
ABSTRACT
Background
Membrane frizzled-related protein (MFRP) plays a critical role in ocular development. MFRP mutations are known to cause nanophthalmos and, in some cases, retinitis pigmentosa, foveoschisis, and/or optic nerve head (ONH) drusen. The broad clinical spectrum of MFRP mutations necessitates further investigation of specific genotype–phenotype relationships.
Materials and Methods
We reviewed ophthalmologic and genetic medical records of two affected siblings and one unaffected sibling.
Results
Genetic testing revealed variants MFRP c.855T>A, p.(Cys285*) and MFRP c.1235T>C, p.(Leu412Pro) in trans in the two affected siblings. In both cases, photopic and scotopic responses were markedly reduced on electroretinogram (ERG), with greater decrease in scotopic function. Optical coherence tomography for both siblings revealed non-cystoid thickening. Blunted foveal reflexes were also observed in both siblings. Notably, foveal avascular zone abnormalities were seen on fundus autofluorescence in only one affected sibling.
Conclusions
MFRP-related ocular disease may be underrecognized due to its presentation with high hyperopia and possibly subtle retinal findings. Presence of variants MFRP c.855T>A, p.(Cys285*) and MFRP c.1235T>C, p.(Leu412Pro) in trans resulted in nanophthalmos and retinitis pigmentosa without associated foveoschisis or ONH drusen in our patients, consistent with the incomplete phenotype previously described in Neri et al. Abnormalities in the foveal avascular zone have been noted in other case studies and were inconsistently associated with the variants described here, representing a potential area for future investigation.
Background
Membrane frizzled-related protein (MFRP) is a type II transmembrane protein localized to the ciliary body and retinal pigment epithelium (RPE) (Citation1). Mutations in MFRP are known to cause autosomal recessive non-syndromic nanophthalmos, a subtype of microphthalmia characterized by short axial length (<20 mm) without other external structural defects (Citation2,Citation3).
While its exact role remains unclear, MFRP is thought to influence ocular development prenatally and aid in emmetropization postnatally. MFRP expression is first detected in the RPE in mid-gestation (14 weeks), precluding a role for MFRP in optic cup formation (Citation4). This delayed expression may account for the lack of external structural abnormalities in nanophthalmos, such as colobomas, that are observed in other microphthalmias. Notably, defects in the fovea, which develops during the third trimester, are seen in nanophthalmos. MFRP also appears to influence ocular development postnatally. Children with MFRP mutations may not experience postnatal axial extension, the main mechanism by which emmetropization occurs in early life. Case studies on young MFRP patients demonstrate no correction of refractive error during childhood, supporting MFRP’s critical role in emmetropization (Citation4).
Whether MFRP is involved in maintaining retinal health later in life is unclear. One case study of four affected siblings with MFRP mutations demonstrated decreased photopic and scotopic responses on electroretinogram (ERG) which was more pronounced in the oldest siblings, potentially indicating progressive degeneration of the rods and cones (Citation5). However, other clinical findings suggest that hyperopia precedes and may directly cause retinal dysfunction, regardless of retinal maintenance by MFRP (Citation6).
In 2012, a review by Neri et al. reported 17 cases of MFRP-related retinitis pigmentosa (RP) or MFRP-related posterior microphthalmos (PM) in the literature. MFRP mutants without evidence of retinal degeneration were excluded from the review. The authors described two distinct MFRP phenotypes: the “complete” phenotype characterized by concurrent PM, RP, foveoschisis, and optic nerve head (ONH) drusen, and the “incomplete” phenotype characterized by presence of PM and RP, but absence of foveoschisis and/or ONH drusen. The authors identified a genotype–phenotype correlation for the complete phenotype, despite failure of past attempts to correlate specific MFRP mutations with clinical phenotypes (Citation7).
Other phenotypes not included in the Neri review, such as nanophthalmos without retinal dystrophy, have been associated with homozygous MFRP mutations (Citation4,Citation8). The broad clinical spectrum of MFRP mutations suggests a need to further investigate specific genotype–phenotype relationships to inform prognosis and guide management.
