
ABSTRACT
Background
The sodium channel and clathrin linker 1 gene (SCLT1) has been involved in the pathogenesis of various ciliopathy disorders such as Bardet-Biedl syndrome, orofaciodigital syndrome type IX, and Senior-Løken syndrome. Detailed exams are warranted to outline all clinical features. Here, we present a family with a milder phenotype of SCLT1-related disease.
Material and Methods
Comprehensive eye examination including fundus images, OCT, color vision, visual fields and electroretinography were performed. Affected individuals were assessed by a pediatrician and a medical geneticist for systemic features of ciliopathy. Investigations included echocardiography, abdominal ultrasonography, blood work-up for diabetes, liver and kidney function. Genetic testing included NGS retinal dystrophy panel, segregation analysis and transcriptome sequencing.
Results
Two male children, age 10 and 8 years, were affected with attention deficit hyperactivity disorder (ADHD), obesity and mild photophobia. The ophthalmic exam revealed reduced best-corrected visual acuity (BCVA), strabismus, hyperopia, astigmatism and moderate red-green defects. Milder changes suggesting photoreceptors disease were found on retinal imaging. Electroretinogram confirmed cone photoreceptors dysfunction. Genetic testing revealed a homozygous likely pathogenic, splice-site variant in SCLT1 gene NM_144643.3: c.1439 + 1del in the proband and in the affected brother. The unaffected parents were heterozygous for the SCLT1 variant. Transcriptome sequencing showed retention of intron 16 in the proband.
Conclusions
In this report, we highlight the importance of further extensive diagnostics in patients with unexplained reduced vision, strabismus, refractive errors and ADHD spectrum disorders. SCLT1-related retinal degeneration is very rare and isolated reduced function of cone photoreceptors has not previously been observed.
1. Introduction
The sodium channel and clathrin linker 1 gene (SCLT1), located on human chromosome 4q28.2, has been reported in the pathogenesis of ciliopathy disorders such as Bardet-Biedl syndrome (BBS) (OMIM # 209900) (Citation1–4), orofaciodigital syndrome type 9 (Citation5) (OMIM # 258865) and Senior-Løken syndrome (OMIM # 266900) (Citation6).
SCLT1 encodes a linker protein associated with the sodium voltage-gated channel alpha subunit 10 and clathrin, which is a core protein involved in ciliogenesis (Citation7). Specifically, SCLT1 is one of the components of the distal appendages (DAPs) of centrioles (Citation7), that anchor the cilia to the plasma membrane. Other DAP components have also been linked to ciliopathy diseases, and similarly to SCLT1, the affected individuals may show different clinical features. For instance, impaired function of CEP164 has been shown to cause nephronophthisis with or without retinal degeneration (OMIM # 614845) (Citation8), but also was reported in BBS (Citation9). Further, alterations in CEP83, which were first described to cause nephronophthisis and intellectual disability (OMIM # 615862) (Citation10), later were shown to cause isolated retinitis pigmentosa (RP) (Citation11).
Ciliopathies are mostly autosomal recessive conditions caused by pathogenic variants in a wide range of genes coding for proteins responsible for ciliogenesis, cilia structure and/or function (Citation12–14). These disorders can be both syndromic and non-syndromic, in which retinal degeneration (RD) is a cardinal clinical feature (Citation15,Citation16). The spectrum of disorders ranges from isolated RD, such as Leber congenital amaurosis due to pathogenic variants in CEP290 (Citation17–19), to complex syndromes with multiple organ dysfunction such as BBS characterised by a combination of RD, obesity, polydactyly, hypogonadism, kidney disease and learning difficulties (Citation20–25). Other examples of syndromic ciliopathies are Alström syndrome (OMIM #203800) (Citation26–30), Senior-Løken syndrome (Citation31–34) and orofaciodigital syndrome (Citation35,Citation36) among others. Furthermore, ciliopathies with milder clinical features have been also recently reported (Citation37–39).
