
ABSTRACT
Purpose
This cross-sectional study describes the ophthalmological and general phenotype of 10 patients from six different families with a comparatively mild form of Zellweger spectrum disorder (ZSD), a rare peroxisomal disorder.
Methods
Ophthalmological assessment included best-corrected visual acuity (BCVA), perimetry, microperimetry, ophthalmoscopy, fundus photography, spectral-domain optical coherence tomography (SD-OCT), and fundus autofluorescence (FAF) imaging. Medical records were reviewed for medical history and systemic manifestations of ZSD.
Results
Nine patients were homozygous for c.2528 G > A (p.Gly843Asp) variants in PEX1 and one patient was compound heterozygous for c.2528 G>A (p.Gly843Asp) and c.2097_2098insT (p.Ile700TyrfsTer42) in PEX1. Median age was 22.6 years (interquartile range (IQR): 15.9 – 29.9 years) at the most recent examination, with a median symptom duration of 22.1 years. Symptom onset was variable with presentations of hearing loss (n = 7) or nyctalopia/reduced visual acuity (n = 3) at a median age of 6 months (IQR: 1.9–8.3 months). BCVA (median of 0.8 logMAR; IQR: 0.6–0.9 logMAR) remained stable over 10.8 years and all patients were hyperopic. Fundus examination revealed a variable retinitis pigmentosa (RP)-like phenotype with rounded hyperpigmentations as most prominent feature in six out of nine patients. Electroretinography, visual field measurements, and microperimetry further established the RP-like phenotype. Multimodal imaging revealed significant intraretinal fluid cavities on SD-OCT and a remarkable pattern of hyperautofluorescent abnormalities on FAF in all patients.
Conclusion
This study highlights the ophthalmological phenotype resembling RP with moderate to severe visual impairment in patients with mild ZSD. These findings can aid ophthalmologists in diagnosing, counselling, and managing patients with mild ZSD.
1. Introduction
Zellweger spectrum disorders (ZSDs) are a group of clinically and genetically heterogeneous autosomal recessive disorders with an overall estimated incidence of 1 in 50,000, but regional variations have been described (Citation1–4). ZSDs are classified as peroxisome biogenesis disorders, in which dysfunction or absence of peroxisomes leads to multiple biochemical abnormalities, including accumulation of C27-bile acid intermediates, very long-chain fatty acids, branched-chain fatty acids phytanic acid and pristanic acid, and plasmalogen deficiency (Citation5,Citation6). Peroxisome biogenesis requires the combined activity of PEX proteins, which are encoded by their respective PEX genes (Citation7,Citation8). Pathogenic variations in 13 PEX genes have been identified in ZSD, of which variants in PEX1 are the most common (Citation9). The most common pathogenic variant in European ZSD patients is c.2528 G>A (p.Gly843Asp) in PEX1, causing a relatively mild phenotype (Citation9–11). Severely affected patients have seizures, severe hypotonia, failure to thrive, renal cysts, and often do not survive the first year of life (Citation12). Patients with intermediate and milder phenotypes typically present with developmental delay, liver dysfunction, adrenocortical dysfunction, leukodystrophy with variable progression, sensorineural hearing loss, and ocular abnormalities (Citation12). However, as a disorder spectrum, there is a wide heterogeneity in symptoms and age at symptom onset, ranging from infancy to adolescence or even adulthood (Citation12–14). Severity is clearly linked to age of presentation (Citation12).
Ocular manifestations in ZSD that have been described include retinopathy, cataracts, macular atrophy, and optic nerve atrophy (Citation15–20). Virtually all ZSD patients develop retinopathy, often leading to blindness, making this a prominent manifestation even in the mildest phenotypes of ZSD (Citation12,Citation21). However, despite the high prevalence of ocular manifestations including retinopathy, these are not well-characterized and the natural history is largely unknown. Recently, a retrospective review of longitudinal data and literature reported on the natural history of vision loss and on retinal structure on optical coherence tomography (OCT) (Citation22). However, an in-depth analysis of ophthalmological findings using current multimodal imaging techniques and outcome measures is lacking. Both are of utmost importance when evaluating clinical benefit in future clinical trials studying potential therapies. Moreover, unlike severely affected patients, patients with mild ZSD are most likely to visit the ophthalmologist during the early stages as other symptoms may be less pronounced or present later in life.
This study aims to provide more insight into the clinical presentation, the natural history, and ophthalmological phenotype in patients with a relatively mild ZSD phenotype. Furthermore, we provide some suggestions for management of the ophthalmological disorders in patients with ZSDs.
2. Methods
2.1. Patients and methods
This cross-sectional study of patients with confirmed ZSD was performed in Amsterdam University Medical Centers (the Netherlands), an expertise centre for peroxisomal disorders. Patients were previously diagnosed with ZSD based on extensive peroxisomal metabolite analysis in blood and enzyme activity analysis in fibroblasts derived from skin biopsies. In addition, biallelic pathogenic variants in the PEX1 gene were confirmed with genetic sequencing. Patients were identified based on the diagnosis of ZSD in combination with ophthalmological abnormalities. Out of the cohort of patients with ZSD patients known in our expertise centre, only those were selected that were likely to be cooperative (i.e. only those with mild or moderate intellectual impairment). The patient or caretaker was then informed of this study and after agreeing to participate, phenotyping was performed during their next ophthalmological consult, which was within their standard clinical care. Patients who did not want to participate or were not deemed sufficiently cooperative would still be evaluated by an ophthalmologist according to our standard care protocol, without publication of their data. Some patients have been previously described by Berendse et al. (Citation23); patient A1 as patient #11, B1 as patient #8, B2 as patient #9, E1 as patient #1, and E2 as patient #3. In addition, patients E1 and E2 were also reported in childhood by Barth et al. (Citation24) However, both reports do not contain a comprehensive ophthalmological analysis. This study adhered to the tenets of the Declaration of Helsinki and informed consent to publish the details from the affected individual was provided by all patients and/or caregivers prior to assessment. Ethics approval for this study was obtained from the Medical Ethics Committee of the Erasmus University Medical Center (MEC-2010-359).