Materials and methods
Medical records of two affected siblings and one unaffected sibling were reviewed. All examinations were performed by a single pediatric ophthalmologist (MAS). Ophthalmic examination included best corrected visual acuity (BCVA), IOL master ocular biometry, slit lamp examination, rebound tonometry (iCare), ocular motility assessment, and dilated fundus examination. Cycloplegia was obtained with instillation of cyclopentolate 2.0% eye drops. Refraction was performed 40 minutes afterward by means of retinoscopy. Spectral domain optical coherence tomography (SD-OCT) was obtained with Zeiss Cirrus systems (Zeiss Meditec, Inc. Dublin, CA). Full field ERG was performed using a RETeval acquisition device (LCK Technologies, Inc. Gaithersburg, MD). Full-field ERGs were recorded per the ISCEV (2015 update) Standard including light-adapted (LA) single and 28 Hz flicker 3.0 cd-s m−2 and dark-adapted (DA) 0.01, 3.0 and 10.0 cd-s m−2 conditions (Citation9). Fundus photographs were obtained with Topcon ImagenetR4 (Topcon Medical Systems, Inc. Oakland, NJ). Genetic analysis for Patient 1 was performed with a 266 gene inherited retinal dystrophy panel. Genetic analysis for Patient 2 was performed via familial variant testing.
Results
Patient 1
Patient 1 is a 7-year-old male who presented at 5 years old with a history of presumed bilateral amblyopia, worsening vision, and high hyperopia at +14.5 OD and +13.5 OS. On review of ocular history, there was found to be a long-term history of poor vision despite introduction of glasses at an early age and good compliance throughout childhood. The patient had been examined by various eye care professionals including a pediatric ophthalmologist. Given his history of unexplained poor vision and high hyperopia, further work-up investigating underlying ocular diseases beyond bilateral amblyopia was pursued. On initial examination, BCVA was 20/80 OD and 20/60 OS at distance, and 20/50 OU near. Slit lamp and fundus examinations were unremarkable. Axial length was 15.55 mm OD and 15.86 mm OS. Anterior chamber depth was 3.59 mm OD and 3.62 mm OS. Horizontal corneal diameter was 12.40 mm OD and 12.00 mm OS. On OCT there was no thickening of the outer retina, no apparent foveal depression, and no foveoschisis (). Fundus photographs revealed a yellowish hue in the macula with loss of foveal reflex as well as radial retinal striations OU and pinpoint white deposits nasal to the fovea OD (). ERG revealed decreased photopic and scotopic responses, with a greater decrease in scotopic function.
Figure 1. Patient 1 at 6 years of age. There is significant disruption of the inner retina with marked thickening and loss of normal foveal contour. Central subfield thickness (ILM-RPE) is 529 microns.
