of ATP to conformational changes. Therefore, this type of enzyme, which converts chemical energy into mechanical energy, is called a mechanochemical enzyme or, colloquially, a motor protein. Myosin is the motor, actin filaments are the tracks along which myosin moves, and ATP is the fuel that powers movement (). Of the more than 18 classes of myosins, only the class VI myosins have been found to be minus-end-directed motors. All other classes are plus-end directed. Myosin IC, IIA, IIIA, IIIB, VI, VIIA, and IXB have all been shown to be present in fish photoreceptor IS (Citation108). Thus, both plus- and minus-end-directed motors are present in or near the regions of elongation and contraction.
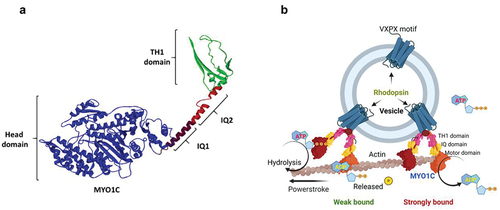
Figure 2. Mode of action for the unconventional motor protein MYO1C. (a) Cartoon model showing the structure of MYO1C with secondary structures and domains. (b) A schematic model showing the possible mechanistic trafficking pathway of Rhodopsin embedded vesicles via MYO1C interaction with actin filaments. The model was created with BioRender.com.
Unconventional motor proteins in protein transport and localization
Myosin VII in retinal cell physiology
The role for unconventional motor proteins in photoreceptor protein transport was first established for Myosin VIIA (Citation109). The unconventional motor protein Myosin VIIa (MYO7A) is a large putative actin-based mechanoenzyme present in various tissues. It is a common component of cilia and actin-rich microvilli, where its critical function appears to be associated with the retina and inner ear. MYO7A has been shown to be expressed in the RPE (Citation31,Citation35,Citation109). MYO7A competitively targets RPE melanosomes and aids in its localization towards the apical membrane. It is also involved in the internalization of phagosomes and their transport to lysosomes in RPE. MYO7A has also been associated with RPE65 an enzyme involved in the formation of the retinoid chromophore, illustrating the involvement of a molecular motor in the spatiotemporal organization of the retinoid cycle in vision (Citation110). Mutations in MYO7A have been linked to retinal cell degeneration in diseases like Usher Syndrome (Citation111). It has recently been reported that in the absence of MYO7A, opsins accumulate in the cilia connecting the photoreceptor cells, although the exact role of MYO7A remains unclear (Citation46).
Usher Syndrome (USH)
Usher Syndrome (USH) is a genetically heterogeneous form of deaf-blindness disease, affecting the neurosensory cells in the cochlea, the retina, and for some clinical sub-types, the vestibular system. USH is an inherited autosomal recessive trait that is characterized by irreversible hearing loss and progressive rod-cone dystrophy due to retinitis pigmentosa (Citation112,Citation113). Early reports indicated a prevalence of 3.6 to 6.2 individuals per 100,000 in the general population. However, the prevalence of USH in the general population is predicted to be as high as 16.7 per 100,000 individuals (Citation114). USH has three clinical subtypes, which are distinguished by their phenotypic differences in the onset and severity of hearing loss and the presence of vestibular dysfunction. The common ophthalmic manifestation of these three subtypes is the retinal degeneration (RD) classified as rod-cone dystrophy (RCD) or RP (Citation31,Citation35). Type 1 (USH1) manifests with profound congenital sensorineural hearing loss (SNHL), peripheral vestibular areflexia, and adolescent onset of RP (Citation31). Type 2 (USH2) manifests as mild to severe congenital SNHL and RP, but not vestibular dysfunction (Citation113,Citation115). Type 3 (USH3) results in progressive SNHL, RP, and variable vestibular dysfunction (Citation113,Citation115). More recently, a less defined clinical subtype termed atypical Usher Syndrome (atypical USH) has been proposed to include USH phenotypes that do not meet the canonical criteria for USH1, USH2 or USH3. While clinical reports of atypical USH vary, several of the cases described involve a combination of hearing loss and cone-rod dystrophy (CRD). Atypical USH is related to the degree of severity of the clinical findings, where USH patients manifest with milder presentations (Citation116).
Unconventional motor protein myosin VII in Usher syndrome
MYO7A is also one of the well-characterized Usher Syndrome 1 (USH1) genes, which has been linked to atypical USH (Citation46). It has been suggested that cellular defects involving the unconventional motor protein, MYO7A, may be responsible for retinal cell degeneration in diseases like Usher syndrome (Citation112,Citation113). Mutations in MYO7A have been associated with the cytoskeletal defects in RPE that could lead to excessive opsin accumulation in photoreceptor cells, defects in RPE phagocytosis, leading to progressive retinal cell degeneration (Citation117).
Unconventional myosins in photoreceptor protein transport and physiology
Role of myosin VIIa in Retinal Physiology
Myosin VIIa is expressed in cochlea, retina, testes, lungs, and kidneys (Citation109). In the retinas of vertebrates, myosin VIIa is localized primarily in the retinal pigment epithelium (Citation109). However, it has also been reported in photoreceptor (PR) cells in lower amounts (Citation118). Within PR cells, myosin VIIa is supposed to localize in connecting cilium region participating in the transport of opsins to the OS (Citation118,Citation119). Interestingly, localization of myosin VIIa in mouse was only in RPE whereas, in humans and macaques, it was additionally found to be localized in IS of photoreceptor (PR) cells as well (Citation120). This phenotypic difference might be the reason for the absence of any major retinal defects in Usher’s mouse model Shaker-1, which has well-studied myosin VIIa mutations (Citation121,Citation122). Furthermore, in RPE of shaker-1 mice, melanosomes abnormally accumulated in the perinuclear region (Citation123–125) but melanocytes from RPE of MYO7A null mice still showed motility indicating compensatory action probably from other myosin proteins (Citation124). In Myo7a-/- mice with GFP tagged Rhodopsin, Rhodopsin was mislocalized in IS of PR cells at 2-month age (Citation126). It has also been demonstrated that myosin VIIa protein from human retinal tissue is a calmodulin binding protein and is stimulated by F-actin (Citation127). While myosin VIIa is known to be in monomer form in Drosophila (Citation128,Citation129), a study by Sakai et.al, showed the ability of myosin VIIa to form a dimer near the membrane in the presence of cargo-molecules, which would demonstrate its ability to transport proteins (Citation130).
(b) Role of other Myosin’s in Retinal Physiology
Mutations in myosin V in mice have been reported to cause anatomical as well as neurological abnormalities. In retinas, myosin V is predominantly expressed PR cells where it is localized to the pre-synaptic terminals and IS (Citation131) indicating a role in neurotransmission. In addition, mutations in myosin V caused defects in ERG in the form of reduced a-wave and b-wave amplitudes (Citation132). Another myosin, myosin 6, mutations of which have been identified in autosomal recessive nonsyndromic deafness (DFNB37). Myosin 6 is highly expressed in the IS of PR cells in mice. Mutations in myosin 6 have been associated with reduced a-wave and b-wave amplitudes in ERG (Citation133) but no degeneration of photoreceptor cells was observed (Citation104). Within RPE, myosin 6 localizes to the periphery where it has been associated with endocytic vesicular trafficking (Citation134,Citation135).