2.2. Ophthalmological assessment
Data were collected in a retrospective and cross-sectional manner. Once patients were included in this study, a retrospective standardized review of previous ophthalmological examinations was completed and included a review of best-corrected visual acuity (BCVA), refraction, slit-lamp biomicroscopy, perimetry, colour fundus photographs, spectral-domain OCT (SD-OCT), fundus autofluorescence (FAF), ophthalmoscopy, and electroretinography (ERG) assessment if available. The following data were collected systematically for this study in one day in the following order: BCVA, refraction, slit-lamp biomicroscopy, semi-automated kinetic perimetry, microperimetry, colour fundus photographs, SD-OCT scans, FAF images, and ophthalmoscopy. Eyes were dilated after microperimetry using tropicamide 0.5% and phenylephrine 5.0%. Clinical assessments were adapted to best fit the intellectual and physical abilities of the patients and not all patients were able to reliably complete all assessments. BCVA was measured and noted in Snellen decimal, as is common in the Netherlands, and later converted to logMAR values. Visual function was determined by a visual field test and microperimetry. Visual field was determined on semi-automated kinetic perimeter Octopus 900 (Haag-Streit AG, Koeniz, Switzerland) using a customized radial testing grid with a size V stimulus on a photopic background of 10 cd/m2 (Citation25). Microperimetry measured retinal sensitivity in a subset of patients with the Macular Integrity Assessment (MAIA, CenterVue, Padova, Italy) and a standard 37-stimuli grid under mesopic conditions in undilated eyes. Fixation stability is assessed based on bivariate contour ellipse area for 95% (BCEA95) which is a measure of 95% of the fixation positions of the eye. Smaller BCEA95 values correspond to higher fixation stability. BCEA95 values were compared to reference values of healthy adults and children, 0.85°2 and 3.00°2, respectively (Citation26). In order to familiarise patients and to minimise learning effects, patients were asked to first perform a practice session (4-level fixed strategy) before the formal measurement (4–2 threshold strategy) (Citation27). Visual function tests, slit-lamp biomicroscopy, and ophthalmoscopy were performed by one examiner to reduce observer bias and to enable inter-patient comparisons.
Multimodal retinal imaging consisted of a 200° photograph and FAF imaging of the fundus on the Optos California device (Optos PLC, Dunfermline, United Kingdom), and SD-OCT and FAF imaging of the macula on the Spectralis device (Heidelberg Engineering, Heidelberg, Germany).
2.3. Clinical assessment
Medical records were retrospectively reviewed for the natural history, as well as for the presence of systemic manifestations of ZSD, including the presence of dysmorphic features, hearing loss, liver abnormalities, white matter abnormalities on MRI of the brain, neurological symptoms, and cognitive functioning. A comparatively mild phenotype of ZSD is defined as surviving past childhood or with relatively little developmental delay, which is uncommon for patients with severe ZSD who typically do not reach adolescence (Citation12).
2.4. Statistical analysis
Data analysis was performed using IBM SPSS 25.0.0.0 (IBM Corp, Armonk, NY, USA). To assess the normality of the data, the Shapiro-Wilk test was used and results were visually represented through plots. Normal distributed data are presented as mean ± standard deviation (SD). Non-normal distributed data is presented as median and interquartile range (IQR).
3. Results
3.1. Patient characteristics
A total of 10 patients with confirmed ZSD were included in this study from six different families. An overview of their clinical and genetical characteristics is given in . Six patients were male (60%) and the median age of our cohort was 22.6 years (IQR: 15.9–29.9 years). Median duration of symptomatic period was 22.1 years (IQR: 15.5–29.4 years) as measured from the first symptom. Nine patients had homozygous missense mutations in PEX1 c.2528 G>A (p.Gly843Asp). Patient D1 was heterozygous for the missense variant c.2528 G>A (p.Gly843Asp) and null-variant c.2097_2098insT (p.Ile700TyrfsTer42) in PEX1. Patients A1 and A2 were initially diagnosed with Usher syndrome, but further DNA investigations and metabolic analysis in blood revealed the association with peroxisome dysfunction.
Table 1. Genetic and clinical characteristics at first presentation and last examination.