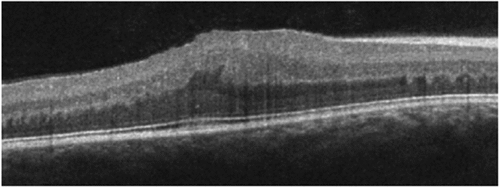
Figure 2. (a) Patient 1 at 7 years of age. SD-OCT reveals grossly abnormal contour and thickening of the outer retina with no apparent foveal depression. Central subfield thickness (ILM-RPE) is 500 microns. SD-OCT one year earlier also showed thickening, loss of contour, and gross contour changes (). Fundus photograph shows yellowish hue within the macula with loss of foveal reflex, radial retinal striations, and pinpoint white deposits nasal to the fovea. The ffERG shows borderline low LA 3.0 and below-normal LA 28 Hz amplitude cone-mediated ERGs. Rod-mediated were markedly depressed to the DA 0.01 flash strength and in mixed rod-cone DA 3.0 and DA 10.0 A- and B-waves. (For all patients, with the exception of the DA 10.0 condition, two successive ERG recordings from the right eye are shown. The rectangles on each recording denote the expected amplitude and implicit time limits for the LA and DA ERGs provided by the manufacturer). (b) Patient 2, the 5-year-old affected sibling. SD-OCT reveals abnormal foveal contour with loss of foveal depression as well as areas of thickening and elevation. Central subfield thickness (ILM-RPE) is 411 microns. Fundus photograph shows blunted foveal reflex, macular striations, and small, pinpoint, hypopigmented macular deposits. The ffERG shows a pattern of loss similar to her 7-year-old sibling (a) with a reduction in cone-mediated LA ERG, most notable in the LA 28 Hz condition. Rod-mediated B-wave ERGs to the DA 0.01 flash strength were relatively less affected in Patient 2, but the amplitude of the A-waves in the mixed DA 3.0 and 10.0 conditions were clearly reduced compared with normal range and the older sibling, Patient 3. (c) Patient 3, the 12-year-old unaffected sibling. SD-OCT reveals a normal, healthy retina. Central subfield thickness (ILM-RPE) is 257 microns. Fundus photograph shows normal macular findings and normal vessel findings. The ffERG shows LA and DA A- and B-waves within expected limits.
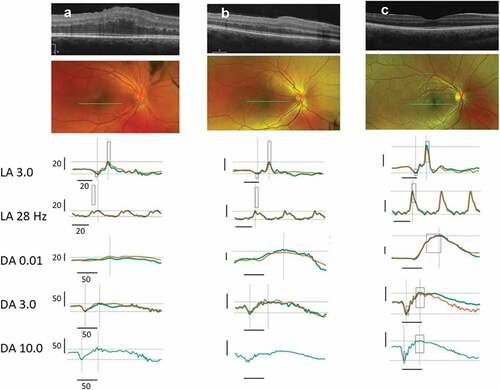
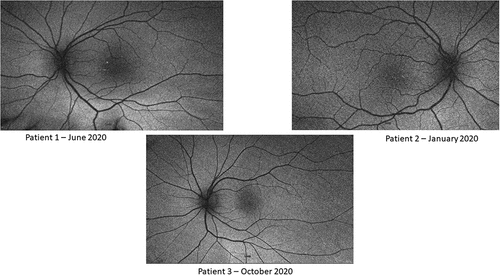
Figure 3. Patient 1: Yellowish hue within the macula with loss of fovea reflex; radial retinal striations OU; pinpoint white deposits noted nasal to the fovea OD. Patient 2: Blunted foveal reflex, macular striations, small pinpoint, hypopigmented macular deposits OU. The deposits are hypo-AF on AF OU. Normal appearing optic nerve and retinal vessels OU. Patient 3: Findings include normal observations. Disc findings include normal observations. Macula findings include normal observations. Periphery findings include normal observations. Vessel findings include normal observations.
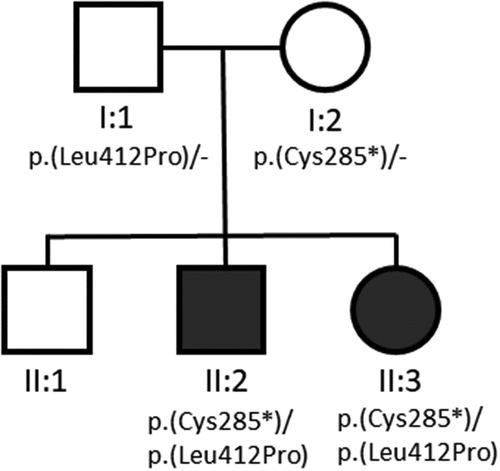
Genetic testing identified a likely pathogenic MFRP c.855T>A, p.(Cys285*) variant (transcript NM_031433.3), as well as a variant of unknown significance, MFRP c.1235T>C, p.(Leu412Pro). Parental testing confirmed that the variants are in trans but this did not upgrade the clinical variant classification of either variant. To our knowledge, the c.1235T>C, p.(Leu412Pro) variant is novel. The c.855T>A, p.(Cys285*) variant was identified through exome sequencing in a patient with a cone dystrophy phenotype. However, as no second MFRP variant was identified, this could easily be a non-diagnostic carrier status finding in that patient (Citation10).