MYO1C expression among various species
As mentioned above, the proper sorting and compartmentalization of proteins to specific cellular compartments of the photoreceptor cell are critical for effective phototransduction and survival. Active transport mechanisms can generate protein concentration gradients within different regions of a contiguous plasma membrane (Citation36). Rhodopsin concentration is high in open discs but low within the PM of connecting cilia, in which molecular motors, such as kinesin-2 (Citation136) and myosin VIIa (Citation119) are proposed to transport Rhodopsin. MYO1C (previously annotated as MYO1β) belongs to the group of proteins called unconventional myosins. Most of the myosins in this group interact with actin and maintain cellular motility and morphology through phagocytosis, endocytosis, vesicular transport and chemotaxis (Citation7,Citation137). The role of different myosins from this family has been studied in photoreceptor protein transport. Recent work in our laboratory has shown that the unconventional motor protein MYO1C is localized within the photoreceptor IS and OS, in vertebrates, such as zebrafish, mouse and humans (Citation84). This indicates that MYO1C might play a role in photoreceptor protein trafficking, localization of proteins, disc formation and therefore for proper visual function.
Motor protein MYO1C in deafness
Previously, MYO1C was found to be expressed near the upper tip link region in the stereocilia of the hair cells in the inner ear (Citation85,Citation89,Citation138). It was therefore suggested that MYO1C participates in the adaption of the hair cells (Citation85,Citation89). Recently, missense mutations in MYO1C have been reported in patients with bilateral sensorineural hearing loss (Citation90,Citation91). Some of these mutations were identified in sites associated with actin binding. Homology modeling showed that the MYO1C mutation V252A was functionally pathogenic due to reduced actin binding (Citation90,Citation91). With the recent establishment of MYO1C in visual function in a murine model, it would be fascinating to investigate in the future if these mice also suffer from bilateral sensorineural hearing loss and establish if the mechanism(s) are correlative to the human condition (Citation84,Citation90,Citation91).
MYO1C function in Rhodopsin localization within photoreceptors
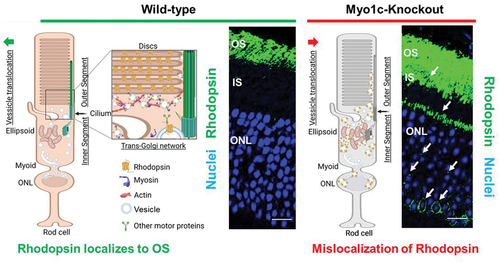
Our recent work identified MYO1C as a novel regulator for proper Rhodopsin localization to rod photoreceptor OS (Citation84). In co-immunoprecipitation assays using retinal lysates from mice, we identified opsins as novel cargo for MYO1C. In the absence of MYO1C, while Rhodopsin was still able to localize to OS, retinas of Myo1c−/− mice showed progressively severe Rhodopsin mislocalization and retention in the photoreceptor IS and the cell bodies (). Significantly, the global genetic deletion of Myo1c affected the retina, while other systemic organs analyzed remained unaffected. Our study identified for the first time an unconventional motor protein, MYO1C, as an essential component of mammalian photoreceptors, where it plays a canonical role in promoting proper opsin localization and for maintaining normal visual function ().
Figure 3. Proposed role for MYO1C in retinal cell physiology. The impact of the unconventional motor protein MYO1C for Rhodopsin localization to photoreceptor outer segments (OS) was established recently (Citation84). The model shows the effects of Myo1c deletion on the distribution of Rhodopsin in photoreceptor inner segments (IS) and outer nuclear layers (ONL). The model was created with BioRender.com. (a) MYO1C regulates the localization of Rhodopsin to the photoreceptor OS. The interaction between MYO1C and Rhodopsin mediates their translocation to photoreceptor compartments using actin as tracks. (b) The loss of MYO1C results in Rhodopsin mislocalization, leading to a retinal diseased phenotype like those observed in C’-terminal Rhodopsin mutants, in relevant mouse models.
MYO1C predictive interaction with Rhodopsin
Protein–protein interactions in vesicle trafficking play an important role in maintaining orientation-based transport of vesicle in the three-dimensional cytoplasmic content. In order to reach vesicles to the targeted region on the plasma membrane or the OS of photoreceptor cells, the cellular positions in cell machinery of adherents like integrins and surface adhesion signaling proteins play an essential role (Citation108). To position the cellular coordinates for this inner vesicle trafficking and to maintain accurate balances, actin filaments in the cytoplasm rearrange based on focal adhesion complexes bound to the integrin cytoplasmic region. The binding of integrin to the extracellular matrixes proteins like fibronectins makes a shift in cytoplasmic signaling and cytoplasmic matrix remodeling. The conventional and unconventional myosins are a family of proteins that participate in the muscle contraction, remodeling the cytoplasmic matrix, and vesicle trafficking. Myosin family proteins have evolutionary conserved sites and domains intra and inter species. Myosin-1 (MYO1) represents a mechanical link between the membrane and actin-cytoskeleton in animal cells. These molecular motors/proteins have an ATPase motor domain that can bind to the ATP and hydrolyze based on the required binding with the actin filament. Besides carrying the motor domain, the C-terminal region of different myosins defines its unique role in cell, by having unique combinations of protein–protein and protein–lipid interacting domains.
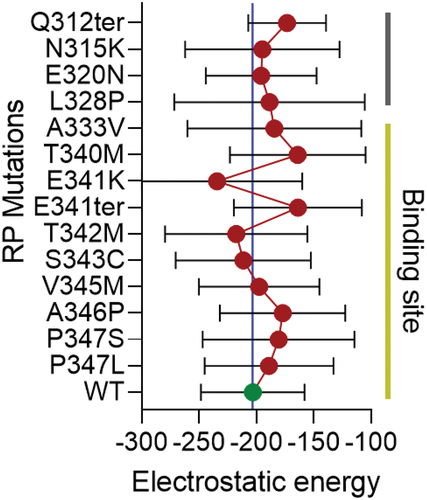
MYO1C protein belongs to the unconventional myosin family protein that contains the evolutionarily conserved motor domain for ATP hydrolysis and as per the UniProt database has two IQ domains, a post IQ domain, and one TH1 domain at its C-terminal domain. IQ domains are around 25 amino acids with consensus sequence of VQxxxRGxxxR, which helps in binding with calmodulin in absence of Ca2+. Whereas the TH1 domain or Class 1 tail homology domain features embedded pleckstrin-homology (PH) domain capable of binding to lipid membranes (). While the mechanisms facilitating the binding of MYO1C with Rhodopsin in photoreceptor function is still under investigation, our in-silico studies suggest that the binding of MYO1C with Rhodopsin likely occurs within the C-terminus sites of both proteins, where Rhodopsin has a conserved VXPX motif that is necessary for its localization to the OS () (Citation27). Our in-silico modeling studies suggest a possible interaction between MYO1C (amino acid residues 779–863) and Rhodopsin (amino acid residues 233–348) occurs at the C-terminal of both proteins ()). The type of interaction they carry is hydrogen bonding and salt bridge formation at the IQ domains of MYO1C and serine and threonine residues near VXPX region of Rhodopsin (). The VXPX region in the cytosolic over-hang of rRhodopsin makes it more available for protein–protein interaction and tethering for the vesicle trafficking. While MYO1C has an N-terminal motor domain for ATP hydrolysis and binding with actin filament, the C-terminal domains IQ and TH1 helps in protein and lipid binding (). Interestingly, known C-terminal Rhodopsin mutations that cause RP, fall within the putative MYO1C-Rhodopsin binding domain and are predicted to alter the electrostatic energy surface potential between the two proteins (). These predictions of protein–protein interaction between MYO1C and rRhodopsin requires a thorough experimental examination to understand the importance and implication of these interactions in human retinal diseases ( and ). There, in-cell condition where proteins form thousands of interaction combinations, it will be fascinating to understand under what circumstances MYO1C recognizes/interacts with the C-terminal end of Rhodopsin and binds to it. Recently, in a zebrafish model, the natural compound pentachloropseudilin (PClP) was shown to act as a reversible and allosteric inhibitor of myosin-I ATPase and motor activity (Citation145,Citation146). Thus, in the future, it would be fascinating to study the in vivo effects of PCIP on MYO1C mediated Rhodopsin trafficking in zebrafish and mouse photoreceptors.