3.2. Ophthalmological assessment
The initial ophthalmological symptoms were nyctalopia (n = 6), nystagmus (n = 4), and reduced visual acuity (n = 3), or a combination thereof (n = 3) (). These symptoms were discovered at a median age of 10.0 months (IQR: 5.5–18.8 months). Full-field ERG (ffERG) test results were available in six patients as part of their diagnostic process, showing scotopic responses that were absent or disrupted earlier and to a greater extent than photopic responses, which is compatible with rod-cone dystrophy (n = 6) ( and Figure S1A). One patient had a follow-up ffERG measurement, which revealed a scotopic electronegative pattern to high-intensity flashes (Figure S1B). Loss of visual acuity was observed in all patients. Those with a measurable BCVA had a median BCVA of 0.8 logMAR (IQR: 0.6–0.9 logMAR), equivalent to 20/125 Snellen acuity (). Based on available retrospective data, visual acuity remained relatively stable with a mean±SD of 0.78 ± 0.28 logMAR over a median follow-up period of 10.8 years (IQR: 8.6–23.7 years) (Figure S2). There were no formal BCVA data of patient D1, besides descriptions of light perception. All patients were hyperopic and median spherical equivalent refraction (SER) was +3.4 dioptres (IQR: 2.5-6.6, n=9) (). Tritanopia was diagnosed in the two oldest siblings, E1 and E2, when they were 17 years old and 20 years old respectively. At the time of assessment, anterior segments had a normal aspect, except for mild nuclear cataract in the two oldest patients (). Patient D1 was reported with the formation of cataract around an age of three years, and mature cataract at the age of nine years. Due to lack of responses to visual stimuli early in life and the associated risk with anaesthesia, cataract surgery was deferred. Five years later, ophthalmological assessment reported mature cataract with liquefication. At her most recent examination, five years later, complete liquefication of the lens was present, as well as posterior synechiae. Intraocular pressure was within normal limits. Other findings of the anterior segment include partial peripheral iris transillumination defects in patient B2.
Table 2. Ophthalmological findings at first and last examination.
Table 3. Visual function at last examination.
Ophthalmoscopy and imaging was not possible in the most severely affected patient D1 due to involuntary movements of the head and eyes. Ophthalmoscopy in the other nine patients revealed varying degrees of the clinical hallmarks of retinitis pigmentosa (RP): bone-spicule hyperpigmentations, round pigment deposits, vascular attenuation, and disc disk pallor (). Round pigment deposits were more prominent in most patients (n = 6), whereas the others had more prominent bone-spicule pigmentation.
Figure 1. Typical findings in patients with mild Zellweger spectrum disorder (ZSD) with biallelic variants c.2528 G>A (p.Gly843Asp) in PEX1 on fundus photographs, fundus autofluorescence (FAF), and spectral-domain optical coherence tomography (SD-OCT).
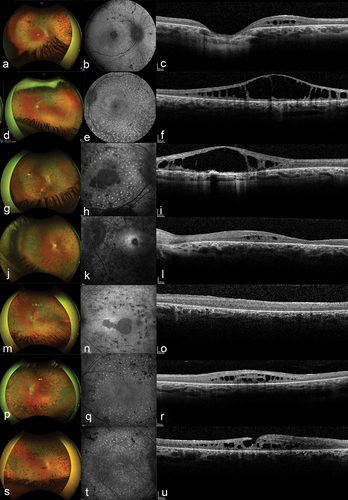
Multimodal imaging consisted of wide-field fundus photographs (n = 6), SD-OCT imaging (n = 9), and FAF imaging (n = 7) (findings are summarized in and ). SD-OCT imaging revealed abnormalities in all nine patients in whom evaluation was possible. Central cystoid fluid collections were seen in 16 eyes and integrity loss of the external limiting membrane (ELM) and ellipsoid zone (EZ) in 15 eyes (). Fluid tended to be mainly present in the outer nuclear layer (ONL) in 14 eyes. Although retinal lamination was lost in patient E1, the cystoid fluid collections were likely located in the ONL as well. In addition, the right eye of patient E1 had a lamellar macular hole (). Patients A2 and B1 had large intraretinal fluid collections with aspect of retinoschisis, with only subtle lines of residual retinal tissue bridging the fluid collection. These fluid collections have an aspect of confluent large cystoid spaces in the central macula (). The same two unrelated patients A2 and B1 received treatment with acetazolamide 500 mg/day and 750 mg/day, respectively, for three months, without a clear effect on the fluid collection. Atrophic changes were present in a few patients and clearly visible on FAF images as hypo-autofluorescent areas (). On FAF of the macula, all patients showed striking round hyperautofluorescent speckles, which did not correspond to the round pigmentation nor to bone-spicule hyperpigmentation or other abnormalities on SD-OCT (). These hyperautofluorescent speckles were present in the macula and midperipheral retina, but not in the far periphery and appear to extent as far as the RP hallmarks (Figure S3).
Psychophysical assessment consisted of visual field tests (n = 7), and determining fixation stability and macular sensitivity on microperimetry (n = 5) ( and ). Due to high fixation losses, patient B1 was not able to complete microperimetry for the right eye. Exhaustion and the accompanying loss of concentration and decreased reaction time, decreased the reliability of the visual field test in patient E1, and her data were excluded from the analysis of the visual field parameters. Generally, the ZSD patients revealed varying degrees of visual field constriction and reduced sensitivity, and some patients, e.g. B2, C1, and E2, only had some central visual field remaining ( and ). The youngest patient who was able to perform the visual field test at the age of 11.0 years (patient C2), had a general reduction in sensitivity of the midperipheral visual field, which could be the start of a ring scotoma (data not shown).
Figure 2. Typical findings on perimetry and microperimetry in patients with mild Zellweger spectrum disorder (ZSD).