On follow-up ERG one year later, Patient 1’s photopic responses were further decreased and DA 0.01 scotopic responses showed almost no activity (). The severity of these decreased responses likely reflects associated RP. Follow-up OCT revealed diffuse non-cystoid thickening with an extra hyporeflective lamination between the outer plexiform and outer nuclear layer through the center of the macula in both eyes (). Follow-up slit lamp examination showed trace anterior vitreous cells and fundus examination revealed blunted foveal reflexes.
Patient 2
Patient 2, the younger sibling of Patient 1, presented at 5 years of age with worsening vision and was screened by the examining ophthalmologist (MAS) due to the findings seen in Patient 1. Initial exam revealed high hyperopia at +8.75 OD and +8.50 OS. BCVA was 20/80 OD and 20/50 OS at distance. Slit lamp examination was unremarkable, but fundus examination revealed blunted foveal reflex and macular striations. Axial length was 17.40 mm OD and 17.55 mm OS. Anterior chamber depth was 3.01 mm OD and 2.93 mm OS. Horizontal corneal diameter was 12.00 mm OD and 11.90 mm OS. On OCT there were several abnormal findings including abnormal foveal contour, areas of thickening and elevation, and loss of normal foveal depression OU (). However, no foveoschisis or retinal edema was noted in either eye. Fundus autofluorescence (FAF) revealed blunted foveal reflex, macular striations, pinpoint hypopigmented macular deposits, and abnormal foveal avascular zone OU (). ERG showed global depression in the amplitude of photopic and scotopic responses consistent with RP, with rod dysfunction exceeding cone dysfunction (). Genetic testing confirmed existence of the same MFRP c.855T>A, p.(Cys285*) and MFRP c.1235T>C, p.(Leu412Pro) variants in trans.
Patient 3
Patient 3 is a 12-year-old older sibling of Patients 1 and 2 (See for pedigree). Given the findings in Patients 1 and 2, Patient 3 underwent a screening eye exam (MAS), imaging and electroretinography. Genetic testing was not performed for this patient, as there have been no complaints of poor vision. BCVA was 20/20 OD and 20/20 OS. Slit lamp and fundus examination were unremarkable. OCT and FAF revealed no abnormalities. Full field ERG was also normal ().
Figure 4. Family pedigree showing two unaffected parents, one unaffected child, and two affected children. Shading represents presence of the nanophthalmos phenotype.
Discussion
High refractive error can be a presenting sign of underlying ophthalmic diseases such as inherited retinal degenerations (IRDs). It is well established that early-onset high myopia is associated with IRDs (Citation11), but high hyperopia may be overlooked as this association is not as frequent (Citation12). Additionally, subnormal visual acuity unresponsive to refractive therapy may be misdiagnosed as bilateral amblyopia, when in reality it may be another presenting sign of IRD, as seen in Patient 1. In the context of high hyperopia and unexplained subnormal vision, a diagnosis of bilateral amblyopia should be questioned, and further work-up with fundus imaging and ERG must be considered.
MFRP-related ocular disease may be underdiagnosed in the general population given its presentation with high hyperopia (as opposed to high myopia) and because the retinal findings may be subtle (as seen in our patients). Notably, Patient 1 had been seen by several eye care professionals including a pediatric ophthalmologist before undergoing further workup for his poor vision. MFRP-related eye disease needs to be on the differential of unexplained subnormal vision in the setting of high hyperopia.