Table 2. Putative MYO1C and rhodopsin active-binding residues. Detailed analysis of protein–protein interacting amino acid residues between the two proteins (see ) is shown below. The respective domains in MYO1C are indicated. The amino acid residues of functional interest in Rhodopsin are indicated. The mutations specific to Rhodopsin are available in the Human Gene Mutation Database http://www.hgmd.cf.ac.uk/ and the post-translational modifications and sequence information of Rhodopsin are available in the UniProt database https://www.uniprot.org/uniprot/P08100. RP, retinitis pigmentosa
Figure 4. Homology modeling of MYO1C and Rhodopsin and their predicted C-terminus interaction. (a) The bovine Rhodopsin structure in lipid bilayer and possible sites of anchoring are shown in distortion view from http://memprotmd.bioch.ox.ac.uk (Citation139). (b) The predicted structure co-ordinates of MYO1C was obtained from the PDB database. (c) Docking of MYO1C to Rhodopsin. The complete structure of both MYO1C and Rhodopsin was obtained from https://alphafold.ebi.ac.uk/ (Citation140).
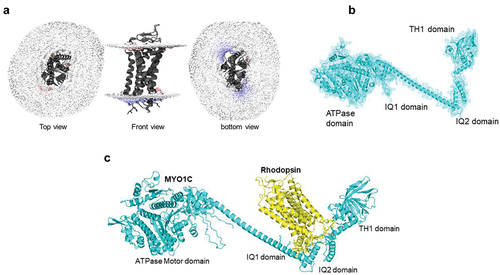
Figure 5. Protein docking and predicted active sites between MYO1C and Rhodopsin. (a, b) MYO1C and Rhodopsin structure shown in cartoon and sticks view. The active interaction interface was marked and domains of MYO1C are indicated by python script and PyMOL command line. The amino acid residues in active site annotated in yellow for Rhodopsin and Cyan for MYO1C. The putative active binding site of MYO1C -Rhodopsin interaction was obtained in the PyMOL Molecular Graphics System, Version 2.0 Schrodinger, LLC. The protein-protein docking analysis was performed using HADDOCK 2.4 (Citation141,Citation142). (c) The active interaction interface amino acid residues of Rhodopsin and MYO1C showing amino acid residues with a spectrum of hydrophilicity and hydrophobicity. The protein-protein docking analysis was performed using HADDOCK 2.4 (Citation141). The hydrogen bond and hydrophobic interactions plays a critical role in stabilizing the complex. (d) To understand the residual contributions in terms of hydrophobicity or hydrophilicity the MYO1C and Rhodopsin molecule complex was surface visualized with Discovery studio visualizer version v21.1.0.20298 (BIOVIA, San Diego, CA, USA) (Citation143). The complete structure of both MYO1C and Rhodopsin was acquired using Swiss homology modeling (Citation144) and target human protein sequences of MYO1C (UniProt: O00159) and Rhodopsin (UniProt: P08100) and template of available PDB structures of Homo sapiens MYO1C (4BYF), Mus musculus MYO1C tail (4R8G), and for Rhodopsin, PDB structure of Bos Taurus Rhodopsin (2PED).
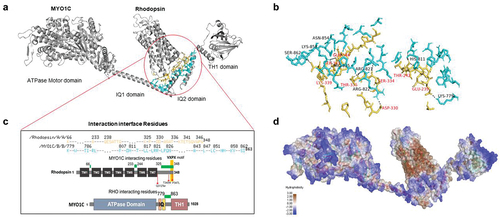
Figure 6. Electrostatic surface potential of individual WT and mutant Rhodopsin molecules and in complex with MYO1C. The bar denotes and marks the surface colors fixed at −5 red and +5 blue, displaying the charge distribution in the surface map of the structure. The composition of amino acid residues determines the final charge distribution, hence it makes a significant contribution in the protein interactions. (a) The C-terminus overhang of WT-Rhodopsin has a conserved VXPX ciliary targeting motif, the panel shows the charge distribution of the entire amino acid residues accessible for cytosolic proteins interaction, on the bottom surface of Rhodopsin. The rotated 3D structure of WT-Rhodopsin shows a side view of it and the overhang with an asterisk symbol for the reference site. The MYO1C-WT Rhodopsin docking structure shows a charge distribution on the surface and the rotated image shows the alignment of Rhodopsin interaction under wild-type conditions. (b) The C-terminus overhang of Rho-T340M mutant has a conserved VXPX ciliary targeting motif in close proximity. The panel shows the charge distribution of the amino acid residues accessible for cytosolic proteins interactions on the bottom surface of T340M. The rotated 3D structure of T340M shows a side view of it and the overhang with an asterisk symbol for the mutation and sidechain of Leucine. The MYO1C-Rho T340M mutant docking structure shows a charge distribution on the surface with the rotated image are showing the alignment difference of T340M interaction. (c) The C-terminus overhang of Rho-P347L has a point mutation at the conserved VXPX ciliary targeting motif on the P residue. The panel shows the charge distribution of the amino acid residues accessible for cytosolic proteins interactions on the bottom surface of Rho-P347L. The rotated 3D structure of Rho-P347L shows a side view of it and the overhang with an asterisk symbol for the mutation and sidechain of Leucine. The MYO1C-P347L docking structure shows a charge distribution on the surface with the rotated image are showing the alignment difference of P347L interaction. (d) The C-terminus overhang deleted from Q312Ter mutation the conserved VXPX chain is not available. The panel shows the charge distribution of the amino acid residues accessible for cytosolic proteins interactions on the bottom surface of Q312Ter. The rotated 3D structure of Q312Ter shows a side view of it and with the reduced overhang. The MYO1C- Q312Ter docking structure shows a charge distribution on the surface with the rotated image are showing the alignment difference of Q312Ter interaction under mutant condition. The mutant Rhodopsin structures generated from the mutagenesis wizard in PyMol.
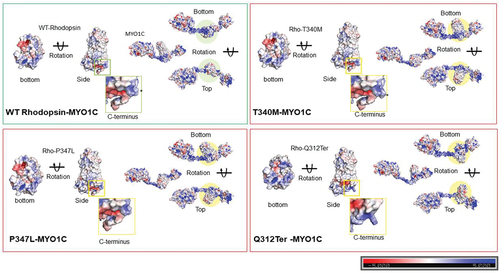
Figure 7. Distribution of electrostatic energy of mutant Rhodopsin interaction with MYO1C. In-silico analysis showed a possible interaction domain of MYO1C and Rhodopsin via the C-terminal overhang of Rhodopsin near the conserved ciliary targeting VXPX motif (). To further understand the contribution of the possible MYO1C and Rhodopsin interaction in disease phenotype, fourteen known C-terminal overhang mutations of Rhodopsin were modeled using the PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC mutagenesis wizard. The docking was performed with HADDOCK 2.4 webserver (Citation141). HADDOCK clustered ~80-100 structures in ~10 cluster(s), and the average electrostatic energy from all 10 clusters with standard deviations plotted for all 15 conditions. The distribution of electrostatic energy implicated in the protein interactions and the changes of electrostatic energy in Rhodopsin mutants in the interactions with MYO1C showed possible distortions and deviations from WT Rhodopsin-WT MYO1C (Citation94). RP, Retinitis Pigmentosa.
