
ABSTRACT
Background
To report a cohort of patients with clinically and genetically diagnosed autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV) and showcase the spectrum of the disease utilizing multimodal imaging and genetic testing. Additionally, the utility of multimodal imaging in guiding treatment will also be illustrated.
Materials/Methods
Five patients from a single-family pedigree in Ohio with clinical signs of ADNIV were evaluated. Medical history, family history, and complete ocular examinations were obtained during regular clinic visits. Multimodal imaging including ocular coherence tomography, fluorescein angiography, wide-field fundus photographs, and Humphrey visual field testing was obtained for all five patients. Additionally, genetic testing for the Calpain-5 (CAPN5) gene was conducted on all patients.
Results
All five patients were noted to have a CAPN5 c.731T > C (p.L244P) mutation on genetic testing. Using multimodal imaging to supplement the clinical examination, pathologic changes such as retinal vascular inflammation, macular edema, and tractional retinal membranes were well illustrated and monitored over time. This allowed for earlier intervention when appropriate such as with intraocular steroid or systemic anti-inflammatory treatments.
Conclusion
Phenotypic presentation varied among patients in this series, but is consistent with the spectrum of pathologic changes previously described in patients with other CAPN5 gene mutations. Monitoring of patients with ADNIV utilizing multimodal imaging can help better assess progression of this disease and guide treatment decisions. Additionally, increased genetic testing in patients with inherited retinal diseases may reveal novel gene mutations that could serve as potential targets for future genetic treatment regimens.
1. Introduction
Autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV, OMIM# 193235) is a rare inherited retinal dystrophy that presents as a bilateral panuveitis that is progressive in nature and typically leads to severe vision loss. It usually appears in the second and third decades of life but has also been identified in infants and children as young as three years old (Citation1–3). It was first described in 1990 in a six-generation family that were noted to have varying degrees of peripheral visual field loss, uveitis, and chorioretinal findings consistent with progressive retinal pigmentary degeneration and lattice-like peripheral thinning (Citation4). A total of 28 related patients were identified out of 61 members of a six-generation family, and the disease was noted to follow an autosomal dominant inheritance pattern (Citation4). Many patients were noted to have findings similar to retinitis pigmentosa in addition to the presence of a significant inflammatory response and neovascularization of both the anterior and posterior segments. This constellation of clinical features and well-defined inheritance pattern was thus termed “ADNIV.” Exome sequencing identified mutations in the Calpain-5 (CAPN5) gene to be the cause (Citation5).
The CAPN5 gene, encoding an intracellular calcium-activated protease, is expressed throughout the retina localizing to the photoreceptor outer segments, the inner plexiform layer, retinal ganglion cells, and retinal mitochondria. Several different mutations associated with increased protease function and mis-localization of the protein have been identified in patients with ADNIV, leading to a variety of deleterious downstream effects (Citation6–8). A large proportion of identified cases have been found in four families of related patients, all of Northern European descent, though other sporadic cases with CAPN5 mutations have been reported (Citation8,Citation9).
The disease can have a variable presentation and severity but is typically characterized by progressively worsening panuveitis and eventual neovascularization (Citation5). Findings can include cystoid macular edema (CME), retinal pigmentary changes, vitreous hemorrhage, preretinal fibrosis, neovascular glaucoma, tractional retinal detachments, and eventual phthisis bulbi (Citation5). A staging system for disease classification was defined by Mahajan et al. (Citation5), ranging from mild inflammation in stage I up to severe tractional retinal detachments and eventual phthisis bulbi in stages IV and V respectively (Citation5). Given the similarity of some findings to those in other forms of uveitis, proliferative diabetic retinopathy, and retinitis pigmentosa, patients’ family history plays a large role in diagnosis. With the identification of the CAPN5 gene, genetic testing can now also be utilized to aid in making this diagnosis.
With the vast majority of cases limited to only four documented family cohorts, there is sparse scientific literature pertaining to this retinal condition. Most prior clinical case descriptions have focused on the CAPN5 c.728 G > T (p.R243L) variant, but as additional mutations have emerged, there are noted genotype–phenotype differences (Citation8). The cases illustrated here showcase the diverse array of clinical presentations for individuals with the CAPN5 c.731T > C (p.L244P) mutation. These cases also underscore the utility of multimodal imaging for continued monitoring and treatment of patients with this condition.
2. Materials and methods
We present five members of a single-family cohort that have clinical signs and symptoms that are suggestive of ADNIV (). Prior to this study, only one member of the family had undergone genetic testing, which was positive for a pathognomonic CAPN5 mutation. Patient’s ages ranged from 15 to 59 years, and disease staging varied across the cohort (). Institutional Review Board Committee approval was obtained, and the research adhered to the tenets of the Declaration of Helsinki. Written informed consent was obtained from all patients for publication of all images and data.
Figure 1. Family pedigree illustrating the relationships among all five members of the patients represented in this case series.
