
A homeobox is a highly conserved DNA sequence of around 180 base pairs that encodes for a homeodomain, i.e. a portion of a protein that has DNA binding properties. Homeobox—containing genes are called homeogenes, and many developmental genes are indeed homeogenes (Citation1). Homeogenes HMX1, HMX2, and HMX3 belong to the homeobox family H6, are expressed during embryogenesis, and encode transcription factors that are involved in the development of sensory organs (Citation1). A homozygous frameshift microdeletion (c.215_240del) in three affected individuals (P1, P2, and P3 in ) from a Swiss consanguineous family (Citation2,Citation3) and the homozygous missense variant p.(Gln217Pro) in two affected cousins (P4 and P5) of a Pakistani consanguineous family (Citation4) were previously reported in the homeogene HMX1 (OMIM# 142992), resulting in the so—called “oculoauricular syndrome” (OAS). Another HMX1 homozygous mutation, the nonsense variant p.(Glu163Ter), was also recently identified in an Egyptian child (P6) born to consanguineous parents and with the same clinical diagnosis (Citation5). This syndrome was first described by Franceschetti and Valerio in 1945 (Citation6) and includes, among other things, colobomatous microphthalmia with corneal opacities, congenital cataract, and symmetric abnormalities of the external ear. Clinically, OAS has overlapping features with other developmental syndromes characterized by eye anterior segment dysgenesis and/or microphthalmia, as well as external ear anomalies. The oculo—auriculo-vertebral spectrum (OAVS), which comprises Goldenhar syndrome, is characterized by hemifacial microsomia, abnormal development of the ear, eye, and vertebral column (Citation7). External ear abnormalities are common and include microtia, anotia, aural atresia, preauricular tags and pits, whereas ocular features are less frequent. Limbal dermoid is the most common ocular finding, but microphthalmia, coloboma of the upper lid or of the optic disk may also be present. Moreover, heart, limb, renal, and central nervous system defects have also been observed in OAVS. Peters plus syndrome is characterized by eye anterior chamber (AC) anomalies, short limbs with broad distal extremities, variable developmental delay/intellectual disability, characteristic facial features, and cleft lip/palate (Citation8). The most common AC anomaly is Peters’ anomaly, consisting of central corneal opacification and posterior corneal defect with or without iridocorneal or lenticulocorneal adhesions. Cataract and glaucoma are common. Ear anomalies, including preauricular pits, are present in more than one third of affected individuals. CHARGE syndrome (acronym for ocular Coloboma, Heart defects, Atresia of the choanae, Retardation of growth and/or of development, Genital anomalies and Ear anomalies) is a multiple congenital anomaly condition, frequently associated with coloboma that can involve retina, optic disk, choroid or iris, as well asmicrophthalmia and outer ear anomalies (Citation9). Another important feature of CHARGE syndrome is the abnormality of semicircular canals.
Table 1. Phenotypic features of OAS patients.
Here we report on a consanguineous family from Pakistan in which we identified a HMX1 nonsense variant [NM_018942.3:c.457C>T,p(Arg153Ter)], present homozygously in two affected siblings and heterozygously in their parents, in agreement with a recessive pattern of heredity for OAS (. This new mutation was found by whole exome sequencing, followed by homozygosity mapping (Citation10) and Sanger sequencing, to validate co—segregation of the genotypes with the phenotype.
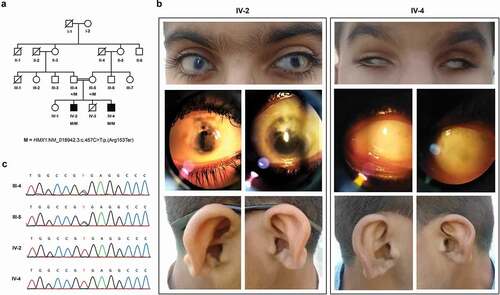
Figure 1. Family structure, clinical and molecular findings: (a) Pedigree showing segregation of the nonsense mutation (M) p(Arg153Ter) in the HMX1 gene. White and black shapes denote healthy and affected individuals, respectively; squares, males; circles, females; symbols with a diagonal line, deceased; double horizontal lines, consanguineous union. (b) Photographs of eyes and ears of the individuals examined (IV-2 and IV-4). (c) Chromatograms showing segregation of p(Arg153Ter) within the family.