The presence of RP and nanophthalmos without associated foveoschisis or ONH drusen in our patients is consistent with the incomplete phenotype described in Neri et al. In that review, all patients with the complete phenotype carried the MFRP c.492delC variant either as a homozygous variant, or as a compound heterozygous variant with another nonsense variant. Patients with the incomplete phenotype did not carry this variant. Consistent with the genotype–phenotype correlations established by Neri et al., the MFRP c.492delC mutation was absent in the cases outlined here. The variants seen in our patients appear to cause retinal degeneration in addition to nanophthalmos in both siblings, but at present there is no foveoschisis or ONH drusen in either.
Notably, the foveal avascular zone was abnormal in Patient 2, but no abnormalities were noted in Patient 1. Other case studies have also noted reduced diameter or absence of a capillary-free zone in MFRP patients and in patients with PM more generally (Citation13,Citation14). However, avascular zone abnormalities do not seem to be a universal finding among MFRP mutants.
The ERGs from Patients 1 and 2 are characterized by reduced A-wave amplitudes in the DA 3.0 and 10.0 flash strengths, reflecting reduced photoreceptor response. Patients 1 and 2 both had borderline reduced cone-mediated single flash LA 3.0 ERG B-wave amplitudes. Amplitude reductions were clearer in the LA 28 Hz photopic flicker ERG, which have a “notched” appearance seen in the incomplete congenital stationary nightblindness (CSNB2), but without the DA 3.0 electronegative B-wave or prolonged LA 3.0 ERG A-wave characteristic of CSNB2 (Citation15). The rod-mediated DA 0.01 B-wave was more depressed in Patient 1 compared with his younger sibling, Patient 2, and, together with retinal changes in SD-OCT, are suggestive of progression. Thus, the ERG findings are consistent with a possible effect of MFRP mutations on photoreceptor function. However, it is not possible to rule out a role that disorganization of the retina architecture might play in altering the waveguide properties of the outer segments, which could result in reduced quantum catch in the outer retina and reduced ERG response.
Therapeutic options for nanophthalmos are limited, and early management focuses on appropriate refractive correction and strabismus management to prevent amblyopia. In adulthood, individuals with nanophthalmos are at higher risk for angle-closure glaucoma as well as complications associated with cataract surgery (Citation16,Citation17).
A recent case report of a 52-year-old MFRP patient demonstrated that a two-month course of topical dorzolamide may be effective in reducing cystoid macular edema and preventing foveoschisis in individuals with MFRP mutations (Citation18). Dorzolamide should be considered in these patients if cystic changes develop. While cystoid macular edema is a common cause of increased retinal thickness in RP, a case study by Dinculescu et al. suggested that Müller cell swelling (potentially due to persistence of tissue that would have moved centrifugally in normal development), rather than cystoid macular edema, may underlie retinal thickness changes in some MFRP patients (Citation19). This possibility, in conjunction with the known phenotypic variability among MFRP patients, makes it unclear whether dorzolamide treatment is indicated in patients without cystic macular changes such as ours.
While no curative treatment for MFRP-associated nanophthalmos exists, gene therapies have successfully prevented photoreceptor degeneration, restored MFRP levels, and normalized electrophysiologic function in several mouse models (Citation19,Citation20). In patient-specific induced pluripotent stem cells, MFRP gene therapy corrected actin filament disorganization and restored apical microvilli, supporting a potential role for interventional gene therapy in treating MFRP-associated nanophthalmos (Citation21).
Our patients’ presentation of nanophthalmos and RP associated with the novel MFRP variants we identified is consistent with previous case studies of MFRP mutations. The avascular zone abnormalities seen in Patient 2 do not appear to be a universal finding among MFRP patients, and OCTA may be indicated to characterize such changes. Describing phenotypic profiles associated with specific MFRP mutations and eliciting MFRP’s exact role in ocular development would be useful in understanding which patients may benefit from dorzolamide therapy. Additionally, this information could be useful in determining which individuals might benefit from emerging gene therapies.