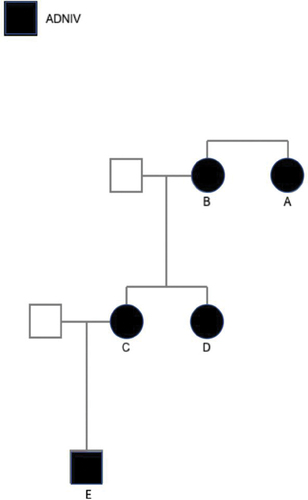
Figure 2. Table highlighting the demographics and relevant past medical history of all five patients. Additionally, staging of the severity of the disease was assigned based on the classification system established by Mahajan et al. 2012.5
In all patients, a complete workup for alternative causes of uveitis was first undertaken to exclude other causes of intraocular inflammation. Laboratory studies including complete blood count, basic metabolic panel, and hemoglobin A1c were unremarkable in all patients. Testing for inflammatory (angiotensin-converting enzyme, anti-nuclear antibody, and antineutrophil cytoplasmic antibody levels) and infectious (QuantiFERON-gold, Rapid Plasma Reagin, Fluorescent Treponemal Antibody-Absorption, and chest x-ray) etiologies were all normal.
Examination included measuring visual acuity using a Snellen chart as well as intraocular pressure using Goldmann applanation tonometry. Slit lamp and indirect biomicroscopy examination of the anterior and posterior segments was performed in all eyes. Further testing included ocular coherence tomography of the macula (OCT) (Zeiss Cirrus HD-OCT, Carl Zeiss Meditec, Dublin, CA, USA), wide-field fundus photographs (Optos California, Optos, Marlborough, MA, USA), Humphrey visual field (HVF) (Carl Zeiss Humphrey Field Analyzer, Carl Zeiss Meditec, Dublin, CA, USA), fundus autofluorescence (Optos California), and fluorescein angiography (FA) (Optos California). Lastly, genetic testing was conducted for all patients, utilizing a commercially available inherited retinal disease (IRD) panel (Spark Therapeutics, Philadelphia, USA). Physical examination findings and all testing were gathered during sequential visits.
3. Results
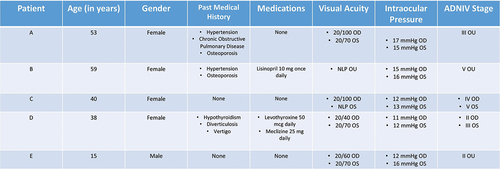
All of the examination data presented were from sequential clinic dates during treatment and evaluation of these patients. Best-corrected visual acuity ranged from 20/40 to no light perception vision, with all 10 eyes showing evidence of inflammation and/or fibrosis on examination. Disease staging ranged from Stage II up to Stage V, with a trend toward worsened best-corrected visual acuity with increased eye disease stage (). Furthermore, all patients carried the identical CAPN5 c.731T > C (p.L244P) mutation (, , , and ).
Figure 3. (A/B) color fundus photograph, OD/OS, with vitreous haze overlying superior traction and temporal pigmentary retinopathy. (C/D) FA, OD/OS, revealing diffuse leakage around the disc as well as along vessels, along with patchy areas of nonperfusion correlating with inner areas of pigmentary retinopathy. (E/F) OCT macula, OD/OS, demonstrating severe macular traction in both eyes and cystoid macular edema. (G) SPARK genetic testing showing gene mutations and variants, consistent with pathogenic CAPN5 mutation. (H/I) HVF 30–2, OD/OS, revealing peripheral constriction in both eyes with the left eye more severely affected and then the right with remaining central island.
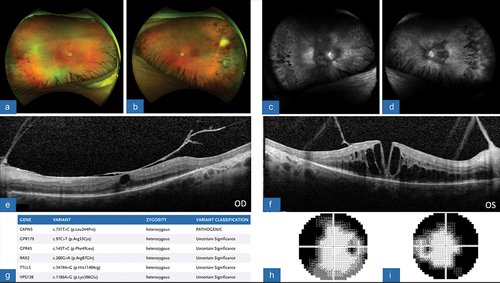
Figure 4. (A) color fundus photograph, OD, showing the presence of a thin posterior retina, mid-peripheral subretinal exudates, and attenuation of the vessels. The superior retina has evidence of crystalline deposits and exudates at the site of previous tractional retinal detachment after undergoing repair with pars plana vitrectomy. (B) FA, OD, revealing vast areas of non-perfusion along the periphery of the retina with leakage and late staining of the optic disc. (C) OCT macula, OD, demonstrating severely diminutive and thin retina, with diffuse thinning of choriocapillaris and a disorganized outer retinal layer with poor foveal contour. (D) SPARK genetic testing showing gene mutations and variants, consistent with pathogenic CAPN5 mutation. (E) HVF 30–2, OD, demonstrating severe depression of all points with some central sparing.