Patient IV-2
The affected individual IV-2 is an 11-year—old boy from a consanguineous Pakistani family (P8 in , ). There was a history of congenital cataract for which he had undergone cataract extraction with posterior capsulotomy followed by anterior vitrectomy (intraocular lenses were not implanted). Later on, he underwent bilateral trabeculectomy because of increased intraocular pressure. He was given anti—glaucoma medications since then, although treatment was discontinued for the last six months. On examination, best corrected visual acuity was counting fingers in both eyes. There was a bilateral horizontal nystagmus, extraocular movements were full, and adnexa were normal (Supplemental Video 1). Sclerocornea of moderate degree with cornea plana was also present (). The AC was shallow with peripheral anterior synechiae; inferior iris coloboma was present in both eyes. The iris was also markedly atrophic and transillumination was positive in both eyes. Opacified margins of posterior capsulotomy were observed in both eyes. The vitreous was clear in both eyes. Fundus view of the right eye was hazy due to opacity, but a pale disc was nevertheless seen. The left eye also had a pale disc and an abnormal foveal reflex. No retinal pigmentary changes or gross vascular abnormalities were noted. Intraocular pressure was high digitally, for which an anti—glaucoma treatment was restarted. The patient presented with the abnormal appearance of external ears and dysplastic auricles ().
Patient IV-4
Another affected individual (IV-4, sibling of IV-2, P9 in ) is a 9-year—old child, blind since birth. On examination, there was microphthalmia with pseudoenophthalmos associated with severe sclerocornea ( and Supplemental Video 2). Retinal examination could not be performed due to severe sclerocornea. The external ears showed hypoplastic lobule and abnormal bridging between the crus of helix and antihelix ().
In this study, we present two patients with homozygous mutations in HMX1 and with OAS, born to healthy parents who are heterozygous for the same mutation. The homeobox transcription factor HMX1 controls the diversification of sympathetic neurons and retinal axon guidance during development (Citation11,Citation12). Its role in the formation of the eye and the external ear has also been described. Expression of HMX1 was observed in the lens of zebrafish, while knockdown animals had delayed retinal development and microphthalmia (Citation13). In addition to the eye, it has also been shown in zebrafish that HMX1 plays a role in the development of the craniofacial region, by controlling the expression of the UHRF1 and DNMT1 genes (Citation14), while in the mouse HMX1 mutations cause enlarged ear pinnae, microphthalmia, and minor craniofacial anomalies (Citation15). However, the exact mechanisms by which HMX1 mutations result in pathological phenotypes in humans is still rather unclear. It is very likely that additional factors besides HMX1 defects are involved in the molecular pathology of the disease since variability of ocular signs can be observed across patients. This phenomenon is well illustrated by the cases presented here, for whom the same genotype results in a different extent of corneal opacity. summarizes the phenotypic features of all OAS cases described so far.
In conclusion, although the number of cases reported to date does not allow for a precise genotype/phenotype association, the identification of a fourth mutation in HMX1 strongly corroborates the hypothesis that this gene is directly involved in OAS.
Supplemental Material
Download MS Power Point (9.8 MB)Acknowledgements
We thank all the family members who have participated in this study.
Disclosure statement
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/13816810.2022.2096242.
Additional information
Funding
References
- Stadler HS, Solursh M. Characterization of the homeobox-containing gene GH6 identifies novel regions of homeobox gene expression in the developing chick embryo. Dev Biol. 1994;161(1):251–62. doi:10.1006/dbio.1994.1025.