To date, only few patients with SCLT1-related retinal degeneration of RP type have been reported (Citation1,Citation2,Citation6). Here, we report two siblings affected with ADHD, obesity, refractive error, esotropia and unexplained reduced vision, and homozygous splice variants in SCLT1.
2. Material and methods
2.1. Clinical assessments
The study adhered to the tenets of the Declaration of Helsinki. Legal guardians have given written consent to participate in this study. The proband (III-1) and his sibling (III-2) underwent comprehensive eye examinations (St. Erik Eye Hospital, Stockholm, Sweden), pediatric assessment (community-based pediatrician), and genetic testing (Department of Clinical Genetics, Karolinska University Hospital, Stockholm, Sweden). The parents (II-2 and II-1) underwent eye exams and genetic testing. Ophthalmological evaluations included: best corrected visual acuity (BCVA), color vision test (HRR), cycloplegic refraction, ophthalmoscopy, Goldman visual fields, fundus photography (Optos Nikon, Tokyo, Japan), optical coherence tomography (Topcon OCT), full field electroretinogram (ERG, Diagnosys, LLC, Lowell, MA, USA) according to the ISCEV standards (Citation40). Pediatric evaluation included comprehensive clinical exam and ancillary tests, such as echocardiography, ultrasonography of abdomen, blood morphology and blood tests to assess liver function (ASAT, ALAT, lipid profile), kidney function (creatinine, electrolytes), glucose and HbA1c. The children were referred for psychological assessment of cognitive function.
2.2. Genetic analysis
The proband (patients III-1) was analysed at the Department of Clinical Genetics, Karolinska University Hospital with massive parallel sequencing of a retinal dystrophy panel of 285 genes (Table S1). Testing of patient III-2 as well as the healthy parents (II-1 and II-2) was performed with polymerase-chain reaction (PCR) and Sanger sequencing of the SCLT1 (NM_144643.3) variant identified. Primers are available on request.
Effects on RNA were evaluated by whole transcriptome sequencing on RNA isolated from blood of the proband according to standard protocols. Briefly, total RNA was isolated and library preparation was done by the Illumina mRNA stranded method with sequencing on Nova Seq S4. The resulting data was aligned to reference genome using the Spliced Transcripts Alignment to a Reference (STAR). The resulting alignment were visualized in Integrated Genomics Viewer (IGV) to detect aberrant splicing.
3. Results
3.1. Patient III-1 (proband)
The male child of Middle East origin from consanguineous parents, who were second cousins, () presented at one and a half years of age at the eye clinic (St. Erik Eye Hospital) due to left esotropia. Cycloplegic refraction revealed significant hyperopia and fundus examination was normal. Between 6- and 8-years of age, vision loss and abnormal foveal reflex were observed (, ). Nystagmus was not present. Extensive diagnostics revealed mild increased autofluorescence in the posterior pole, and abnormalities in the outer nuclear layer corresponding to photoreceptors (). The patient had moderate colour vision defects. Full field ERG showed normal function of rod photoreceptors and reduced function of cone photoreceptors (, Table S2). Systemic manifestations included well-controlled obesity and ADHD-spectrum disorders. Dysmorphic features or polydactyly were not present. Despite mild phenotype (reduced function of cone photoreceptors, obesity and ADHD), ciliopathy was suggested as a possible disease causing mechanism. The proband was referred for comprehensive pediatric evaluation regarding ciliopathy (). A psychological assessment of cognitive function was normal. Genetic testing with a retinal dystrophy gene panel (Table S1) revealed a homozygous splice variant in SCLT1 gene NM_144643.3: c.1439 + 1del, which is novel (). Segregation analysis in the affected younger brother showed the same genotype, and the unaffected parents were heterozygous carriers. The proband was assessed clinically and obtained genetic counselling by a medical geneticist at the Karolinska University Hospital.