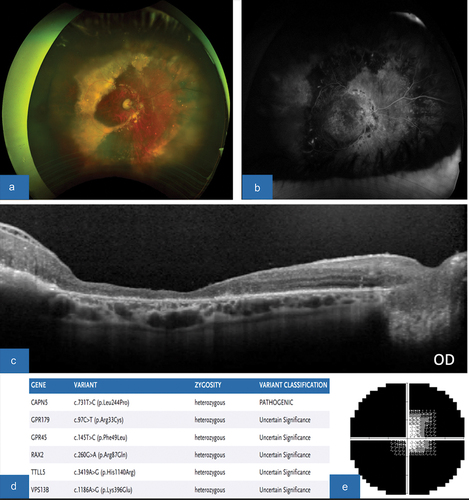
Figure 5. (A/B) color fundus photograph, OD/OS, showing neovascularization and fibrosis of the disc and periphery OU, severe exudative lesions OD with subretinal fibrosis, and areas of peripheral tractional membrane with surrounding atrophy OU. (C/D) FA, OD/OS, revealing areas of severe leakage around disc and extensive nonperfusion. (E/F) OCT macula, OD/OS, demonstrates prominent epiretinal membrane severe CME with areas of subretinal exudative lesions and loss of IS/OS junction. (G) SPARK genetic testing showing gene mutations and variants, consistent with pathogenic CAPN5 mutation. (H/I) HVF 30–2, OD/OS, revealing severe constriction with central island OD>OS.
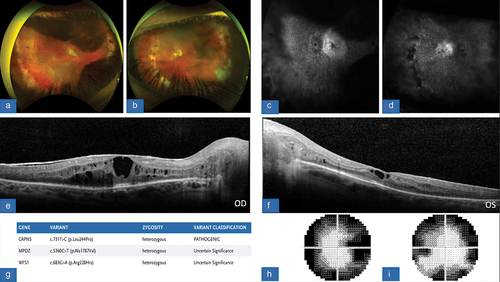
Figure 6. (A/B) color fundus photograph, OD/OS, with evidence of RPE disruption along with a tractional membrane, greater in temporal OS>OD. (C/D) FA, OD/OS, revealing of areas of non-perfusion along nasal and temporal peripheries of the retina. There is global inner retinal and outer retinal non-perfusion with leakage. (E/F) OCT macula, OD/OS, showing diffuse retinal thickening and enlarged outer plexiform and outer nuclear layer. Vitreous debris is noted OS as well as small intra-retinal cysts. (G) SPARK genetic testing showing gene mutations and variants, consistent with pathogenic CAPN5 mutation as well as BBS1 and ABCA4. (H/I) HVF 30–2, OD/OS, demonstrates defects in visual field mostly involving the superior aspect in an arcuate pattern.
Patient A was a 53-year-old female who presented with bilateral findings with moderate vision loss, including best-corrected visual acuities of approximately 20/80 and 20/70 in the right and left eyes, respectively. The clinical examination was notable for mild vitritis, the appearance of retinal traction, optic nerve pallor, retinal vessel attenuation, and peripheral pigmentary changes in both eyes, which was also noted and documented on ultra-wide-field fundus imaging of both eyes (). FA showed evidence of persistent inflammation with the presence of small vessel leakage and optic disc leakage in both eyes, as well as findings consistent with the vascular attenuation and peripheral pigmentary changes (). OCT imaging demonstrated the presence of macular edema and vitreomacular traction present in both eyes, with the more severe traction in the right eye appearing to correspond with increased intraretinal fluid and retinal thickening (). HVF testing demonstrated bilateral generalized constriction and genetic testing confirmed presence of the CAPN5 c.731T > C (p.L244P) mutation (). The patient was started on topical prednisolone four times per day in both eyes due to the presence of CME and inflammation. On follow up one month later, the CME on OCT imaging had improved in the left eye but had worsened in the right. Elevated intraocular pressure in the same eye precluded the use of sub-Tenon’s or intravitreal steroids consequently the patient was referred to the Rheumatology service for planned initiation of systemic immune-modulating therapy with continued monitoring in clinic. Vitrectomy surgery was also considered, particularly if the vitreomacular traction in the right eye progressed with further monitoring utilizing OCT imaging during her subsequent visits.
Patient B was a 59-year-old female who presented with bilateral phthisis and no light perception vision in both eyes, noting that she had been blind in both eyes for approximately 23 years. While previously treated, all drops had been discontinued in the past per patient preference given her lack of discomfort or pain. Due to end-stage phthisis, no imaging is included. Genetic testing, however, confirmed the presence of the CAPN5 gene mutation.
Patient C is a 42-year-old female who was noted to have a phthisical left eye secondary to end-stage ADNIV. She had been receiving regular treatment with intraocular corticosteroids to both eyes preoperatively, as well as recurrent placement of intravitreal 0.59 milligram (mg) fluocinolone acetonide implants (Retisert, Bausch and Lomb, Laval, Canada). She is pseudophakic in both eyes. After being lost to follow-up for six months, she presented with decreased in vision in her right eye to hand motion. On examination, she was noted to have a tractional retinal detachment involving the central macula, as well as an anterior cyclitic membrane overlying the pupil. Given the vision threatening nature of the retinal detachment, surgical intervention was advised. She underwent 25-gauge pars plana vitrectomy for retinal detachment repair as well as intravitreal injection of triamcinolone acetonide (Supplementary Video 1). Her postoperative vision gradually improved in the right eye to 20/100, which was maintained at the reported one-year follow-up visit following her surgery. Her multimodal imaging findings postoperatively showed areas of crystalline deposits and exudates near the area of the previous retinal detachment in addition to a severely diminutive retina evident on OCT (). Despite successful vitrectomy surgery, recalcitrant posterior uveitis eventually recurred requiring a 0.18 mg intravitreal implant injection of fluocinolone acetonide (Yutiq, Eyepoint Pharmaceuticals, Watertown, MA, USA) in the right eye for control of inflammation. Vision was noted to be stable one month after with a decrease in the degree of inflammatory reaction.