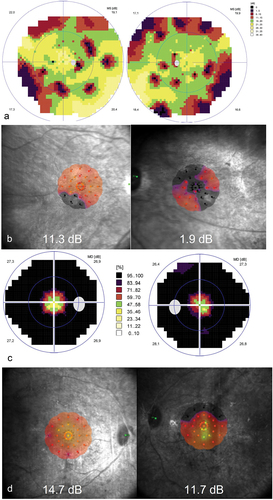
The median of mean sensitivities as measured on the visual field was 9.8 dB (IQR: 4.7–19.7 dB) for the right eye and 9.4 dB (IQR: 5.7–18.3 dB) for the left eye. Median of mean macular sensitivity on microperimetry was 11.3 dB (IQR: 8.3–13.8 dB, n = 4) and 7.6 dB (IQR: 4.7–11.9, n = 5), for the right and left eye respectively. Using BCEA95 as a measure of fixation stability, we found a poor fixation stability with medians of 22.3°2 (IQR: 13.3–38.5°2, n = 4) in the right eye and 21.5°2 (IQR: 10.9–26.6°2, n = 5) in the left eye.
3.3. Clinical features
Systematic clinical assessments were performed on the same day as the ophthalmological assessment if possible or otherwise retrieved on retrospective review of medical records (findings are summarized in ). All patients with the homozygous pathogenic c.2528 G>A variants (p.Gly843Asp) in the PEX1 gene followed a similar pattern in onset and severity of symptoms. The median age at symptom onset was at 6 months (IQR: 1.9–8.3 months). One of the earliest symptoms, and the presenting symptom for most patients, was sensorineural hearing loss which presented within the first two years of life (n = 7), whereas three patients had ophthalmological symptoms (nystagmus and reduced visual acuity) as initial symptoms. One patient received cochlear implants, and the other nine patients used hearing aids.
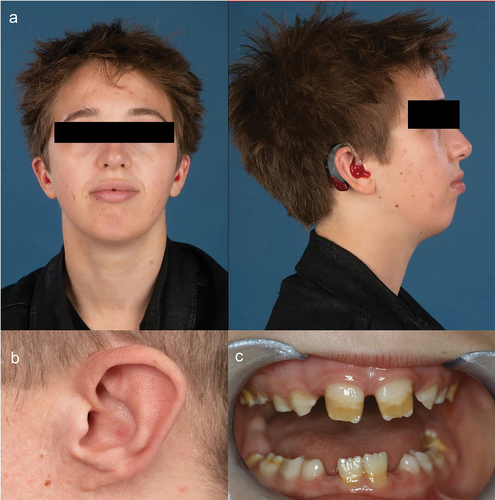
Like most ZSD patients, all patients in our study displayed mild dysmorphic features at the earliest presentation including high forehead (n = 7), low-set ears and/or attached earlobes (n = 6), broad nasal bridge (n = 5), and epicanthic folds (n = 5) (). However, some features, such as epicanthi, become less distinct and have disappeared over time. Two siblings and two unrelated patients had a transverse palmar crease and one patient (D1) presented with syndactyly of the second and third toe. At the time of writing, all patients developed amelogenesis imperfecta in the secondary teeth, resulting in small, pitted, and discoloured teeth (). One patient, F1, had not developed secondary teeth yet.
Figure 3. Mild craniofacial dysmorphic features and amelogenesis imperfecta in adults with mild Zellweger spectrum disorder (ZSD). Patients with comparatively mild ZSD had varying degrees of.
3.4. Cognitive function and neurologic findings
Patient D1, who carried heterozygous variants c.2528 G>A (p.Gly843Asp) and c.2097_2098insT (p.Ile700TyrfsTer42) in PEX1, had severe hypotonia and psychomotor disability, resulting in little to no contact. Of the patients with homozygous c.2528 G>A (p.Gly843Asp) mutations in PEX1, eight patients presented in early childhood with a global delay in motor and communication development, especially in the development of fine motor skills (). Communication problems were characterized by a slow progression in speech-language development, even when adjusting for the additional visual and auditive impairment. One patient had a moderate to severe intellectual deficit. Mild cognitive impairment was present in six patients, whilst two siblings and one other patient showed no intellectual deficit.
At 24 years of age, patient A1 was diagnosed with autism spectrum disorder.
All patients underwent MRI of the brain and regular neurological examination (). MRI scans were normal in five patients, while white matter abnormalities were present in the other five patients at a median age of 12.0 years (IQR: 6.5–36 years). White matter abnormalities were mainly localized in the hilus of the dentate nucleus, cerebellar white matter, and/or parieto-occipital white matter. On examination, most patients had poor or absent myostatic reflexes (n = 4), and mild to moderate ataxia (n = 4).
3.5. Systemic screening
As part of the follow-up in our centre of expertise, patients undergo frequent screening for liver fibrosis/cirrhosis or liver dysfunction by regular ultrasounds and by performing liver function tests in blood (). Five patients presented with (spleno-)hepatomegaly during childhood of which two patients also had signs of liver cirrhosis (ages: 13 and 43 years). Patient C2 was diagnosed with hepatosplenomegaly at her first visit to our centre at the age of two years, but at seven years of age, her liver regained normal proportions. Five patients (aged 20–39 years) did not show liver abnormalities on screening to date. One patient, F1, was found to have a uretropelvic junction stenosis. Patient C2 developed diabetes type 1 at the age of five years.