Figure 1. Pedigree and segregation analysis. Proband (III-1) and affected younger brother (III-2) are homozygous for splice-site variant in SCLT1 gene NM_144643.3: c.1439 + 1del while parents (II-1 and II-2) are heterozygous. The arrow depicts proband, horizontal lines—parents examined clinically. Black squares: affected male individuals. Double horizontal lines: consanguinity.
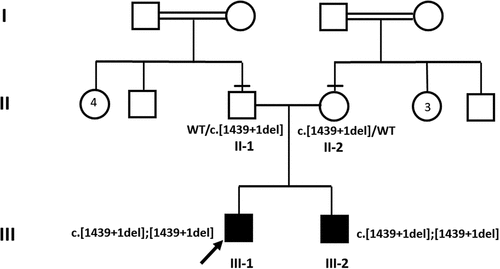
Figure 2a. Ophthalmological phenotype (Patient III-1). (a): fundus images and (b): OCT shows increased autofluorescence signal in the posterior pole and disruptions in the outer nuclear layer corresponding to photoreceptors; (c): Goldman visual fields (tested to II4E isopter) present well- preserved fields; (d): upper panel—full filed ERG shows reduced function of cone photoreceptors with preserved function of rod photoreceptors, lower panel—age-adjusted normative data.
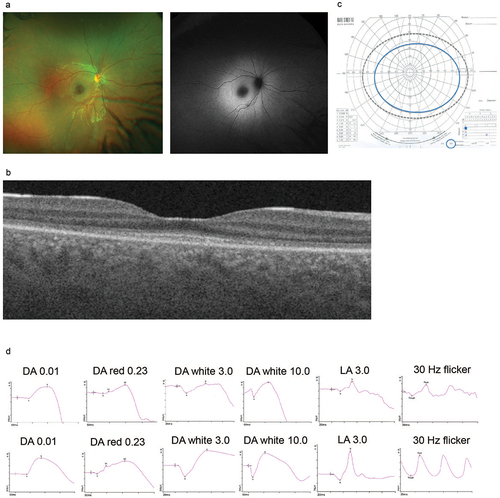
Table 1. Clinical features.
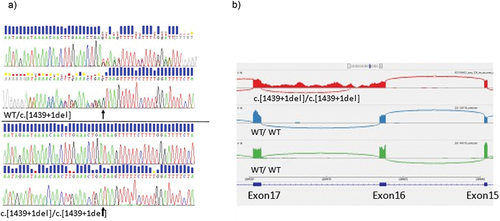
The homozygous splice-site variant in SCLT1 gene (c.1439 + 1del) () was novel (not present in ClinVar (Citation41) or gnomAD (Citation42)) and affected the 5’ canonical splice site of exon 16 at the first position of intron 16. Splicing predictions in Alamut Visual Plus v1.2 (Sophia genetics, Boston, USA) predicted a loss of donor site with score 100%. Transcriptome sequencing of the proband verified the presence of aberrant splicing, with retention of intron 16 sequences (). Translation of this aberrant transcript is predicted to result in a protein with a pre-mature stop codon 29 amino acids downstream of p.D480.
Figure 3. DNA and RNA sequencing results. A) Electropherogram from genomic DNA sequence in parent II-1 (top) and proband III-1 (bottom). Arrow indicates heterozygote position in parent, and deletion in proband. B) Sashimi-plot from transcriptome sequencing showing region Exon15 to exon 17 in SCLT1-gene. Top trace in red represents proband III-1, and traces in blue and green, are normal controls with wild-type genotype.
3.2. Patient III-2
The proband’s younger brother was referred to the eye clinic at the age of 5 years due to reduced vision. His eye exam revealed esophoria, and cycloplegic refraction showed moderate hyperopia and astigmatism (). His vision remained stable over the course of 3 years. Comprehensive eye exam including structural and functional retinal parameters showed similar results to his brother (Patient III-1), however Patient III-2 was affected to a lesser extent (, Table S2). Similarly to his brother, this patient also underwent an extensive pediatric evaluation regarding ciliopathy, which was unremarkable except for ADHD and well-controlled obesity as seen in the proband. DNA sequencing revealed the same homozygous SCLT1 splice variant as observed in the proband ().