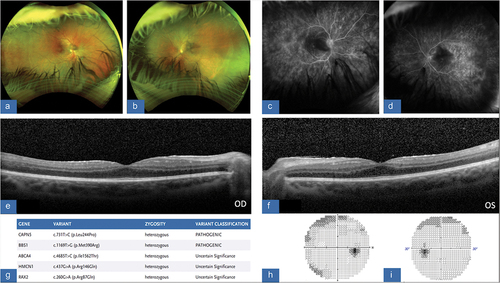
Patient D is a 12 -year-old female who was noted to have a visual acuity of 20/40 in the right eye and 20/70 in the left eye. She is pseudophakic in both eyes and had a moderate amount of vitritis. On funduscopic examination, she was found to have temporal pigmentary changes on examination in conjunction with CME (). FA also revealed leakage around the optic disc with extensive areas of non-perfusion again correlating with the areas of pigmentary changes in the far periphery (). Consequently, patient D received a 0.18 mg fluocinolone acetonide implant in the left eye given the significant amount of inflammation and CME noted on OCT. She was followed closely and after one month was found to have stable examination findings, specifically no change in CME, flare, and no progression of peripheral retinopathy.
Patient E is a 17 -year-old male who presented with several months of decreasing vision. His visual acuity during this visit was found to be 20/60 in the right eye and 20/70 in the left eye. His anterior chamber was noted to be deep and quiet, with the presence of a mild posterior subcapsular cataract in both eyes. He was found to have vitreoretinal traction developing in the far temporal periphery of both fundi with areas of leakage and non-perfusion also in the far periphery (). Due to the prominent vascular leakage seen on FA, out of proportion to the relatively unaffected fundus appearance, more aggressive steroid management with topical difluprednate was instituted as a “bridge” to long-term treatment with systemic antimetabolites. Patient E was consequently referred to rheumatology for co-management with systemic methotrexate.
4. Discussion
ADNIV is a rare clinical entity that has thus far only been diagnosed in a small cohort of families and individuals worldwide. As evident by the imaging findings, these patients typically develop progressively worsening intraocular inflammation and neovascularization, which frequently results in severe vision loss by the fifth and sixth decades of life, although there can be variable penetrance (Citation5,Citation8,Citation10,Citation11). A strong risk factor for poor visual outcomes is the presence of tractional retinal detachment, which develops as a result of worsening posterior segment fibrosis and neovascularization (Citation11,Citation12). There are specific genotype–phenotype correlations where disease can occur earlier. The CAPN5 c.865C>T (p.Arg289Trp) variant causes a severe, early-onset pediatric ADNIV, and in the CAPN5 c.799 G>A, p. (Gly267Ser) and CAPN5 c.1126 G>A, p. (Gly376Ser) disease onset is earlier with severe vision loss in the 20s (Citation1,Citation2). The development of anterior segment neovascularization and glaucoma has also been associated with poor outcomes (Citation11,Citation13). While these complications can be severe, they typically develop after decades of disease activity and early identification and appropriate management may delay or prevent severe vision loss. Regular, frequent examination and imaging for these patients to monitor for progression is therefore of significant importance and may allow for delaying or preventing some of the most serious sequelae of this disease.
In the earliest stages of ADNIV examination, findings can be nonspecific, with only mild signs of inflammation similar to a non-infectious posterior uveitis (Citation5). As noted, however, certain variants can lead to earlier and more severe presentations, such as the c.865C>T (p.Arg289Trp) variant associated with pediatric NIV (Citation1,Citation2). Even in mild, early disease patients can begin to show changes on testing with gradual reduction in the B-wave on electroretinography (ERG) (Citation14). As the disease progresses, patients begin to develop findings such as optic disc edema and CME, which can be apparent on OCT and FA testing (Citation5). Mild retinal vasculitis may be present, which can also be identified with wide-angle FA (Citation5,Citation8). Dilated fundus examination and wide-angle fundus imaging are often notable for peripheral retinal pigmentary changes, and ERG testing can show complete loss of the B-wave, most frequently in the scotopic bright-flash ERG (Citation14).