4. Discussion
In this cross-sectional study, we describe the ophthalmological features of 10 patients with a comparatively mild phenotype of ZSD caused by pathogenic variants in the PEX1 gene. We provide insights in the RP-like rod-cone dystrophy phenotype of patients with mild ZSD with descriptions of the symptom onset, ophthalmological assessment, analyses of visual function, and multimodal imaging.
We found an earlier age at onset of ophthalmological symptoms at a median age 10 months, compared to 4.4 years as was found in a recent meta-analysis (Citation28). The cause of this difference is unclear, but may for instance be related to differences in underlying genotype, different organisation of health care systems, such as screening for hearing loss and visual function, and access to diagnostic strategies. Within our cohort, all patients had a phenotype similar to RP with night blindness and loss of visual acuity as initial symptoms, followed by visual field restriction. BCVA was poor, but appeared to remain relatively stable on a median follow-up of 10 years. We found that hyperopia may be an ophthalmological characteristic of ZSD. Like in our study cohort, the team of Yergeau found that all 30 patients, with at least one c.2528 G>A (p.Gly843Asp) allele in PEX1, were hyperopic (Citation22). Despite the common description of cataract in patients with ZSD (Citation16,Citation18,Citation29,Citation30), only three patients in our study had (a history of) cataract, including one patient with early-onset mature cataract. Ophthalmoscopy revealed varying RP hallmarks: waxy-pale optic disc, narrowed vasculature, and bone-spicule hyperpigmentation. Interestingly, besides bone-spicule hyperpigmentation, the ZSD patients in this study had extensive round hyperpigmentations as a more prominent feature, which appear comparable to those found in some patients with non-syndromic CRB1-associated inherited retinal degeneration (Citation31). Two previously reported patients with a relatively mild ZSD phenotype also had patchy RPE lesions and round pigment clumps, as well as optic nerve head drusen (Citation19,Citation32).
In seven patients, we were also able to assess visual fields, which has not been reported to date. Visual fields were variable, ranging from a relatively mild reduction in sensitivity with a small peripheral scotoma to severe visual field constriction. Microperimetry, as a sensitive marker of central retinal function that is also used in clinical trials for RP and related disorders (Citation33), could be performed in five patients. Microperimetry in these patients showed a markedly reduced macular sensitivity even in patients with relatively little visual field constriction, indicating early macular involvement and dysfunction.
Multimodal imaging on SD-OCT and FAF revealed striking retinal abnormalities in this patient group. First, nearly all patients who underwent SD-OCT showed marked cystoid fluid collections that in some cases were reminiscent of X-linked retinoschisis (XLRS) (Citation34). Furthermore, like in XLRS, most of the cystoid fluid collections were parafoveal and central, and appear to mainly reside in the ONL, which contains the nuclei of the cone and rod photoreceptors (Citation34,Citation35). Efforts to treat the cystoid fluid collections with oral carbonic anhydrase inhibitors did not results in anatomical or functional improvement, similar to other reported attempts with dorzolamide, brinzolamide, and nepafenac eye drops (Citation36). Before starting therapy with carbonic anhydrase inhibitors or other systemic therapies, the risks of nephrolithiasis should be considered with a metabolic specialist as patients with ZSD are at risk for increased levels of oxalate acid in urine. Consistent with previous reports, all patients had loss of integrity of the 415 ELM and EZ, as well as atrophic RPE changes, which is also found in both XLRS and RP (Citation32,Citation35,Citation37,Citation38).
Another striking feature were the extensive round hyperautofluorescent speckles in the macula and midperipheral retina on FAF. Interestingly, these hyperautofluorescent lesions on FAF were not associated with clear abnormalities on SD-OCT imaging. When comparing the wide-field FAF images with the fundus images, the specks appear to extent up until the RP hallmarks. Coincidently, an ffERG performed at an age of 18 years, had an electronegative aspect, which is a common finding in XLRS patients (Citation34). Describing the ophthalmological phenotype may aid in understanding the natural history of ZSD, which is essential for developing relevant clinical outcome measures for future therapeutic clinical trials for ZSD.
Besides the ophthalmological phenotype, we also report on the other features in this relatively mild ZSD group. All patients in this study had hearing impairment, which was diagnosed within the first two years of life and was often the presenting symptom with a median age at diagnosis of 6 months. In contrast, previous reports showed hearing impairment was present in approximately 52% of mildly affected ZSD patients (Citation23,Citation28). Furthermore, all patients showed enamel abnormalities of the teeth. The relatively mild phenotype of the patients in this cohort is further underlined, given that a substantial portion of the patients in this cohort did not show any liver disease or abnormalities on MRI of the brain and survived past childhood. The patients with homozygous variations in PEX1 were able to attend school (adapted for hearing-impaired children) and maintain a job or study with the support of low-vision aids and hearing aids, which is uncommon for those patients with a more severe ZSD form.
Previously, it has been described that patients with missense mutations such as homozygous c.2528 G>A (p.Gly843Asp) mutations in the PEX1 gene, are less severely affected compared to those with null mutations like c.2097_2098insT (p.Ile700TyrfsTer42) in the same gene (Citation10,Citation39–41). An intermediate phenotype is found in those patients who are compound heterozygous for c.2528 G>A (p.Gly843Asp) and c.2097_2098insT (p.Ile700TyrfsTer42) in PEX1. Our findings underline this general genotype-phenotype correlation, as the patients with homozygous c.2528 G>A (p.Gly843Asp) variants in PEX1 had a mild phenotype, whereas the compound heterozygous patient with the aforementioned mutations was comparatively severely affected, but had still survived past childhood. Unfortunately, due to the limited ophthalmological assessment of the compound heterozygous patient, we can only speculate on such a potential ophthalmological genotype-phenotype correlation. Although, Yergeau et al. recently found that patients with compound heterozygous variants in PEX1 have a significantly worse visual acuity compared to those with homozygous mutations in the same gene, which corroborates our findings (Citation22).