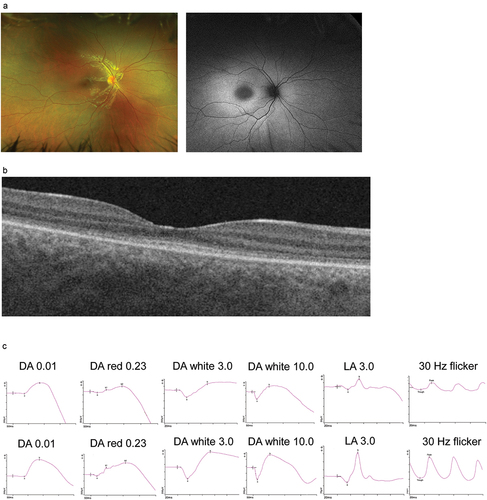
Figure 2b. Ophthalmological phenotype (Patient III-2). (a): fundus images and (b): OCT shows increased autofluorescence signal in the posterior pole and disruptions in the outer nuclear layer corresponding to photoreceptors; (c): upper panel—full filed ERG presents reduced function of cone photoreceptors with preserved function of rod photoreceptors, lower panel—age-adjusted normative data.
3.3. Patient II-1 and II-2
The 39-year-old father (II-1) and 38-year-old mother (II-2) did not report any clinical symptoms, and their eye exams were normal. Segregation analysis confirmed that both parents were heterozygous carriers of pathogenic splice-site variant in SCLT1 ().
4. Discussion
We report two siblings from a consanguineous family affected with SCLT1-related disease presenting with refractive errors, strabismus, reduced function of cone photoreceptors, ADHD and obesity. Compared to previous reports, the affected individuals in the family presented here have milder systemic and ocular phenotype.
4.1. The SCLT1-related condition is a very rare form of ciliopathy and presents with heterogenous phenotype
Animal studies showed that Sclt1-/- mice exhibit typical ciliopathy phenotypes, including cystic kidney, cleft palate and polydactyly (Citation43).
The first published case with pathogenic variants in SCLT1 gene was a severely affected male child diagnosed with orofaciodigital syndrome type 9 (OMIM #258865) (Citation5) who passed away at the age of 3 months due to a cardiorespiratory arrest. Since then, six individuals from five families with pathogenic SCLT1 variants have been reported. The clinical picture was variable with all individuals displaying retinal symptoms and many systemic features, such as liver and kidney disease, cognitive impairment, hypogonadism, obesity and polydactyly () (Citation1,Citation2,Citation4,Citation6). In contrast, the two siblings presented here had mild systemic features, such as ADHD and obesity ().
Table 2. Clinical features of individuals with pathogenic variants in SCLT1.
4.2. SCLT1-related retinal degeneration spans from generalised, advanced retinal degeneration of RP type to milder condition affecting only cone photoreceptors
All previously reported patients with SCLT1-related retinal disease had advanced retinal degeneration (Citation1,Citation2,Citation4,Citation6) at an early age with non-recordable electroretinogram (Citation2,Citation6), whereas the two individuals presented here had milder ocular phenotype ( , ). Both children had subtle fundus changes, which could had been missed with ophthalmoscopic exam. Electroretinogram revealed normal function of rod photoreceptors and reduced function of cone photoreceptors, as seen in cone dystrophies. Importantly, the visual functions remained relatively stable and well-preserved until the most recent visit at the age of 8 and 10 years, respectively. This is the first report of milder clinical phenotype caused by pathogenic variants in SCLT1 gene, thereby providing evidence of both systemic and ocular heterogeneity.