Given that calpains are postulated to be vital proteins for cell regulation in photoreceptors, it is thought that the earliest ERG change with loss of the B-wave is due to a photoreceptor synaptic dysfunction (Citation7,Citation15). As the disease progresses over many years, the pathology begins to become more apparent on clinical examination, with worsening anterior segment inflammation and cataract formation (Citation5). In the posterior segment epiretinal membranes, macular holes, vitreoretinal traction and CME can be seen on examination, fundus photography, and OCT imaging (Citation5,Citation11). Fundus examination and wide-angle fundus imaging can also show retinal fibrosis, neovascularization, tractional retinal detachments, worsening retinal pigmentary changes, and the development of bands of fibrosis within the vitreous (Citation5).
The retinal degeneration that typifies this disease is particularly apparent on wide-angle fundus auto-fluorescence displaying RPE and photoreceptor degeneration, which is not always apparent on older fundus images with a narrow field of view (Citation7,Citation9,Citation16). FA typically shows continued evidence of inflammatory changes, and wide-field angiography is useful in highlighting any subtle neovascular changes throughout the retina (Citation5). Finally, ERG changes become even more pronounced, with worsening reduction in the A-wave in addition to the previous loss of the B-wave (Citation5,Citation14). In the severe stages of the disease, much of the key pathology is readily apparent on examination, with severe tractional retinal detachments, vitreous hemorrhages, and possible angle closure or neovascular glaucoma (Citation5). Utilizing B-scan ultrasonography can be useful in some cases to better assess the posterior segment, as these patients frequently develop severe cataracts, vitreous hemorrhages, anterior tractional retinal detachments, and cyclitic membranes that preclude a complete posterior examination. The ERG is typically completely extinguished at this point, with severe dysfunction of both rods and cones (Citation7,Citation14).
The examination and imaging findings in the patients in this case series are illustrative of the findings and progression of moderate to advanced disease in ADNIV. This includes the presence of cataracts, CME, posterior segment inflammation, progressive retinal traction, retinal detachments, and eventual phthisis. As seen in this case series, the disease can cause significant retinal changes and vision loss, even from a relatively young age. At 15 years old, patient E required topical steroids and systemic anti-inflammatory medication due to the significant amount of posterior segment inflammation. Additionally, there was presence of other related findings such as retinal traction and bilateral cataracts. Patient B had already developed bilateral phthisis with no light perception vision in both eyes by the age of 59. This underscores the importance of imaging in these patients, as in this series it allowed for improved monitoring and earlier detection of disease changes. The detection of such changes such as the cystoid macular edema in patients A and D, retinal traction in patient A, and vascular leakage in patients D and E played a key role in treatment decisions for these patients and likely allowed for earlier intervention as opposed to relying solely on subtle examination findings.
Notably, patient E had subtle findings on clinical examination and was of relatively young age for the development of more serious pathology in ADNIV. However, he was found to have more severe changes such as extensive non-perfusion and vascular leakage on wide-field FA requiring prompt treatment. Disease monitoring is also aided by regular imaging, such as the retinal traction in patient A being monitored with serial OCT scans, ensuring even mild progression can be detected. The imaging findings in this cohort are typical of those reported elsewhere and underscore the progressive nature of this disease.
These patients also demonstrate the difficulty in treating patients with ADNIV given the often relentless progression to severe vision loss in most patients. Given the rarity of the disease, there are currently no criteria for specific treatment procedures that are considered standard-of-care. However, given the triad of uveitis, neovascularization, and fibrosis that are hallmarks of this disease, multiple treatments targeting different aspects of these disease processes should be considered.
Topical, periocular, and intravitreal injections of steroids are the cornerstones of medical treatment for active inflammation, underscoring the inflammatory reaction that typically prevails in this disease process (Citation11). Of note, intraocular steroids including fluocinolone acetonide implants have been shown to decrease the degree of inflammation and neovascularization but do not stabilize vision long term, mitigate end-stage fibrosis, decrease retinal thickening, or inhibit photoreceptor degeneration (Citation12).
Systemic immunosuppressants are often used as an adjunct to steroid treatment. It has been demonstrated from proteomic analysis of vitreous biopsies in ADNIV patients that major T-cell signaling pathways are implicated including MTOR and PI3K (Citation11,Citation17). Notably, analysis of vitreous samples from these patients revealed the absence of the inflammatory cytokine TNF-alpha. This likely explains the lack of treatment response to infusions of the anti-TNF alpha therapeutic infliximab (Citation18). Methotrexate, through inhibition of T-cell activation, may be utilized for treatment of the uveitic component of this disease process. The anti-metabolite is also thought to potentially mitigate the fibrotic changes that can develop, based on its utility in treating other fibrotic disease processes such as proliferative vitreoretinopathy (Citation11,Citation19). Similar to the use of methotrexate, azathioprine has also been used in an attempt to control the inflammatory changes present in this disease (Citation11).
Additionally, overexpression of cytokines including IL-6 has been noted in patients with ADNIV which likely play a role in exacerbating fibrotic disease and inflammation (Citation18,Citation20). Medications that target IL-6 include tocilizumab and sarilumab, which are more commonly used in the treatment of systemic autoimmune conditions such as rheumatoid arthritis. Intravenous infusions of tocilizumab have been trialed for ADNIV, showing promise for the purposes of controlling the severe intraocular proliferation that can develop in these patients postoperatively (Citation18).