We found a relative high variability between patients with regard to visual function, cystoid cavity volume on SD-OCT, as well as the amount of bone-spicule and round hyperpigmentations, even between siblings. This is in line with previous reports that reported phenotypic variability in other organ systems in ZSD patients with the same genotype, suggesting variable penetrance of PEX1 mutations or genetic modifiers. It should further be noted that this study only included patients with PEX1-mediated ZSD, whereas ZSD is also associated with pathogenic variants in 12 other PEX-genes. Patients with pathogenic variants in other PEX genes may present differently.
Tissue-specific vulnerability in peroxisomal disorders is poorly understood, even despite the high prevalence of retinopathy in ZSD. Some studies have demonstrated a high concentration of peroxisomes within the retina, which may explain the high vulnerability of the retina in patients with peroxisomal dysfunction, even in mildly affected patients (Citation42,Citation43). But patients diagnosed with other peroxisomal disorders, such as X-linked adrenoleukodystrophy and rhizomelic chondrodysplasia punctata, typically do not develop retinopathy. The difference in retinal manifestation of peroxisomal dysfunction may be caused by the accumulation of one specific metabolite, e.g. polyunsaturated fatty acids (Citation44), but can also be the result of a combination of different metabolite changes. In light of the close involvement of peroxisomes with the metabolism of lipids and fatty acids, pathogenic changes in PEX1 will inevitably have an effect on retinal metabolism. Previous studies have investigated the cellular and molecular basis of retinopathy in a mouse model for mild ZSD with pathogenic variants in Pex1, equivalent to human homozygous c.2528 G>A (p.Gly843Asp) variants in the PEX1 gene as seen in most patients in our study (Citation42,Citation45). The mouse model demonstrated a more severe degeneration of cone photoreceptors compared to rods, suggesting a cone-rod dystrophy (Citation42). This aligns with the different lipid requirements of rod and cone receptors (Citation44) and with some of our findings as reduced visual acuity was noticed in three patients before or parallel with nyctalopia. Despite this, most of the other findings in our study are more suggestive of RP based on the ffERG, visual field, and natural history. Also, peroxin 1 protein levels were not as strongly reduced in the mouse model compared to fibroblasts of patients with homozygous c.2528 G>A (p.Gly843Asp) variants in PEX1, making it difficult to draw strong parallels to the human retinal situation. The altered lipid metabolism as a consequence of peroxisome dysfunction may also underlie the hyperautofluorescent deposits seen on FAF imaging, which likely correspond to lipofuscin components (Citation46). However, there may also be a role for peroxisome dysfunction in the anti-oxidant pathway or ciliary function as a cause of these deposits and other retinal abnormalities (Citation7,Citation47).
In summary, we report on the ophthalmological phenotype of patients with mild ZSD based on biallelic pathogenic variants in the PEX1 gene. Although there can be considerable inter- and intrafamilial variability, we found a relatively uniform retinal dystrophy phenotype resembling a form of RP, with prominent round pigmentations and relatively sparse bone-spicule hyperpigmentations, striking retinoschisis-like cavities on SD-OCT, and a remarkable pattern of hyperautofluorescent deposits on FAF. Hyperopia is a common finding in ZSD, especially in patients with a c.2528 G>A (p.Gly843Asp) variant in PEX1. Ophthalmological symptoms may present within the first two years, and ophthalmologists should thus be mindful of symptoms in other organ systems during childhood (especially sensorineural hearing loss, liver abnormalities, developmental delay), or later in life (poor dental status with enamel abnormalities) as signs of relatively mild ZSD. These findings can help ophthalmologists with early referrals to a multidisciplinary team comprising of at least a metabolic paediatrician, neurologist, and clinical geneticist. Moreover, the combination of early hearing loss and the RP-like phenotype may mimic Usher syndrome, but the absence of a genetic cause for Usher syndrome should spur clinicians to screen for ZSD. In addition, adult patients with mild ZSD should be followed up regularly by an ophthalmologist.
Supplemental Material
Download Zip (2 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/13816810.2024.2330389.
Additional information
Funding
References
- Shimozawa N, Nagase T, Takemoto Y, Ohura T, Suzuki Y, Kondo N. Genetic heterogeneity of peroxisome biogenesis disorders among Japanese patients: evidence for a founder haplotype for the most common PEX10 gene mutation. American J Med Genet. 2003;120A:40–3. doi:10.1002/ajmg.a.20030.
- Levesque S, Morin C, Guay S-P, Villeneuve J, Marquis P, Yik WY, Jiralerspong S, Bouchard L, Steinberg S, Hacia JG, et al. A founder mutation in the PEX6 gene is responsible for increased incidence of Zellweger syndrome in a French Canadian population. BMC Med Genet. 2012;13(1):72–72. doi:10.1186/1471-2350-13-72.
- Scriver CR, Childs B. The metabolic & molecular bases of inherited disease. 8th/assoc. : Barton Childs … [et al.] edn. New York [etc.]: McGraw-Hill, Medical Publishing Division; 2001.