Possible mechanism of photoreceptor cell phenotypes in Myo1c−/− animals
The carboxyl-terminus of Rhodopsin has been shown to contain a VXPX ciliary targeting/sorting motif, which has been proposed to associate with cytoplasmic proteins that support its transport and trafficking from the site of synthesis the photoreceptor IS to the OS (Citation25,Citation52). In vitro evidence in transgenic mice, together with the occurrence of naturally occurring mutations in humans with RP point to the carboxyl-terminal VXPX motif for interaction with other transport proteins (Citation24,Citation26,Citation147,Citation148). Consistent with this idea, C-terminal Rhodopsin mutants described above (Q344ter, P347S, P347L), either lacking this motif or having variants, exhibit a trafficking defect, where rRhodopsin localized not only to the rod OS but also abnormally accumulated in the IS, cell bodies, and ONL compartments. Based on these previous findings and our observations that absence of MYO1C causes Rhodopsin mislocalization, allow us to formulate the hypothesis that mislocalized and un-liganded Rhodopsin in MYO1C deficient retinas would become accessible to proteins not normally encountered by Rhodopsin and initiate a aberrant signaling pathway in the IS/ONL compartments that leads to accelerated OS degeneration. Such examples could be the activation of transducin or an unknown G-protein. Photoreceptor OS loss and eventual cell death in the Myo1c−/− mice retina could be attributed to either one of the two consequences of this primary defect, or their combination. The rate of photoreceptor disc renewal may be reduced due to the progressive loss of material to the extracellular space (in this case Rhodopsin, the most abundant OS protein), hence the progressive shortening of rod OS observed in Myo1c−/− retinas (Citation84). Maintenance of the photoreceptor OS at a proper length is likely to be required for cell survival as illustrated by the photoreceptor cell degeneration in rds mutant mice. Loss of cellular contents to the extracellular space may additionally contribute to the photoreceptor phenotypes in the absence of MYO1C. Thus, the source of photoreceptor phenotypes in the Myo1c−/− retinas could be related to the excessive mislocalization of opsin protein in the cell body, perhaps leading to excessive metabolic burden associated with its mistrafficking. Thus, in line with other Rhodopsin C-terminus mutant models, we predict that any opsin mislocalization/mistrafficking that result in accumulation in the photoreceptor cell body would impair cell viability and long-term survival (Citation26,Citation147,Citation148). To test these hypotheses and to understand the cellular events in Myo1c−/− mice retinas, we have begun to profile the retinas of 2- and 6-month-old Myo1c−/− mice using RNA-seq transcriptome analysis. We also aim to confirm the transcriptome data in rod specific (Rho-Cre+) and cone specific (HGRP-Cre+)—conditional MYO1C deficient mice.
Conclusions
For proteins to perform specific biological functions, they must first target to the proper subcellular compartment. Therefore, revealing the mechanisms that control proper protein localization and trafficking in the photoreceptor cells is critical for the understanding of visual retina function and pathologies involved in inherited retinal cell degeneration. The recent discovery of an unconventional motor protein, MYO1C, as an essential component of Rhodopsin (RHO) localization to rod OS, uncovers a previously unknown role for unconventional motor proteins in protein transport in photoreceptor cells. Based on our recent work and those in other laboratories, it appears that unconventional myosins are dispensable for the protein transport of most outer-segment resident proteins, but critical for proper Rhodopsin localization to photoreceptor OS. In a similar manner, in Myo1c−/− mice, Rhodopsin was the only OS resident protein found to be mislocalized in rod photoreceptors. Our in silico analysis further suggests that the C’-terminal domain in MYO1C that contains IQ-protein interacting domains might interact with the C’-terminal domain of Rhodopsin, which contains a previously identified VXPX OS targeting sequence ( and ) that may explain why in Myo1c−/− animals significant amounts of Rhodopsin was found to be mislocalized. Consistent with this hypothesis, mouse models of C-terminal Rhodopsin mutants, described in this manuscript, exhibit a trafficking defect, where Rhodopsin mislocalized and abnormally accumulated in the photoreceptor IS, cell bodies, and ONL compartments. Although, this hypothesis requires further experimental evidence, in the future it will be appropriate to screen for Myo1c mutations and variants, specifically in its C’-terminal IQ domain, in patients diagnosed with RP and/or Usher Syndrome.
Author contributions
Conceptualization, G.P.L.; methodology, G.P.L., R.R.; software, R.R., A.K.S.; validation, R.R., V.R.D., G.P.L.; formal analysis, R.R., V.R.D., G.P.L., A.A.K., G.S., S.C., E.V-K., H.R., S.R.M., S.W., R.B.H; investigation, R.R., V.R.D., G.P.L., G.S., S.C., E.V-K., S.W., S.R.M., R.B.H; resources, G.P.L., G.S., S.C., E.V-K., S.R.M., R.B.H.; data curation, G.P.L.; writing-original draft preparation, G.P.L., H.-J.K., R.M., R.R., V.R.; manuscript writing, review, and editing, G.P.L., A.A.K., H.-J.K., V.R.D., S.C., E.V-K., S.R.M., R.B.H; supervision, G.P.L.; project administration, G.P.L.; funding acquisition, G.P.L. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
We thank Dr Beata Jastrzebska, Ph.D. (Department of Pharmacology, Case Western Reserve University, OH, USA) for a critical review of this manuscript. We also thank Dr Deepak Nihalani, Ph.D. (NIH/NIDKK) for helpful discussions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Mustafi D, Engel AH, Palczewski K. Structure of cone photoreceptors. Prog retin eye res. 2009;28(4):289–302. doi:https://doi.org/10.1016/j.preteyeres.2009.05.003.
- Wensel TG. Signal transducing membrane complexes of photoreceptor outer segments. Vision Res. 2008;48(20):2052–61.
- Österberg G. Topography of the layer of rods and cones. Acta Ophthalmol. 1935;13(6):1–96.
- Curcio CA, Sloan KR, Kalina RE, Hendrickson AE. Human photoreceptor topography. J Comparative Neurol. 1990;292(4):497–523.
- Kryger Z, Galli-Resta L, Jacobs GH, Reese BE. The topography of rod and cone photoreceptors in the retina of the ground squirrel. Vis neurosci. 1998;15(4):685–91.
- Hofmann L, Palczewski K. Advances in understanding the molecular basis of the first steps in color vision. Prog retin eye res. 2015;49:46–66.
- Stewart EEM, Valsecchi M, Schütz AC. A review of interactions between peripheral and foveal vision. J Vis. 2020;20(12):2. doi:https://doi.org/10.1167/jov.20.12.2
- Wikler KC, Rakic P. Distribution of photoreceptor subtypes in the retina of diurnal and nocturnal primates. J Neurosci. 1990;10(10):3390–401.
- Sun C, Zhou J, Meng X. Primary cilia in retinal pigment epithelium development and diseases. J Cell Mol Med. 2021 Oct;25(19):9084–88. doi:https://doi.org/10.1111/jcmm.16882.
- Sjostrand FS. The ultrastructure of the inner segments of the retinal rods of the Guinea pig eye as revealed by electron microscopy. J Cell Comparative Physiol. 1953;42(1):45–70. doi:https://doi.org/10.1002/jcp.1030420104.
- Wang JS, Kefalov VJ. The cone-specific visual cycle. Prog retin eye res. 2011;30(2):115–28.
- Carter-Dawson LD, Lavail MM. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comparative Neurol. 1979;188(2):245–62.
- Stone J, van Driel D, Valter K, Rees S, Provis J. The locations of mitochondria in mammalian photoreceptors: relation to retinal vasculature. Brain Res. 2008;1189:58–69.
- Imanishi Y. Protein sorting in healthy and diseased photoreceptors. Ann Rev Vis Sci. 2019;5:73–98.
- Bales KL, Gross AK. Aberrant protein trafficking in retinal degenerations: the initial phase of retinal remodeling. Exper Eye Res. 2016;150:71–80.
- Young RW. Biogenesis and renewal of visual cell outer segment membranes. Exper Eye Res. 1974;18(3):215–23.
- Young RW, Bok D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol. 1969;42(2):392–403.
- Long KO, Fisher SK, Fariss RN, Anderson DH. Disc shedding and autophagy in the cone-dominant ground squirrel retina. Exper Eye Res. 1986;43(2):193–205.