4.3. Pathogenic variants in SCLT1 gene may affect splicing
A few case reports of individuals with BBS/ciliopathy disorder and loss of function genetic variants in SCLT1 have been reported of which three are splice variants with the loss of exons 5, 14 and 17 respectively (Citation1,Citation2,Citation5,Citation6). All variants resulted in severely affected individuals with a range of systemic manifestations typical for ciliopathies (). In contrast, our patients had mild clinical phenotype due to a novel splice-site variant (NM_144643.3: c.1439 + 1del) in SCLT1 gene () in a homozygous state leading to retention of intron 16. Splice-site variants have been reported as common disease causing variations, with estimated frequency as high as 5.9–8.5% of all currently known pathogenic variants (Citation44). Also, splice-site variants have been detected in a wide range of genes causative for RD (Citation45) and they may lead to modified explanations of disease mechanisms. The variant identified in this family, c.1439 + 1del, was predicted to cause a loss of donor-site and was shown by RNA sequencing to cause a retention of intron 16. Control samples did not show any inclusion of sequences from intron 16 (n = 10). The resulting transcript would include a pre-mature stop codon 29 amino acids downstream, which could putatively lead to an alternative truncated protein, although other mechanisms cannot be ruled out.
In this report, we highlight the importance of extensive diagnostics in patients with unexplained reduced vision, refractive errors, strabismus, obesity and autism/ADHD spectrum disorders. We and other authors (Citation16,Citation37,Citation46) showed that ciliopathy diseases may present with mild systemic and ocular phenotypes, and thereby can be underdiagnosed, especially in young children. SCLT1-related retinal degeneration is an ultra-rare condition, and isolated cone photoreceptors dysfunction has not been previously observed. Regular follow-up of our patients is important to determine natural history of SCLT1-related retinal degeneration and to clarify if this is a case of cone dystrophy or an early stage of disease leading to cone-rod dystrophy. Also, in line with previous studies (Citation47,Citation48), we would like to emphasize the need of extensive clinical examinations together with genetic diagnostics in inherited retinal conditions, which enables identification of systemic associations early on. These actions have not only a role in preventive management strategies for the disease but also an individual’s general health, well-being and family planning (Citation49,Citation50).
Supplemental Material
Download PDF (76.8 KB)Acknowledgments
We greatly thank all the family members and patients who participated in this study.
Disclosure statement
AL has received honoraria from Illumina, MKGP from Novartis (unrelated to this study).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/13816810.2023.2215332.
Additional information
Funding
References
- Horiuchi K, Kogiso T, Sagawa T, Ito T, Taniai M, Miura K, Hattori M, Morisada N, Hashimoto E, Tokushige K. Bardet-Biedl syndrome caused by skipping of SCLT1 complicated by microvesicular steatohepatitis. Intern Med. 2020;59(21):2719–24. doi:10.2169/internalmedicine.5045-20.
- Morisada N, Hamada R, Miura K, Ye MJ, Nozu K, Hattori M, Iijima K. Bardet–biedl syndrome in two unrelated patients with identical compound heterozygous SCLT1 mutations. CEN Case Rep. 2020;9(3):260–5. doi:10.1007/s13730-020-00472-y.
- Sakakibara N, Nozu K, Yamamura T, Horinouchi T, Nagano C, Ye MJ, Ishiko S, Aoto Y, Rossanti R, Hamada R, et al. Comprehensive genetic analysis using next-generation sequencing for the diagnosis of nephronophthisis-related ciliopathies in the Japanese population. J Hum Genet. 2022;67(7):427–40. doi:10.1038/s10038-022-01020-5.
- Shamseldin HE, Shaheen R, Ewida N, Bubshait DK, Alkuraya H, Almardawi E, Howaidi A, Sabr Y, Abdalla EM, Alfaifi AY, et al. The morbid genome of ciliopathies: an update. Genet Med. 2020;22(6):1051–60. doi:10.1038/s41436-020-0761-1.