- Klouwer FC, Huffnagel I, Ferdinandusse S, Waterham H, Wanders R, Engelen M, Poll-The B. Clinical and biochemical pitfalls in the diagnosis of peroxisomal disorders. Neuropediatrics. 2016;47:205–20. doi:10.1055/s-0036-1582140.
- Waterham HR, Ebberink MS. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim Biophys Acta. 2012;1822(9):1430–41. doi:10.1016/j.bbadis.2012.04.006.
- Waterham HR, Ferdinandusse S, Wanders RJA. Human disorders of peroxisome metabolism and biogenesis. Biochimica et Biophysica Acta (BBA) - Mol Cell Res. 2016;1863:922–33. doi:10.1016/j.bbamcr.2015.11.015.
- Fujiki Y, Okumoto K, Honsho M, Abe Y. Molecular insights into peroxisome homeostasis and peroxisome biogenesis disorders. Biochimica et Biophysica Acta (BBA) - Mol Cell Res. 2022;1869:119330. doi:10.1016/j.bbamcr.2022.119330.
- Wanders RJA, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. doi:10.1146/annurev.biochem.74.082803.133329.
- Reuber BE, Germain-Lee E, Collins CS, Morrell JC, Ameritunga R, Moser HW, Valle D, Gould SJ. Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat Genet. 1997;17: 445–8. doi:10.1038/ng1297-445.
- Collins CS, Gould SJ. Identification of a common PEX1 mutation in Zellweger syndrome. Hum Mutat. 1999;14:45–53. doi:10.1002/(SICI)1098-1004(1999)14:1<45:AID-HUMU6>3.0.CO;2-J.
- Walter C, Gootjes J, Mooijer PA, Portsteffen H, Klein C, Waterham HR, Barth PG, Epplen JT, Kunau W-H, Wanders RJA, et al. Disorders of peroxisome biogenesis due to mutations in PEX1: phenotypes and PEX1 protein levels. Am J Hum Genet. 2001;69(1):35–48. doi:10.1086/321265.
- Klouwer FCC, Berendse K, Ferdinandusse S, Wanders RJA, Engelen M, Poll-The BT. Zellweger spectrum disorders: clinical overview and management approach. Orphanet J Rare Dis. 2015;10:151–151. doi:10.1186/s13023-015-0368-9.
- Braverman NE, D’Agostino MD, Maclean GE. Peroxisome biogenesis disorders: biological, clinical and pathophysiological perspectives. Dev Disabil Res Rev. 2013;17(3):187–196. doi:10.1002/ddrr.1113.
- Braverman NE, Raymond GV, Rizzo WB, Moser AB, Wilkinson ME, Stone EM, Steinberg SJ, Wangler MF, Rush ET, Hacia JG, et al. Peroxisome biogenesis disorders in the Zellweger spectrum: an overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. 2016;117(3):313–21. doi:10.1016/j.ymgme.2015.12.009.
- Stanescu B, Dralands L. Cerebro-hepato-renal (Zellweger’s) syndrome: ocular involvement. Arch Ophthalmol. 1972;87:590–2. doi:10.1001/archopht.1972.01000020592018.
- Hittner HM, Kretzer FL, Mehta RS. Zellweger syndrome: lenticular opacities indicating carrier status and lens abnormalities characteristic of homozygotes. Arch Ophthalmol. 1981;99:1977–82. doi:10.1001/archopht.1981.03930020853008.
- O’Neill DP. The eye and liver disorders. Eye. 1992;6:366–70. doi:10.1038/eye.1992.75.
- Folz SJ, Trobe JD. The peroxisome and the eye. Surv Ophthalmol. 1991;35(5):353–68. doi:10.1016/0039-6257(91)90185-i.
- Majewski J, Wang Z, Lopez I, Al Humaid S, Ren H, Racine J, Bazinet A, Mitchel G, Braverman N, Koenekoop RK, et al. A new ocular phenotype associated with an unexpected but known systemic disorder and mutation: novel use of genomic diagnostics and exome sequencing. J Med Genet. 2011;48(9):593–96. doi:10.1136/jmedgenet-2011-100288.
- Ratbi I, Jaouad IC, Elorch H, Al-Sheqaih N, Elalloussi M, Lyahyai J, Berraho A, Newman WG, Sefiani A. Severe early onset retinitis pigmentosa in a Moroccan patient with Heimler syndrome due to novel homozygous mutation of PEX1 gene. Eur J Med Genet. 2016;59:507–11. doi:10.1016/j.ejmg.2016.09.004.
- Ratbi I, Falkenberg K, Sommen M, Al-Sheqaih N, Guaoua S, Vandeweyer G, Urquhart J, Chandler K, Williams S, Roberts N, et al. Heimler syndrome is caused by hypomorphic mutations in the peroxisome-biogenesis genes PEX1 and PEX6. Am J Hum Genet. 2015;97(4):535–45. doi:10.1016/j.ajhg.2015.08.011.
- Yergeau C, Coussa RG, Antaki F, Argyriou C, Koenekoop RK, Braverman NE. Zellweger spectrum disorder: ophthalmic findings from a new natural history study cohort and scoping literature review. Ophthalmology. 2023;130: 1313–26. doi:10.1016/j.ophtha.2023.07.026.