- Hogan MJ, Wood I, Steinberg RH. Phagocytosis by pigment epithelium of human retinal cones. Nature. 1974;252(5481):305–07.
- Jastrzebska B, Fotiadis D, Jang GF, Stenkamp RE, Engel A, Palczewski K. Functional and structural characterization of Rhodopsin oligomers. J Biol Chem. 2006;281(17):11917–22.
- Jastrzebska B. GPCR: g protein complexes–the fundamental signaling assembly. Amino Acids. 2013;45(6):1303–14.
- Nemet I, Ropelewski P, Imanishi Y. Rhodopsin trafficking and mistrafficking: signals, molecular components, and mechanisms. Prog Molecular Biol Translational Sci. 2015;132:39–71.
- Orban T, Jastrzebska B, Palczewski K. Structural approaches to understanding retinal proteins needed for vision. Current Opinion Cell Biol. 2014;27:32–43.
- Deretic D, Schmerl S, Hargrave PA, Arendt A, McDowell JH. Regulation of sorting and post-Golgi trafficking of Rhodopsin by its C-terminal sequence QVS(A)PA. Proc Nat Academy Sci USA. 1998;95(18):10620–25.
- Mazelova J, Astuto-Gribble L, Inoue H, Tam BM, Schonteich E, Prekeris R, Moritz OL, Randazzo PA, Deretic D. Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 2009;28(3):183–92. doi:https://doi.org/10.1038/emboj.2008.267
- Sung CH, Makino C, Baylor D, Nathans J. A Rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci. 1994;14(10):5818.
- Hollingsworth TJ, Gross AK. Chapter one - Defective trafficking of rhodopsin and its role in retinal degenerations. In: Jeon KW, editor. International review of cell and molecular biology. Vol. 293. Cambridge (MA): Academic Press; 2012. p. 1–44.
- Fanelli F, Felline A, Marigo VP. Structural aspects of rod opsin and their implication in genetic diseases. Flugers Arch. 2021 Sep;473(9):1339–59. PMID: 33728518. Review.
- Wang J, Morita Y, Mazelova J, Deretic D. The Arf GAP ASAP1 provides a platform to regulate Arf4- and Rab11-Rab8-mediated ciliary receptor targeting. EMBO J. 2012;31(20):4057–71.
- Arshavsky VY, Burns ME. Photoreceptor signaling: supporting vision across a wide range of light intensities. J Biol Chem. 2012;287(3):1620–26.
- Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11:273–84.
- Jones BW, Marc RE. Retinal remodeling during retinal degeneration. Exper Eye Res. 2005;81(2):123–37.
- Gorbatyuk MS, Starr CR, Gorbatyuk OS. Endoplasmic reticulum stress: new insights into the pathogenesis and treatment of retinal degenerative diseases. Prog retin eye res. 2020;79:100860.
- Concepcion F, Chen J. Q344ter mutation causes mislocalization of rhodopsin molecules that are catalytically active: a mouse model of Q344ter-Induced retinal degeneration. PLOS ONE. 2010;5(6):e10904.
- Athanasiou D, Aguila M, Bellingham J, Li W, McCulley C, Reeves PJ, Cheetham ME. The molecular and cellular basis of Rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog Retin Eye Res. 2018;62:1–23. doi:https://doi.org/10.1016/j.preteyeres.2017.10.002.
- Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, Hoyng CB, Roepman R, Klevering BJ. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–86. doi:https://doi.org/10.1016/j.preteyeres.2018.03.005.
- Sengillo JD, Cabral T, Schuerch K, Duong J, Lee W, Boudreault K, Xu Y, Justus S, Sparrow JR, Mahajan VB, et al. Electroretinography Reveals difference in cone function between syndromic and nonsyndromic USH2A patients. Sci Reports. 2017;7(1):11170. doi:https://doi.org/10.1038/s41598-017-11679-y.
- Ortega JT, Jastrzebska B. Rhodopsin as a molecular target to mitigate retinitis pigmentosa. Adv Exp Med Biol. 2022;1371:61–77. doi:https://doi.org/10.1007/5584_2021_682.
- Malanson KM, Lem J. Rhodopsin-mediated retinitis pigmentosa. Prog Molecular Biol Translational Sci. 2009;88:1–31.
- Gross AK, Xie G, Oprian DD. Slow binding of retinal to Rhodopsin mutants G90D and T94D. Biochemistry. 2003;42(7):2002–08.
- Kiser PD, Palczewski K. Retinoids and retinal diseases. Ann Rev Vis Sci. 2016;2(1):197–234.
- Wan A, Place E, Pierce EA, Comander J. Characterizing variants of unknown significance in Rhodopsin: a functional genomics approach. Human Mutation. 2019;40(8):1127–44.
- Tam BM, Moritz OL, Papermaster DS. The C terminus of peripherin/rds participates in rod outer segment targeting and alignment of disk incisures. Molecular Biol Cell. 2004;15(4):2027–37.
- Sung CH, Davenport CM, Hennessey JC, Maumenee IH, Jacobson SG, Heckenlively JR, Nowakowski R, Fishman G, Gouras P, Nathans J. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Nat Academy Sci USA. 1991;88(15):6481–85. doi:https://doi.org/10.1073/pnas.88.15.6481.
- Sung CH, Davenport CM, Nathans J. Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa. Clustering of functional classes along the polypeptide chain. J Biol Chem. 1993;268(35):26645–49.
- Liu XZ, Hope C, Walsh J, Newton V, Ke XM, Liang CY, Xu LR, Zhou JM, Trump D, Steel KP, et al. Mutations in the myosin VIIA gene cause a wide phenotypic spectrum, including atypical Usher syndrome. Am J Human Genetics. 1998;63(3):909–12. doi:https://doi.org/10.1086/302026.
- Sakami S, Maeda T, Bereta G, Okano K, Golczak M, Sumaroka A, Roman AJ, Cideciyan AV, Jacobson SG, Palczewski K. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem 2011;286(12):10551–67. doi:https://doi.org/10.1074/jbc.M110.209759.
- Saliba RS, Munro PM, Luthert PJ, Cheetham ME. The cellular fate of mutant Rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002;115(Pt 14):2907–18.
- Athanasiou D, Kosmaoglou M, Kanuga N, Novoselov SS, Paton AW, Paton JC, Chapple JP, Cheetham ME. BiP prevents rod opsin aggregation. Molecular Biol Cell. 2012;23(18):3522–31. doi:https://doi.org/10.1091/mbc.E12-02-0168.
- Maeda A, Okano K, Park PSH, Lem J, Crouch RK, Maeda T, Palczewski K. Palmitoylation stabilizes unliganded rod opsin. Proc Nat Academy Sci. 2010;107(18):8428. doi:https://doi.org/10.1073/pnas.1000640107.
- Tam BM, Noorwez SM, Kaushal S, Kono M, Moritz OL. Photoactivation-induced instability of Rhodopsin mutants T4K and T17M in rod outer segments underlies retinal degeneration in X. laevis transgenic models of retinitis pigmentosa. J Neurosci. 2014;34(40):13336–48.
- Deretic D, Williams AH, Ransom N, Morel V, Hargrave PA, Arendt A. Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4). Proc Nat Academy Sci USA. 2005;102(9):3301.
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Molecular Cell Biol. 2002;3(11):813–25.
- Luby-Phelps K, Fogerty J, Baker SA, Pazour GJ, Besharse JC. Spatial distribution of intraflagellar transport proteins in vertebrate photoreceptors. Vision Research. 2008;48(3):413–23.
- Tsujikawa M, Malicki J. Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron. 2004;42(5):703–16.
- Crouse JA, Lopes VS, Sanagustin JT, Keady BT, Williams DS, Pazour GJ. Distinct functions for IFT140 and IFT20 in opsin transport. Cytoskeleton (Hoboken). 2014;71(5):302–10.