Despite their benefits, the systemic use of immunosuppressive agents has many well-documented risks and potential side effects. There has been recent exploration of the use of intravitreal immune-modulating agents in order to achieve high local drug concentrations while minimizing systemic risks. Velez et al (Citation18) explored the use of intravitreal methotrexate, finding significant decreases in vitreous inflammation after intravitreal methotrexate injections (400 µg/0.1 mL).
Given the neovascularization and vitreous hemorrhage that is frequently seen in advanced disease, adjunctive intravitreal anti-vascular endothelial growth factor injections have also been explored (Citation11,Citation18,Citation21). These injections have shown some promise in managing some of the neovascular sequelae of this disease when used in conjunction with treatment of the inflammation but have also been associated with poor visual outcomes long term (Citation11,Citation18,Citation21). It is unclear if this is due to patients requiring such treatment representing more severe disease progression overall, or if these injections can cause worsening of retinal traction due to contraction of proliferative neovascular membranes (Citation11). Laser photocoagulation has also been explored for the management of retinal neovascularization but has not been shown to lead to improvement (Citation4).
Surgical intervention with vitrectomy is often necessitated in the case of progressive retinal traction and retinal detachment and can help minimize or delay severe vision loss. These patients, however, frequently develop recurrent retinal detachments and cyclitic membranes, and surgical intervention may be only partially beneficial in the long term (Citation1).
As these patients often also develop elevated glaucoma secondary to chronic steroids, neovascularization, or inflammation, close monitoring and treatment of intraocular pressure is frequently necessary. Topical intraocular pressure-lowering medications are frequently used, but given the diverse array of etiologies of glaucoma in patients with this disease, surgical management such as trabeculectomy or tube shunt placement is often required (Citation13).
Overall, the vast array of treatment modalities reflects the three-prong disease process that is present in patients with this disease including inflammation, neovascularization, and fibrosis. These cases demonstrate the key role that both local and systemic anti-inflammatory and/or immune-modulating therapies play in the management of this disease. All patients in this series received or had received local steroids in the past, with even phthisical eyes frequently continued on topical steroids to minimize inflammation and ocular discomfort. As noted, given the many manifestations of this disease, there are many options used to address sequelae, and obtaining an assortment of testing and imaging to allow for early identification and treatment is essential for these patients.
Of note, all five patients tested positive for the same mutation of the CAPN5 gene. The mutation was found to be a p.Leu244Pro which is the same mutation found in the ADNIV-3 family mutation (Citation5). The calpain family of proteases has also been implicated in a number of other systemic disease processes and ocular disease (Citation22). For example, the gene encoding for CAPN3 has been implicated in causing limb-muscle girdle dystrophy, while CAPN10 and CAPN5 variants have been associated with development of 2 Diabetes M”>type 2 diabetes mellitus (Citation5). Calpains are known to be calcium dependent and are involved in mediating limited amounts of intracellular proteolysis. Mutations in these genes that regulate cellular homeostasis lead to increased expression of numerous inflammatory cytokines (Citation8,Citation23). This, in turn, causes unchecked inflammation, neovascularization, and fibrosis demonstrated in patients with ADNIV. Even among members of the same family who share the same CAPN5 mutation, there exists a large amount of phenotypic heterogeneity. This may indicate the presence of disease modifiers that may benefit from further investigation that genetic testing may aid in identifying.
The role of gene therapy with adeno-associated viral (AAV) vectors for IRD has rapidly evolved in the last decade (Citation24). In 2017, the FDA approved coretigene neparvovec-ryzl (Luxturna, Spark Therapeutics). This medication is a gene therapy for the treatment of diallelic RPE65 mutations utilizing an engineered adeno-associated virus (AAV) as a vector for treatment. Unlike other retinal genetic diseases where there is loss of function such as RPE65 variant-associated retinal dystrophy, the postulated pathophysiology of ADNIV involves gain of function in the CAPN5 gene (Citation9,Citation25,Citation26). There has been sparse research done on dominant gain of function genetic treatments, and this may be a direction for future research.
While ADNIV is a rare disease that affects a small number of patients globally, multimodal imaging is an invaluable tool for clinicians to aid in diagnosis, treatment, and monitoring for patients that have this condition. Further scientific inquiry and research in this disease’s pathogenesis and treatment may have implications for better management and understanding of a variety of other uveitic and retinal degenerative conditions. Additionally, better understanding of the neovascular conditions encountered in patients with ADNIV may have implications for the more commonly treated and evaluated retinal conditions such as diabetic retinopathy and neovascular age-related macular degeneration.
The increased utility of genetic testing for retinal diseases aids in discovering future therapeutic targets as the depth and breadth of treatment of retinal diseases continues to evolve. As more cases of rare retinal dystrophies with genetic testing are reported, we speculate that many variants of unknown significant may become better understood and potentially allow for subsequent development of novel gene therapies.
Supplemental Material
Download MP4 Video (95.3 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/13816810.2023.2255257.