- Adly N, Alhashem A, Ammari A, Alkuraya FS. Ciliary genes TBC1D32/C6orf170 and SCLT1 are mutated in patients with OFD type IX. Hum Mutat. 2014;35(1):36–40. doi:10.1002/humu.22477.
- Katagiri S, Hayashi T, Yoshitake K, Murai N, Matsui Z, Kubo H, Satoh H, Matsufuji S, Takamura T, Yokoo T, et al. Compound heterozygous splice site variants in the SCLT1 gene highlight an additional candidate locus for Senior-Løken syndrome. Sci Rep. 2018;8(1):16733. doi:10.1038/s41598-018-35152-6.
- Tanos BE, Yang HJ, Soni R, Wang WJ, Macaluso FP, Asara JM, Tsou MFB. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 2013;27(2):163–8. doi:10.1101/gad.207043.112.
- Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd T, Zhou W, Cluckey A, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150(3):533–48. doi:10.1016/j.cell.2012.06.028.
- Shamseldin HE, Al Mogarri I, Alqwaiee MM, Alharbi AS, Baqais K, AlSaadi M, AlAnzi T, Alhashem A, Saghier A, Ameen W, et al. An exome-first approach to aid in the diagnosis of primary ciliary dyskinesia. Hum Genet. 2020;139(10):1273–83. doi:10.1007/s00439-020-02170-2.
- Failler M, Gee HY, Krug P, Joo K, Halbritter J, Belkacem L, Filhol E, Porath J, Braun D, Schueler M, et al. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am J Hum Genet. 2014;94(6):905–14. doi:10.1016/j.ajhg.2014.05.002.
- Veldman BCF, Kuper WFE, Lilien M, Schuurs-Hoeijmakers JHM, Marcelis C, Phan M, Hettinga Y, Talsma HE, Hasselt PM, Haijes HA. Beyond nephronophthisis: retinal dystrophy in the absence of kidney dysfunction in childhood expands the clinical spectrum of CEP83 deficiency. Am J Med Genet A. 2021;185(7):2204–10. doi:10.1002/ajmg.a.62225.
- Adams NA, Awadein A, Toma HS. The retinal ciliopathies. Ophthalmic Genet. 2007;28(3):113–25. doi:10.1080/13816810701537424.
- de Castro-Miro M, Tonda R, Escudero-Ferruz P, Andres R, Mayor-Lorenzo A, Castro J, Ciccioli M, Hidalgo DA, Rodríguez-Ezcurra JJ, Farrando J, et al. Novel candidate genes and a wide spectrum of structural and point mutations responsible for inherited retinal dystrophies revealed by exome sequencing. PLoS One. 2016;11(12):e0168966. doi:10.1371/journal.pone.0168966.
- Madhivanan K, Aguilar RC. Ciliopathies: the trafficking connection. Traffic. 2014;15(10):1031–56. doi:10.1111/tra.12195.
- Bujakowska KM, Liu Q, Pierce EA. Photoreceptor cilia and retinal ciliopathies. Cold Spring Harb Perspect Biol. 2017;9(10):a028274. doi:10.1101/cshperspect.a028274.
- Estrada-Cuzcano A, Roepman R, Cremers FP, den Hollander AI, Mans DA. Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum Mol Genet. 2012;21(R1):R111–24. doi:10.1093/hmg/dds298.
- Leroy BP, Birch DG, Duncan JL, Lam BL, Koenekoop RK, Porto FBO, Russell SR, Girach A. Leber congenital amaurosis due to cep290 mutations—severe vision impairment with a high unmet medical need. Retina. 2021;41(5):898–907. doi:10.1097/IAE.0000000000003133.
- Feldhaus B, Weisschuh N, Nasser F, den Hollander AI, Cremers FPM, Zrenner E, Kohl S, Zobor D. CEP290 mutation spectrum and delineation of the associated phenotype in a large German cohort: a monocentric study. Am J Ophthalmol. 2020;211:142–50. doi:10.1016/j.ajo.2019.11.012.