- Banerjee P, Kleyn PW, Knowles JA, Lewis CA, Ross BM, Parano E, Kovats SG, Lee JJ, Penchaszadeh GK, Ott J, et al. TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nat Genet. 1998;18(2):177–79. doi:https://doi.org/10.1038/ng0298-177.
- Hagstrom SA, North MA, Nishina PM, Berson EL, Dryja TP. Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with Retinitis pigmentosa. Nature Genetics. 1998;18(2):174–76.
- Hagstrom SA, Watson RF, Pauer GJ, Grossman GH. Tulp1 is involved in specific photoreceptor protein transport pathways. Adv Exper Med Biol. 2012;723:783–89.
- Hagstrom SA, Adamian M, Scimeca M, Pawlyk BS, Yue G, Li T. A role for the tubby-like protein 1 in Rhodopsin transport. Investigative Ophthalmol Vis Sci. 2001;42(9):1955–62.
- Lobo GP, Au A, Kiser PD, Hagstrom SA. Involvement of endoplasmic reticulum stress in TULP1 induced retinal degeneration. PLoS One. 2016;11(3):e0151806.
- Lobo GP, Ebke LA, Au A, Hagstrom SA. TULP1 missense mutations induces the endoplasmic reticulum unfolded protein response stress complex (ER-UPR). Adv Exper Med Biol. 2016;854:223–30.
- Tai AW, Chuang J-Z, Bode C, Wolfrum U, Sung C-H. Rhodopsin’s carboxy-terminal cytoplasmic tail acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain tctex-1. Cell. 1999;97(7):877–87.
- Insinna C, Baye LM, Amsterdam A, Besharse JC, Link BA. Analysis of a zebrafish dync1h1mutant reveals multiple functions for cytoplasmic dynein 1 during retinal photoreceptor development. Neural Development. 2010;5(1):12.
- Krock BL, Mills-Henry I, Perkins BD. Retrograde intraflagellar transport by cytoplasmic dynein-2 is required for outer segment extension in vertebrate photoreceptors but not arrestin translocation. Investigative Ophthalmol Vis Sci. 2009;50(11):5463–71.
- Kong S, Du X, Peng C, Wu Y, Li H, Jin X, Hou L, Deng K, Xu T, Tao W. Dlic1 deficiency impairs ciliogenesis of photoreceptors by destabilizing dynein. Cell Res 2013;23(6):835–50. doi:https://doi.org/10.1038/cr.2013.59.
- Jiang M, Paniagua AE, Volland S, Wang H, Balaji A, Li DG, Lopes VS, Burgess BL, Williams DS. Microtubule motor transport in the delivery of melanosomes to the actin-rich apical domain of the retinal pigment epithelium. J Cell Sci. 2020;133(15):jcs242214. doi:https://doi.org/10.1242/jcs.242214.
- Sukumaran S, Perkins BD. Early defects in photoreceptor outer segment morphogenesis in zebrafish ift57, ift88 and ift172 Intraflagellar Transport mutants. Vision Res. 2009;49(4):479–89.
- Verhey KJ, Kaul N, Soppina V. Kinesin assembly and movement in cells. Ann Rev Biophysics. 2011;40(1):267–88.
- Jimeno D, Feiner L, Lillo C, Teofilo K, Goldstein LSB, Pierce EA. Analysis of kinesin-2 function in photoreceptor cells using synchronous Cre-loxP knockout of Kif3a with RHO-Cre. Investigative Ophthalmol Vis Sci. 2006;47(11):5039–46. doi:https://doi.org/10.1167/iovs.06-0032.
- Marszalek JR, Liu X, Roberts EA, Chui D, Marth JD, Williams DS, Goldstein LSB. Genetic evidence for selective transport of opsin and arrestin by Kinesin-II in mammalian photoreceptors. Cell 2000;102(2):175–87. doi:https://doi.org/10.1016/S0092-8674(00)00023-4.
- Lopes VS, Jimeno D, Khanobdee K, Song X, Chen B, Nusinowitz S, Williams DS. Dysfunction of heterotrimeric kinesin-2 in rod photoreceptor cells and the role of opsin mislocalization in rapid cell death. Molecular Biol Cell. 2010;21(23):4076–88. doi:https://doi.org/10.1091/mbc.e10-08-0715.
- Feng D, Chen Z, Yang K, Miao S, Xu B, Kang Y, Xie H, Zhao C. The cytoplasmic tail of Rhodopsin triggers rapid rod degeneration in kinesin-2 mutants. J Biol Chem. 2017;292(42):17375–86. doi:https://doi.org/10.1074/jbc.M117.784017.
- Avasthi P, Watt CB, Williams DS, Le YZ, Li S, Chen C-K, Marc RE, Frederick JM, Baehr W. Trafficking of membrane proteins to cone but not rod outer segments is dependent on heterotrimeric kinesin-II. J Neurosci. 2009;29(45):14287. doi:https://doi.org/10.1523/JNEUROSCI.3976-09.2009.
- Gillespie PG, Albanesi JP, Bähler M, Bement WM, Berg JS, Burgess DR, Burnside B, Cheney RE, Corey DP, Coudrier E, et al. Myosin-I nomenclature. J Cell Biol. 2001;155(5):703–04. doi:https://doi.org/10.1083/jcb.200110032.
- Ruppert C, Godel J, Müller RT, Kroschewski R, Reinhard J, Bähler M. Localization of the rat myosin I molecules myr 1 and myr 2 and in vivo targeting of their tail domains. J Cell Sci. 1995;108(Pt 12):3775–86.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biol Evolution. 2018;35(6):1547–49.
- Krendel M, Kim SV, Willinger T, Wang T, Kashgarian M, Flavell RA, Mooseker MS. Disruption of Myosin 1e promotes podocyte injury. J Am Soc Nephrol: JASN 2009;20(1):86–94. doi:https://doi.org/10.1681/asn.2007111172.
- Tyska MJ, Mackey AT, Huang JD, Copeland NG, Jenkins NA, Mooseker MS. Myosin-1a is critical for normal brush border structure and composition. Molecular Biol Cell. 2005;16(5):2443–57.
- Durrbach A, Collins K, Matsudaira P, Louvard D, Coudrier E. Brush border myosin-I truncated in the motor domain impairs the distribution and the function of endocytic compartments in an hepatoma cell line. Proc Nat Academy Sci USA. 1996;93(14):7053–58.
- Sherr EH, Joyce MP, Greene LA. Mammalian myosin I alpha, I beta, and I gamma: new widely expressed genes of the myosin I family. J Cell Biol. 1993;120(6):1405–16.
- Cordonnier MN, Dauzonne D, Louvard D, Coudrier E. Actin filaments and myosin I alpha cooperate with microtubules for the movement of lysosomes. Molecular Biol Cell. 2001;12(12):4013–29.
- Bose A, Robida S, Furcinitti PS, Chawla A, Fogarty K, Corvera S, Czech MP. Unconventional myosin Myo1c promotes membrane fusion in a regulated exocytic pathway. Molecular Cell Biol. 2004;24(12):5447–58. doi:https://doi.org/10.1128/mcb.24.12.5447-5458.2004.
- Solanki AK, Biswal MR, Walterhouse S, Martin R, Kondkar AA, Knölker H-J, Rahman B, Arif E, Husain S, Montezuma SR, et al. Loss of motor protein MYO1C causes rhodopsin mislocalization and results in impaired visual function. Cells. 2021;10(6):353–71. doi:https://doi.org/10.3390/cells10061322.
- Steyger PS, Gillespie PG, Baird RA. Myosin Ibeta is located at tip link anchors in vestibular hair bundles. J Neurosci. 1998 Jun 15;18(12):4603–15.