Additional information
Funding
References
- O’Keefe G, Hanif AM, Mahajan VB, Jain N. Early onset neovascular inflammatory vitreoretinopathy due to a De Novo CAPN5 mutation: report of a case. Ocul Immunol Inflamm. 2019;27(5):706–8. doi:10.1080/09273948.2019.1582783. Epub 2019 Apr 15. PMID: 30986125; PMCID: PMC7173350.
- Velez G, Bassuk AG, Schaefer KA, Brooks B, Gakhar L, Mahajan M, Kahn P, Tsang SH, Ferguson PJ, Mahajan VB. A novel de novo CAPN5 mutation in a patient with inflammatory vitreoretinopathy, hearing loss, and developmental delay. Cold Spring Harb Mol Case Stud. 2018 Jun 1;4(3):a002519. doi:10.1101/mcs.a002519. PMID: 29472286; PMCID: PMC5983175.
- Wang Y, Li H, Zang S, Li F, Chen Y, Zhang X, Song Z, Peng Q, Gu F. Photoreceptor cell-derived CAPN5 regulates retinal pigment epithelium cell proliferation through direct regulation of SLIT2 cleavage. Invest Ophthalmol Visual Sci. 2018 Apr 1;59(5):1810–21. doi:10.1167/iovs.17-22689. PMID: 29610848.
- Bennett SR, Folk JC, Kimura AE, Russell SR, Stone EM, Raphtis EM. Autosomal dominant neovascular inflammatory vitreoretinopathy. Ophthalmology. 1990 Sep;97(9):1125–35. discussion 1135-6. doi:10.1016/s0161-6420(90)32447-8. PMID: 2234842.
- Mahajan VB, Skeie JM, Bassuk AG, Fingert JH, Braun TA, Daggett HT, Folk JC, Sheffield VC, Stone EM, Barsh GS. Calpain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS Genet. 2012;8(10):e1003001. doi:10.1371/journal.pgen.1003001. Epub 2012 Oct 4. PMID: 23055945; PMCID: PMC3464205.
- Velez G, Sun YJ, Khan S, Yang J, Herrmann J, Chemudupati T, MacLaren RE, Gakhar L, Wakatsuki S, Bassuk AG, Mahajan VB. Structural insights into the unique activation mechanisms of a non-classical calpain and its disease-causing variants. Cell Rep. 2020 Jan 21;30(3):881–92.e5. doi:10.1016/j.celrep.2019.12.077. PMID: 31968260; PMCID: PMC7001764.
- Wert KJ, Bassuk AG, Wu WH, Gakhar L, Coglan D, Mahajan M, Wu S, Yang J, Lin CS, Tsang SH, Mahajan VB. CAPN5 mutation in hereditary uveitis: the R243L mutation increases calpain catalytic activity and triggers intraocular inflammation in a mouse model. Hum Mol Genet. 2015 Aug 15;24(16):4584–98. doi:10.1093/hmg/ddv189. Epub 2015 May 20. PMID: 25994508; PMCID: PMC4512628.
- Velez G, Mahajan VB. Molecular surgery: proteomics of a rare genetic disease gives insight into common causes of blindness. iScience. 2020 Oct 13;23(11):101667. doi:10.1016/j.isci.2020.101667. PMID: 33134897; PMCID: PMC7586135.
- Bassuk AG, Yeh S, Wu S, Martin DF, Tsang SH, Gakhar L, Mahajan VB, Wen R. Structural modeling of a novel CAPN5 mutation that causes uveitis and neovascular retinal detachment. PLoS One. 2015 Apr 9;10(4):e0122352. doi:10.1371/journal.pone.0122352. PMID: 25856303; PMCID: PMC4391918.
- Tang PH, Chemudupati T, Wert KJ, Folk JC, Mahajan M, Tsang SH, Bassuk AG, Mahajan VB. Phenotypic variance in calpain-5 retinal degeneration. Am J Ophthalmol Case Rep. 2020 Feb 24;18:100627. doi:10.1016/j.ajoc.2020.100627. PMID: 32274441; PMCID: PMC7132063.
- Boyce TM, Whitmore SS, Varzavand K, Russell SR, Sohn EH, Folk JC, Stone EM, Han IC. Long-term outcomes and risk factors for severe vision loss in autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV). Am J Ophthalmol. 2022 Jan;233:144–52. doi:10.1016/j.ajo.2021.07.015. Epub 2021 Jul 21. PMID: 34302771; PMCID: PMC9177238.
- Tlucek PS, Folk JC, Sobol WM, Mahajan VB. Surgical management of fibrotic encapsulation of the fluocinolone acetonide implant in CAPN5-associated proliferative vitreoretinopathy. Clin Ophthalmol. 2013;7:1093–8. doi:10.2147/OPTH.S43939. Epub 2013 Jun 10. PMID: 23785231; PMCID: PMC3682853.
- Cham A, Bansal M, Banda HK, Kwon Y, Tlucek PS, Bassuk AG, Tsang SH, Sobol WM, Folk JC, Yeh S, et al. Secondary glaucoma in CAPN5-associated neovascular inflammatory vitreoretinopathy. Clin Ophthalmol. 2016 Jun 27;10:1187–97. doi:10.2147/OPTH.S103324. PMID: 27390515; PMCID: PMC4930228.