- Cideciyan AV, Jacobson SG. Leber congenital amaurosis (LCA): potential for improvement of vision. Invest Ophthalmol Vis Sci. 2019;60(5):1680–95. doi:10.1167/iovs.19-26672.
- Heon E, Westall C, Carmi R, Elbedour K, Panton C, Mackeen L, Stone EM, Sheffield VC. Ocular phenotypes of three genetic variants of Bardet-Biedl syndrome. Am J Med Genet A. 2005;132A(3):283–7. doi:10.1002/ajmg.a.30466.
- Billingsley G, Deveault C, Heon E. BBS mutational analysis: a strategic approach. Ophthalmic Genet. 2011;32(3):181–7. doi:10.3109/13816810.2011.567319.
- Campo RV, Aaberg TM. Ocular and systemic manifestations of the Bardet-Biedl syndrome. Am J Ophthalmol. 1982;94(6):750–6. doi:10.1016/0002-9394(82)90299-9.
- Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, Heath O, McManamon PJ, O’Leary E, Pryse-Phillips W. The cardinal manifestations of Bardet–Biedl syndrome, a form of Laurence–Moon–Biedl syndrome. N Engl J Med. 1989;321(15):1002–9. doi:10.1056/NEJM198910123211503.
- Riise R, Andreasson S, Wright AF, Tornqvist K. Ocular findings in the Laurence-Moon-Bardet-Biedl syndrome. Acta Ophthalmol Scand. 1996;74(6):612–7. doi:10.1111/j.1600-0420.1996.tb00746.x.
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437–46.
- Millay RH, Weleber RG, Heckenlively JR. Ophthalmologic and systemic manifestations of Alstrom’s disease. Am J Ophthalmol. 1986;102(4):482–90.
- Michaud JL, Heon E, Guilbert F, Weill J, Puech B, Benson L, Smallhorn JF, Shuman, CT, Buncic, JR, Levin, AV, Weksberg R, et al. Natural history of Alström syndrome in early childhood: onset with dilated cardiomyopathy. J Pediatr. 1996;128(2):225–9. doi:10.1016/S0022-3476(96)70394-3.
- Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, Beck S, Boerkoel, CF, Sicolo N, Martin M, Nishina, PM, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002;31(1):74–8. doi:10.1038/ng867.
- Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, Carey C, MacDermott, S, Russell-Eggitt I, Shea SE, Davis J, et al. New Alström syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165(6):675–83. doi:10.1001/archinte.165.6.675.
- Marshall JD, Maffei P, Collin GB, Naggert JK. Alstrom syndrome: genetics and clinical overview. Curr Genomics. 2011;12(3):225–35.
- Ronquillo CC, Bernstein PS, Baehr W. Senior-Loken syndrome: a syndromic form of retinal dystrophy associated with nephronophthisis. Vision Res. 2012;75:88–97.
- Sergouniotis PI, Hadfield KD, Black GC. Fundus examination pointing to the diagnosis of Senior-Loken syndrome. JAMA Ophthalmol. 2016;134(8):e161299.
- Tay SA, Vincent AL. Senior-Loken syndrome and intracranial hypertension. Ophthalmic Genet. 2020;41(4):354–7.
- Yahalom C, Volovelsky O, Macarov M, Altalbishi A, Alsweiti Y, Schneider N, Hanany M, Khan MI, Cremers Frans PM, Anteby I, Banin E, et al. Senior-Loken syndrome: a case series and review of the renoretinal phenotype and advances of molecular diagnosis. Retina. 2021;41(10):2179–87. doi:10.1097/IAE.0000000000003138.
- Whelan DT, Feldman W, Dost I. The oro-facial-digital syndrome. Clin Genet. 1975;8(3):205–12.
- Erickson RP, Bodensteiner JB. Oro-facial-digital syndrome IX with severe microcephaly: a new variant in a genetically isolated population. Am J Med Genet A. 2007;143A(24):3309–13.