- Skeie JM, Mahajan VB. Proteomic Interactions in the mouse vitreous-retina complex. PLOS ONE. 2013;8(11):e82140.
- Steyger PS, Gillespie PG, Baird RA. Myosin Ibeta is located at tip link anchors in vestibular hair bundles. J Neurosci. 1998;18(12):4603–15.
- García JA, Yee AG, Gillespie PG, Corey DP. Localization of myosin-Ibeta near both ends of tip links in frog saccular hair cells. J Neurosci. 1998;18(21):8637–47.
- Holt JR, Gillespie SKH, Provance DW, Shah K, Shokat KM, Corey DP, Mercer JA, Gillespie PG. A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell 2002;108(3):371–81. doi:https://doi.org/10.1016/S0092-8674(02)00629-3.
- Adamek N, Geeves MA, Coluccio LM. Myo1c mutations associated with hearing loss cause defects in the interaction with nucleotide and actin. Cell Mol Life Sci. 2011;68(1):139–50.
- Zadro C, Alemanno MS, Bellacchio E, Ficarella R, Donaudy F, Melchionda S, Zelante L, Rabionet R, Hilgert N, Estivill X, et al. Are MYO1C and MYO1F associated with hearing loss? Biochimica Et Biophysica Acta (BBA) - Molecular Basis of Disease. 2009;1792(1):27–32. doi:https://doi.org/10.1016/j.bbadis.2008.10.017.
- Phillips KR, Tong S, Goodyear R, Richardson GP, Cyr JL. Stereociliary myosin-1c receptors are sensitive to calcium chelation and absent from cadherin 23 mutant mice. J Neurosci. 2006 Oct 18;26(42):10777–88. doi:https://doi.org/10.1523/JNEUROSCI.1847-06.2006. PMID: 17050716.
- Bähler M, Kroschewski R, Stöffler HE, Behrmann T. Rat myr 4 defines a novel subclass of myosin I: identification, distribution, localization, and mapping of calmodulin-binding sites with differential calcium sensitivity. J Cell Biol. 1994;126(2):375–89.
- Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, Buelli S, Tomasoni S, Piras R, Krendel M, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. New England J Med. 2011;365(4):295–306. doi:https://doi.org/10.1056/NEJMoa1101273.
- Baek JI, Oh SK, Kim DB, Choi SY, Kim UK, Lee KY, Lee SH. Targeted massive parallel sequencing: the effective detection of novel causative mutations associated with hearing loss in small families. Orphanet J Rare Diseases. 2012;7:60. doi:https://doi.org/10.1186/1750-1172-7-60.
- Chen AH, Stephan DA, Hasson T, Fukushima K, Nelissen CM, Chen AF, Jun AI, Ramesh A, Van Camp G, Smith RJ. MYO1F as a candidate gene for nonsyndromic deafness, DFNB15. Archives Otolaryngol–head Neck Surgery. 2001;127(8):921–25. doi:https://doi.org/10.1001/archotol.127.8.921.
- Kim Sangwon V, Mehal Wajahat Z, Dong X, Heinrich V, Pypaert M, Mellman I, Dembo M, Mooseker Mark S, Wu D, Flavell Richard A. Modulation of cell adhesion and motility in the immune system by myo1f. Science 2006;314(5796):136–39. doi:https://doi.org/10.1126/science.1131920.
- Maravillas-Montero JL, López-Ortega O, Patiño-López G, Santos-Argumedo L. Myosin 1g regulates cytoskeleton plasticity, cell migration, exocytosis, and endocytosis in B lymphocytes. Eur J Immunol. 2014;44(3):877–86.
- Tassopoulou-Fishell M, Deeley K, Harvey EM, Sciote J, Vieira AR. Genetic variation in myosin 1H contributes to mandibular prognathism. Am J Orthodontics Dentofacial Orthopedics. 2012;141(1):51–59.
- Donaudy F, Ferrara A, Esposito L, Hertzano R, Ben-David O, Bell RE, Melchionda S, Zelante L, Avraham KB, Gasparini P. Multiple mutations of MYO1A, a cochlear-expressed gene, in sensorineural hearing loss. Am J Human Genetics. 2003;72(6):1571–77. doi:https://doi.org/10.1086/375654.
- Skowron JF, Bement WM, Mooseker MS. Human brush border myosin-I and myosin-Ic expression in human intestine and Caco-2BBe cells. Cell Motility the Cytoskeleton. 1998;41(4):308–24.
- Mazzolini R, Dopeso H, Mateo-Lozano S, Chang W, Rodrigues P, Bazzocco S, Alazzouzi H, Landolfi S, Hernández-Losa J, Andretta E, et al. Brush border myosin IA has tumor suppressor activity in the intestine. Proc Nat Academy Sci USA. 2012;109(5):1530–35. doi:https://doi.org/10.1073/pnas.1108411109.
- Taki T, Akiyama M, Saito S, Ono R, Taniwaki M, Kato Y, Yuza Y, Eto Y, Hayashi Y. The MYO1F, unconventional myosin type 1F, gene is fused to MLL in infant acute monocytic leukemia with a complex translocation involving chromosomes 7, 11, 19 and 22. Oncogene 2005;24(33):5191–97. doi:https://doi.org/10.1038/sj.onc.1208711.
- Kitamoto J, Libby RT, Gibbs D, Steel KP, Williams DS. Myosin VI is required for normal retinal function. Exper Eye Res. 2005;81(1):116–20.
- Fishilevich S, Zimmerman S, Kohn A, Iny Stein T, Olender T, Kolker E, Safran M, Lancet D. Genic insights from integrated human proteomics in genecards. Database: J Biol Databases Curation. 2016;2016:1–17. doi:https://doi.org/10.1093/database/baw030.
- Ross JL, Shuman H, Holzbaur EL, Goldman YE. Kinesin and dynein-dynactin at intersecting microtubules: motor density affects dynein function. Biophysical J. 2008;94(8):3115–25.
- Foth BJ, Goedecke MC, Soldati D. New insights into myosin evolution and classification. Proc Nat Academy Sci USA. 2006;103(10):3681–86.
- Lin-Jones J, Sohlberg L, Dosé A, Breckler J, Hillman DW, Burnside B. Identification and localization of myosin superfamily members in fish retina and retinal pigmented epithelium. J Comparative Neurol. 2009;513(2):209–23.
- Hasson T, Heintzelman MB, Santos-Sacchi J, Corey DP, Mooseker MS. Expression in cochlea and retina of myosin VIIa, the gene product defective in Usher syndrome type 1B. Proc Nat Academy Sci USA. 1995;92(21):9815–19.
- Lopes VS, Gibbs D, Libby RT, Aleman TS, Welch DL, Lillo C, Jacobson SG, Radu RA, Steel KP, Williams DS. The Usher 1B protein, MYO7A, is required for normal localization and function of the visual retinoid cycle enzyme, RPE65. Human Molecular Genetics. 2011;20(13):2560–70. doi:https://doi.org/10.1093/hmg/ddr155.
- Williams DS, Lopes VS. The many different cellular functions of MYO7A in the retina. Biochem Soc Trans. 2011;39(5):1207–10.
- Vernon M. Usher’s syndrome—deafness and progressive blindness: clinical cases, prevention, theory and literature survey. J Chronic Diseases. 1969;22(3):133–51.
- Mathur P, Yang J. Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochimica et biophysica acta. 2015;1852(3):406–20.
- Davenport SLH, Omenn GS. The heterogeneity of Usher syndrome (Abstract). Vth International Conference on Birth Defects. Montreal 1977.
- Smith RJH, Berlin CI, Hejtmancik JF, Keats BJB, Kimberling WJ, Lewis RA, Möller CG, Pelias MZ, Tranebjærǵ L. Clinical diagnosis of the Usher syndromes. Am J Med Genetics 1994;50(1):32–38. doi:https://doi.org/10.1002/ajmg.1320500107.