- Tang PH, Kinnick TR, Folk JC, Mahajan M, Bassuk AG, Tsang SH, Mahajan VB. Progression of scotopic single-flash electroretinography in the stages of capn5 vitreoretinopathy. Retin Cases Brief Rep. 2021 Jul 1;15(4):473–8. doi:10.1097/ICB.0000000000000828. PMID: 30300311; PMCID: PMC6453748.
- Schaefer KA, Toral MA, Velez G, Cox AJ, Baker SA, Borcherding NC, Colgan DF, Bondada V, Mashburn CB, Yu CG, Geddes JW, Tsang SH, Bassuk AG, Mahajan VB. Calpain-5 expression in the retina localizes to photoreceptor synapses. Invest Ophthalmol Visual Sci. 2016 May 1;57(6):2509–21. doi:10.1167/iovs.15-18680. PMID: 27152965; PMCID: PMC4868102.
- Randazzo NM MBBCh, BSc(Hons), Shanks ME PhD, Clouston P PhD, FRCPath, MacLaren RE MB ChB, DPhil, FRCOphth. Two novel CAPN5 variants associated with mild and severe autosomal dominant neovascular inflammatory vitreoretinopathy phenotypes. Ocul Immunol Inflamm. 2019;27(5):693–8. doi:10.1080/09273948.2017.1370651. Epub 2017 Oct 17. PMID: 29040051; PMCID: PMC6711405.
- Mahajan VB, Vallone JG, Lin JH, Mullins RF, Ko AC, Folk JC, Stone EM. T-cell infiltration in autosomal dominant neovascular inflammatory vitreoretinopathy. Mol Vis. 2010 Jun 8;16:1034–40. PMID: 20596252; PMCID: PMC2893052.
- Velez G, Bassuk AG, Colgan D, Tsang SH, Mahajan VB. Therapeutic drug repositioning using personalized proteomics of liquid biopsies. JCI Insight. 2017 Dec 21;2(24):e97818. doi:10.1172/jci.insight.97818. PMID: 29263305; PMCID: PMC5752263.
- Amarnani D, Machuca-Parra AI, Wong LL, Marko CK, Stefater JA, Stryjewski TP, Eliott D, Arboleda-Velasquez JF, Kim LA. Effect of methotrexate on an in vitro Patient-derived model of proliferative vitreoretinopathy. Invest Ophthalmol Visual Sci. 2017 Aug 1;58(10):3940–9. doi:10.1167/iovs.16-20912. PMID: 28777835; PMCID: PMC5544356.
- Chen X, Yang W, Deng X, Ye S, Xiao W. Interleukin-6 promotes proliferative vitreoretinopathy by inducing epithelial-mesenchymal transition via the JAK1/STAT3 signaling pathway. Mol Vis. 2020 Jul 29;26:517–29. PMID: 32818015; PMCID: PMC7406861.
- Mangla D, Par G, Saggau D, Alliman K, Nielsen J. Management of CAPN5 NIV with intensive combination anti-VEGF and corticosteroid therapy. Invest Ophthalmol Visual Sci. 2019;60(9):3500.
- Vu JT, Wang E, Wu J, Sun YJ, Velez G, Bassuk AG, Lee SH, Mahajan VB. Calpains as mechanistic drivers and therapeutic targets for ocular disease. Trends Mol Med. 2022 Aug;28(8):644–61. doi:10.1016/j.molmed.2022.05.007. Epub 2022 May 29. PMID: 35641420; PMCID: PMC9345745.
- Velez G, Yang J, Li AS, Tsang SH, Bassuk AG, Mahajan VB. Proteomic insight into the pathogenesis of CAPN5-vitreoretinopathy. Sci Rep. 2019 May 20;9(1):7608. doi:10.1038/s41598-019-44031-7. PMID: 31110225; PMCID: PMC6527583.
- DiCarlo JE, Mahajan VB, Tsang SH. Gene therapy and genome surgery in the retina. J Clin Invest. 2018 Jun 1;128(6):2177–88. doi:10.1172/JCI120429. Epub 2018 Jun 1. PMID: 29856367; PMCID: PMC5983345.
- Wert KJ, Koch SF, Velez G, Hsu CW, Mahajan M, Bassuk AG, Tsang SH, Mahajan VB. CAPN5 genetic inactivation phenotype supports therapeutic inhibition trials. Hum Mutat. 2019 Dec;40(12):2377–92. doi:10.1002/humu.23894. Epub 2019 Aug 26. PMID: 31403230; PMCID: PMC7493429.
- Wert KJ, Skeie JM, Bassuk AG, Olivier AK, Tsang SH, Mahajan VB. Functional validation of a human CAPN5 exome variant by lentiviral transduction into mouse retina. Hum Mol Genet. 2014 May 15;23(10):2665–77. doi:10.1093/hmg/ddt661. Epub 2013 Dec 30. PMID: 24381307; PMCID: PMC3990166.