- Grudzinska Pechhacker MK, Jacobson SG, Drack AV, Scipio MD, Strubbe I, Pfeifer W, Duncan JL, Dollfus H, Goetz N, Muller J, Vincent AL, et al. Comparative natural history of visual function from patients with biallelic variants in BBS1 and BBS10. Invest Ophthalmol Vis Sci. 2021;62(15):26. doi:10.1167/iovs.62.15.26.
- Rafalska A, Tracewska AM, Turno-Krecicka A, Szafraniec MJ, Misiuk-Hojlo M. A mild phenotype caused by two novel compound heterozygous mutations in CEP290. Genes (Basel). 2020;11(11): 1240 doi:10.3390/genes11111240.
- Kakar N, Horn D, Decker E, Sowada N, Kubisch C, Ahmad J, Borck G, Bergmann C. Expanding the phenotype associated with biallelic WDR60 mutations: siblings with retinal degeneration and polydactyly lacking other features of short rib thoracic dystrophies. Am J Med Genet A. 2018;176(2):438–42. doi:10.1002/ajmg.a.38562.
- McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015;130(1):1–12. doi:10.1007/s10633-014-9473-7.
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, Karapetyan K, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–7. doi:10.1093/nar/gkx1153.
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43. doi:10.1038/s41586-020-2308-7.
- Li J, Lu D, Liu H, Williams BO, Overbeek PA, Lee B, Zheng L, Yang T. Sclt1 deficiency causes cystic kidney by activating ERK and STAT3 signaling. Hum Mol Genet. 2017;26(15):2949–60. doi:10.1093/hmg/ddx183.
- Sakaguchi N, Suyama M. Pervasive occurrence of splice-site-creating mutations and their possible involvement in genetic disorders. NPJ Genom Med. 2022;7(1):22.
- Qian X, Wang J, Wang M, Igelman AD, Jones KD, Li Y, Wang K, Goetz KE, Birch DG, Yang P, Pennesi ME, et al. Identification of deep-intronic splice mutations in a large cohort of patients with inherited retinal diseases. Front Genet. 2021;12:647400. doi:10.3389/fgene.2021.647400.
- Khan AO, Decker E, Bachmann N, Bolz HJ, Bergmann C. C8orf37 is mutated in Bardet-Biedl syndrome and constitutes a locus allelic to non-syndromic retinal dystrophies. Ophthalmic Genet. 2016;37(3):290–3.
- Black GC, Sergouniotis P, Sodi A, Leroy BP, Van Cauwenbergh C, Liskova P, Grønskov K, Klett A, Kohl S, Taurina G, Sukys M, et al. The need for widely available genomic testing in rare eye diseases: an ERN-EYE position statement. Orphanet J Rare Dis. 2021;16(1):142. doi:10.1186/s13023-021-01756-x.
- Eden M, Payne K, Jones C, Wright SJ, Hall G, McAllister M, Black G. Identifying variation in models of care for the genomic-based diagnosis of inherited retinal dystrophies in the United Kingdom. Eye (Lond). 2016;30(7):966–71. doi:10.1038/eye.2016.74.
- Gong J, Cheung S, Fasso-Opie A, Galvin O, Moniz LS, Earle D, Durham T, Menzo J, Li N, Duffy S, Dolgin J, et al. The impact of inherited retinal diseases in the United States of America (US) and Canada from a cost-of-illness perspective. Clin Ophthalmol. 2021;15:2855–66. doi:10.2147/OPTH.S313719.
- Galvin O, Chi G, Brady L, Hippert C, Del Valle Rubido M, Daly A, Michaelides, M. The impact of inherited retinal diseases in the Republic of Ireland (ROI) and the United Kingdom (UK) from a cost-of-illness perspective. Clin Ophthalmol. 2020;14:707–19. doi:10.2147/OPTH.S241928.