- Nolen RM, Hufnagel RB, Friedman TB, Turriff AE, Brewer CC, Zalewski CK, King KA, Wafa TT, Griffith AJ, Brooks BP, et al. Atypical and ultra-rare Usher syndrome: a review. Ophthalmic Genetics. 2020;41(5):401–12. doi:https://doi.org/10.1080/13816810.2020.1747090.
- Well D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, et al. Defective myosin VIIA gene responsible for Usher syndrome type IB. Nature. 1995;374(6517):60–61. doi:https://doi.org/10.1038/374060a0.
- Liu X, Vansant G, Udovichenko IP, Wolfrum U, Williams DS. Myosin VIIa, the product of the Usher 1B syndrome gene, is concentrated in the connecting cilia of photoreceptor cells. Cell Motility Cytoskeleton. 1997;37(3):240–52. doi:https://doi.org/10.1002/(SICI)1097-0169(1997)37:3<240::AID-CM6>3.0.CO;2-A.
- Liu X, Udovichenko IP, Brown SD, Steel KP, Williams DS. Myosin VIIa participates in opsin transport through the photoreceptor cilium. J Neurosci. 1999;19(15):6267–74.
- El-Amraoui A, Sahly I, Picaud S, Sahel J, Abitbol M, Petit C. Human Usher 1B/Mouse shaker-1: the retinal phenotype discrepancy explained by the presence/absence of myosin VIIA in the photoreceptor cells. Human Molecular Genetics. 1996;5(8):1171–78.
- Gibson F, Walsh J, Mburu P, Varela A, Brown KA, Antonio M, Beisel KW, Steel KP, Brown SD. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature. 1995;374(6517):62–64. doi:https://doi.org/10.1038/374062a0.
- Mburu P, Liu XZ, Walsh J, Saw D, Cope M, Gibson F, Kendrick-Jones J, Steel K, Brown S. Mutation analysis of the mouse myosin VIIA deafness gene. Genes Funct. 1997;1(3):191–203. doi:https://doi.org/10.1046/j.1365-4624.1997.00020.x.
- Liu X, Ondek B, Williams DS. Mutant myosin VIIa causes defective melanosome distribution in the RPE of shaker-1 mice. Nature Genetics. 1998;19(2):117–18.
- Gibbs D, Azarian SM, Lillo C, Kitamoto J, Klomp AE, Steel KP, Libby RT, Williams DS. Role of myosin VIIa and Rab27a in the motility and localization of RPE melanosomes. J Cell Sci. 2004;117(26):6473–83. doi:https://doi.org/10.1242/jcs.01580.
- Futter CE, Ramalho JS, Jaissle GB, Seeliger MW, Seabra MC. The role of Rab27a in the regulation of melanosome distribution within retinal pigment epithelial cells. Molecular Biol Cell. 2004;15(5):2264–75.
- Calabro KR, Boye SL, Choudhury S, Fajardo D, Peterson JJ, Li W, Crosson SM, Kim M-J, Ding D, Salvi R, et al. A novel mouse model of MYO7A USH1B reveals auditory and visual system haploinsufficiencies. Frontiers Neurosci. 2019;13:1255.
- Udovichenko IP, Gibbs D, Williams DS. Actin-based motor properties of native myosin VIIa. J Cell Sci. 2002;115(Pt 2):445–50.
- Umeki N, Jung HS, Watanabe S, Sakai T, Li XD, Ikebe R, Craig R, Ikebe M. The tail binds to the head-neck domain, inhibiting ATPase activity of myosin VIIA. Proc Nat Academy Sci USA. 2009;106(21):8483–88. doi:https://doi.org/10.1073/pnas.0812930106.
- Yang Y, Baboolal TG, Siththanandan V, Chen M, Walker ML, Knight PJ, Peckham M, Sellers JR. A ferm domain autoregulates drosophila myosin 7a activity. Proc Nat Academy Sci USA. 2009;106(11):4189–94. doi:https://doi.org/10.1073/pnas.0808682106.
- Sakai T, Umeki N, Ikebe R, Ikebe M. Cargo binding activates myosin VIIA motor function in cells. Proc Nat Academy Sci USA. 2011;108(17):7028–33.
- Schlamp CL, Williams DS. Myosin V in the retina: localization in the rod photoreceptor synapse. Exper Eye Res. 1996;63(6):613–19.
- Libby RT, Lillo C, Kitamoto J, Williams DS, Steel KP. Myosin Va is required for normal photoreceptor synaptic activity. J Cell Sci. 2004;117(19):4509–15.
- Mochizuki E, Okumura K, Ishikawa M, Yoshimoto S, Yamaguchi J, Seki Y, Wada K, Yokohama M, Ushiki T, Tokano H, et al. Phenotypic and expression analysis of a novel spontaneous myosin VI null mutant mouse. Exp Anim. 2010;59(1):57–71. doi:https://doi.org/10.1538/expanim.59.57.
- Aschenbrenner L, Lee T, Hasson T. Myo6 facilitates the translocation of endocytic vesicles from cell peripheries. Molecular Biol Cell. 2003;14(7):2728–43.
- Aschenbrenner L, Naccache SN, Hasson T. Uncoated endocytic vesicles require the unconventional myosin, Myo6, for rapid transport through actin barriers. Molecular Biol Cell. 2004;15(5):2253–63.
- Trivedi D, Colin E, Louie CM, Williams DS. Live-cell imaging evidence for the ciliary transport of rod photoreceptor opsin by heterotrimeric kinesin-2. J Neurosci. 2012;32(31):10587–93.
- Münnich S, Manstein DJ. Expression, purification, crystallization and preliminary X-ray crystallographic analysis of human myosin 1c in complex with calmodulin. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013 Sep;69(Pt 9):1020–22.
- García JA, Yee AG, Gillespie PG, Corey DP. Localization of myosin-Ibeta near both ends of tip links in frog saccular hair cells. J Neurosci. 1998 Nov 1;18(21):8637–47.
- Newport TD, P SMS, Stansfeld PJ. The MemProtMD database: a resource for membrane-embedded protein structures and their lipid interactions. Nucleic Acids Res. 2018;47(D1):D390–D7.
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–89. doi:https://doi.org/10.1038/s41586-021-03819-2.
- van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, Bonvin AMJJ. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J Molecular Biol. 2016;428(4):720–25 doi:https://doi.org/10.1016/j.jmb.2015.09.014.
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–32.
- Biovia DS. Discovery studio modeling environment. 21.1.0.20298 ed. San Diego, CA, USA: Dassault Systèmes; 2017.
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. Swiss-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–w303. doi:https://doi.org/10.1093/nar/gky427.
- Chinthalapudi K, Taft MH, Martin R, Heissler SM, Preller M, Hartmann FK, et al. Mechanism and specificity of pentachloropseudilin-mediated inhibition of myosin motor activity. J Biol Chem 2011;286:29700–08.
- Gupta P, Martin R, Knölker H-J, Nihalani D, Kumar Sinha D. Myosin-1 inhibition by PClP affects membrane shape, cortical actin distribution and lipid droplet dynamics in early Zebrafish embryos. PLOS ONE. 2017;12(7):e0180301. doi:https://doi.org/10.1371/journal.pone.0180301.
- Li T, Snyder WK, Olsson JE, Dryja TP. Transgenic mice carrying the dominant Rhodopsin mutation P347S: evidence for defective vectorial transport of Rhodopsin to the outer segments. Proc Nat Academy Sci. 1996;93(24):14176.
- Bales KIL, Kennedy, Sweatt JD, Gross AK. Autosomal dominant retinitis pigmentosa Rhodopsin mutant Q344X drives specific alterations in chromatin complex gene transcription. Molecular Vis. 2018;24:153–64.