Acknowledgements
We thank the family for their enthusiastic participation.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Mandal MN, Vasireddy V, Jablonski MM, Wang X, Heckenlively JR, Hughes BA, Reddy GB, Ayyagari R. Spatial and temporal expression of MFRP and its interaction with CTRP5. Invest Ophthalmol Vis Sci. 2006 Dec;47(12):5514–21. doi:10.1167/iovs.06-0449. PMID: 17122143.
- O’Grady RB. Nanophthalmos. Am J Ophthalmol. 1971 June;71(6):1251–53. doi:10.1016/0002-9394(71)90971-8. PMID: 5091124.
- Cross HE, Yoder F. Familial nanophthalmos. Am J Ophthalmol. 1976 Mar;81(3):300–06. doi:10.1016/0002-9394(76)90244-0. PMID: 1258954.
- Sundin OH, Dharmaraj S, Bhutto IA, Hasegawa T, McLeod DS, Merges CA, Silval ED, Maumenee IH, Lutty GA. Developmental basis of nanophthalmos: MFRP is required for both prenatal ocular growth and postnatal emmetropization. Ophthalmic Genet. 2008 Mar;29(1):1–9. doi:10.1080/13816810701651241. PMID: 18363166; PMCID: PMC2739627.
- Ayala-Ramirez R, Graue-Wiechers F, Robredo V, Amato-Almanza M, Horta-Diez I, Zenteno JC. A new autosomal recessive syndrome consisting of posterior microphthalmos, retinitis pigmentosa, foveoschisis, and optic disc drusen is caused by a MFRP gene mutation. Mol Vis, 2006 Dec 4;12:1483–89. PMID: 17167404.
- Velez G, Tsang SH, Tsai YT, Hsu CW, Gore A, Abdelhakim AH, Mahajan M, Silverman RH, Sparrow JR, Bassuk AG, et al. Gene therapy restores MFRP and corrects axial eye length. Sci Rep. 2017 Nov 23;7(1):16151. doi:10.1038/s41598-017-16275-8. PMID: 29170418; PMCID: PMC5701072.
- Neri A, Leaci R, Zenteno JC, Casubolo C, Delfini E, Macaluso C. Membrane frizzled-related protein gene-related ophthalmological syndrome: 30-month follow-up of a sporadic case and review of genotype-phenotype correlation in the literature. Mol Vis. 2012;18:2623–32. Epub 2012 Oct 26. PMID: 23112574; PMCID: PMC3482175.
- Sundin OH, Leppert GS, Silva ED, Yang JM, Dharmaraj S, Maumenee IH, Santos LC, Parsa CF, Traboulsi EI, Broman KW, et al. Extreme hyperopia is the result of null mutations in MFRP, which encodes a frizzled-related protein. Proc Natl Acad Sci U S A. 2005 July 5;102(27):9553–58. doi:10.1073/pnas.0501451102. Epub 2005 Jun 23. PMID: 15976030; PMCID: PMC1172243.
- McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, Bach M. ISCEV standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015 Aug;130(1):1–12. doi:10.1007/s10633-014-9473-7. Epub 2014 Dec 14. Erratum in: Doc Ophthalmol. 2015 Aug;131(1):81-3. PMID: 25502644.
- Haer-Wigman L, van Zelst-Stams WA, Pfundt R, van den Born LI, Klaver CC, Verheij JB, Hoyng CB, Breuning MH, Boon CJ, Kievit AJ, et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur J Hum Genet. 2017 May;25(5):591–99. doi:10.1038/ejhg.2017.9. Epub 2017 Feb 22. PMID: 28224992; PMCID: PMC5437915.
- Sieving PA, Fishman GA. Refractive errors of retinitis pigmentosa patients. Br J Ophthalmol. 1978 Mar;62(3):163–7. doi:10.1136/bjo.62.3.163. PMID: 638108; PMCID: PMC1043172.