- Schorderet DF, Nichini O, Boisset G, Polok B, Tiab L, Mayeur H, Raji B, de la Houssaye G, Abitbol MM, Munier FL. Mutation in the human homeobox gene NKX5-3 causes an oculo-auricular syndrome. Am J Hum Genet. 2008;82(5):1178–84. doi:10.1016/j.ajhg.2008.03.007.
- Vaclavik V, Schorderet DF, Borruat FX, Munier FL. Retinal dystrophy in the oculo-auricular syndrome due to HMX1 mutation. Ophthalmic Genet. 2011;32(2):114–17. doi:10.3109/13816810.2011.562955.
- Gillespie RL, Urquhart J, Lovell SC, Biswas S, Parry NR, Schorderet DF, Lloyd IC, Clayton-Smith J, Black GC. Abrogation of HMX1 function causes rare oculoauricular syndrome associated with congenital cataract, anterior segment dysgenesis, and retinal dystrophy. Invest Ophthalmol Vis Sci. 2015;56(2):883–91. doi:10.1167/iovs.14-15861.
- Abdel-Salam GMH, Abdel-Hamid MS, Mehrez MI, Kamal AM, Taher MB, Afifi HH. Further delineation of the oculoauricular syndrome phenotype: a new family with a novel truncating HMX1 mutation. Ophthalmic Genet. 2018;39(2):215–20. doi:10.1080/13816810.2017.1401089.
- Franceschetti A, Valerio M. Malformations associées des yeux et des oreilles [Associated malformations of the eyes and ears]. Confin Neurol. 1944-1945;6(5):255–57. doi:10.1159/000105978.
- Beleza-Meireles A, Clayton-Smith J, Saraiva JM, Tassabehji M. Oculo-auriculo-vertebral spectrum: a review of the literature and genetic update. J Med Genet. 2014;51(10):635–45. doi:10.1136/jmedgenet-2014-102476.
- Lesnik Oberstein SAJ, Ruivenkamp CAL, Hennekam RC. Peters plus syndrome. In: Adam M, Ardinger H, Pagon R, Wallace S, Bean L, Gripp K, Mirzaa G, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2007 [updated 2017]. pp. 1993–2022.
- Sanlaville D, Verloes A. CHARGE syndrome: an update. Eur J Hum Genet. 2007;15(4):389–99. doi:10.1038/sj.ejhg.5201778.
- Quinodoz M, Peter VG, Bedoni N, Royer Bertrand B, Cisarova K, Salmaninejad A, Sepahi N, Rodrigues R, Piran M, Mojarrad M, et al. AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. Nat Commun. 2021;12(1):518. doi:10.1038/s41467-020-20584-4.
- Furlan A, Lübke M, Adameyko I, Lallemend F, Ernfors P. The transcription factor HMX1 and growth factor receptor activities control sympathetic neurons diversification. Embo J. 2013;32(11):1613–25. doi:10.1038/emboj.2013.85.
- Boulling A, Wicht L, Schorderet DF. Identification of HMX1 target genes: a predictive promoter model approach. Mol Vis. 2013;19:1779–94.
- Boisset G, Schorderet DF. Zebrafish HMX1 promotes retinogenesis. Exp Eye Res. 2012;105:34–42. doi:10.1016/j.exer.2012.10.002.
- El Fersioui Y, Pinton G, Allaman-Pillet N, Schorderet DF. HMX1 regulates urfh1 expression in the craniofacial region in zebrafish. PLoS One. 2021;16(1):e0245239. doi:10.1371/journal.pone.0245239.
- Munroe RJ, Prabhu V, Acland GM, Johnson KR, Harris BS, O’-Brien TP, Welsh IC, Noden DM, Schimenti JC. Mouse H6 Homeobox 1 (HMX1) mutations cause cranial abnormalities and reduced body mass. BMC Dev Biol. 2009;9:27. doi:10.1186/1471-213X-9-27.