- Hendriks M, Verhoeven VJM, Buitendijk GHS, Polling JR, Meester-Smoor MA, Hofman A, RD5000 Consortium, Kamermans M, Ingeborgh van den Born L, Klaver CCW. Development of refractive errors-what can we learn from inherited retinal dystrophies? Am J Ophthalmol. 2017 Oct;182:81–89. doi:10.1016/j.ajo.2017.07.008. Epub 2017 Jul 25. PMID: 28751151.
- Morillo Sánchez MJ, Llavero Valero P, González-Del Pozo M, Ponte Zuñiga B, Antiñolo G, Ramos Jiménez M, Rodríguez De La Rúa Franch E. Posterior microphthalmos, retinitis pigmentosa, and foveoschisis caused by a mutation in the MFRP gene: a familial study. Ophthalmic Genet. 2019 June;40(3):288–92. doi:10.1080/13816810.2019.1633547. Epub 2019 Jul 2. PMID: 31264930.
- Khairallah M, Messaoud R, Zaouali S, Ben Yahia S, Ladjimi A, Jenzri S. Posterior segment changes associated with posterior microphthalmos. Ophthalmology. 2002 Mar;109(3):569–74. doi:10.1016/s0161-6420(01)00996-4. PMID: 11874763.
- Audo I, Robson AG, Holder GE, Moore AT. The negative ERG: clinical phenotypes and disease mechanisms of inner retinal dysfunction. Surv Ophthalmol. 2008 Jan-Feb;53(1):16–40. doi:10.1016/j.survophthal.2007.10.010. PMID: 18191655.
- Sharan S, Grigg JR, Higgins RA. Nanophthalmos: ultrasound biomicroscopy and Pentacam assessment of angle structures before and after cataract surgery. J Cataract Refract Surg. 2006 June;32(6):1052–55. doi:10.1016/j.jcrs.2006.02.051. PMID: 16814070.
- Wladis EJ, Gewirtz MB, Guo S. Cataract surgery in the small adult eye. Surv Ophthalmol. 2006 Mar-Apr;51(2):153–61. doi:10.1016/j.survophthal.2005.12.005. PMID: 16500215.
- Zacharias LC, Susanna R, Jr, Sundin O, Finzi S, Susanna BN, Takahashi WY. Efficacy of topical dorzolamide therapy for cystoid macular edema in a patient with MFRP-related nanophthalmos-retinitis pigmentosa-foveoschisis-optic disk drusen syndrome. Retin Cases Brief Rep. 2015 Winter;9(1):61–63. doi:10.1097/ICB.0000000000000088. PMID: 25383852; PMCID: PMC4272654.
- Dinculescu A, Estreicher J, Zenteno JC, Aleman TS, Schwartz SB, Huang WC, Roman AJ, Sumaroka A, Li Q, Deng WT, et al. Gene therapy for retinitis pigmentosa caused by MFRP mutations: human phenotype and preliminary proof of concept. Hum Gene Ther. 2012 Apr;23(4):367–76. doi:10.1089/hum.2011.169. Epub 2012 Jan 26. PMID: 22142163; PMCID: PMC3327606.
- Chekuri A, Sahu B, Chavali VRM, Voronchikhina M, Soto-Hermida A, Suk JJ, Alapati AN, Bartsch DU, Ayala-Ramirez R, Zenteno JC, et al. Long-term effects of gene therapy in a novel mouse model of human MFRP-associated retinopathy. Hum Gene Ther. 2019 May;30(5):632–50. doi:10.1089/hum.2018.192. Epub 2019 Jan 16. PMID: 30499344; PMCID: PMC6534092.
- Li Y, Wu WH, Hsu CW, Nguyen HV, Tsai YT, Chan L, Nagasaki T, Maumenee IH, Yannuzzi LA, Hoang QV, et al. Gene therapy in patient-specific stem cell lines and a preclinical model of retinitis pigmentosa with membrane frizzled-related protein defects. Mol Ther. 2014 Sep;22(9):1688–97. doi:10.1038/mt.2014.100. Epub 2014 Jun 4. PMID: 24895994; PMCID: PMC4435479.