- Berendse K, Engelen M, Ferdinandusse S, Majoie CBLM, Waterham HR, Vaz FM, Koelman JHTM, Barth PG, Wanders RJA, Poll‐The BT, et al. Zellweger spectrum disorders: clinical manifestations in patients surviving into adulthood. J Inherit Metab Dis. 2016;39(1):93–106. doi:10.1007/s10545-015-9880-2.
- Barth PG, Schutgens RBH, Wanders RJA, Heymans HSA, Moser AE, Moser HW, Bleeker‐Wagemakers EM, Jansonius‐Schultheiss K, Derix M, Nelck GF, et al. A sibship with a mild variant of Zellweger syndrome. J Inherit Metab Dis. 1987;10(3):253–59. doi:10.1007/bf01800071.
- Buckley TMW, Josan AS, Taylor LJ, Jolly JK, Cehajic-Kapetanovic J, MacLaren RE. Characterizing visual fields in RPGR related retinitis pigmentosa using octopus static-automated perimetry. Transl Vis Sci Technol. 2022;11:15. doi:10.1167/tvst.11.5.15.
- Molina-Martín A, Piñero DP, Pérez-Cambrodí RJ. Normal values for microperimetry with the MAIA microperimeter: sensitivity and fixation analysis in healthy adults and children. Eur J Ophthalmol. 2017;27(5):607–13. doi:10.5301/ejo.5000930.
- Talib M, Jolly JK, Boon CJF. Measuring Central Retinal Sensitivity Using Microperimetry. In: Camiel JFBoon, Jan Wijnholds, editors. Retinal gene therapy: methods and protocols. Springer New York; 2018. pp. 339–49. doi:10.1007/978-1-4939-7522-8_25.
- Bose M, Yergeau C, D’Souza Y, Cuthbertson DD, Lopez MJ, Smolen AK, Braverman NE. Characterization of severity in Zellweger spectrum disorder by clinical findings: a scoping review, meta-analysis and medical chart review. Cells. 2022;11:1891. doi:10.3390/cells11121891.
- Ebberink MS, Csanyi B, Chong WK, Denis S, Sharp P, Mooijer PAW, Dekker CJM, Spooner C, Ngu LH, De Sousa C, et al. Identification of an unusual variant peroxisome biogenesis disorder caused by mutations in the PEX16 gene. J Med Genet. 2010;47(9):608–15. doi:10.1136/jmg.2009.074302.
- Lipiński P, Stawiński P, Rydzanicz M, Wypchło M, Płoski R, Stradomska TJ, Jurkiewicz E, Ferdinandusse S, Wanders RJA, Vaz FM, et al. Mild Zellweger syndrome due to functionally confirmed novel PEX1 variants. J Appl Genet. 2020;61(1):87–91. doi:10.1007/s13353-019-00523-w.
- Talib M, van Schooneveld MJ, Wijnholds J, van Genderen MM, Schalij‐Delfos NE, Talsma HE, Florijn RJ, ten Brink JB, Cremers FPM, Thiadens AAHJ, et al. Defining inclusion criteria and endpoints for clinical trials: a prospective cross-sectional study in CRB1-associated retinal dystrophies. Acta Ophthalmol. 2021;99:402–14. doi:10.1111/aos.14597.
- Pakzad-Vaezi KL, Maberley DA. Infantile refsum disease in a young adult: case presentation and brief review. Retin Cases Brief Rep. 2014;8:56–9. doi:10.1097/icb.0000000000000004.
- Pfau M, Jolly JK, Wu Z, Denniss J, Lad EM, Guymer RH, Fleckenstein M, Holz FG, Schmitz-Valckenberg S. Fundus-controlled perimetry (microperimetry): application as outcome measure in clinical trials. Prog Retin Eye Res. 2021;82:100907. doi:10.1016/j.preteyeres.2020.100907.
- Hahn LC, van Schooneveld MJ, Wesseling NL, Florijn RJ, ten Brink JB, Lissenberg-Witte BI, Strubbe I, Meester-Smoor MA, Thiadens AA, Diederen RM, et al. X-Linked Retinoschisis: novel clinical observations and genetic spectrum in 340 patients. Ophthalmology. 2022;129(2):191–202. doi:10.1016/j.ophtha.2021.09.021.
- Georgiou M, Finocchio L, Fujinami K, Fujinami-Yokokawa Y, Virgili G, Mahroo OA, Webster AR, Michaelides M. X-Linked retinoschisis: deep phenotyping and genetic characterization. Ophthalmology. 2022;129:542–51. doi:10.1016/j.ophtha.2021.11.019.
- Miranda V, Cortez L, Rosmaninho-Salgado J, Ramos F, Paiva C. Ophthalmic manifestations of heimler syndrome in two siblings with PEX1 variants. J Pediat Ophth Strab. 2023;1–8. doi:10.3928/01913913-20230220-01.
- Ventura MJ, Wheaton D, Xu M, Birch D, Bowne SJ, Sullivan LS, Daiger SP, Whitney AE, Jones RO, Moser AB, et al. Diagnosis of a mild peroxisomal phenotype with next-generation sequencing. Mol Genet Metab Rep. 2016;9:75–78.
- Barillari MR, Karali M, Di Iorio V, Contaldo M, Piccolo V, Esposito M, Costa G, Argenziano G, Serpico R, Carotenuto M, et al. Mild form of Zellweger Spectrum Disorders (ZSD) due to variants in PEX1: detailed clinical investigation in a 9-years-old female. Mol Genet Metab Rep. 2020;24:100615.
- Rosewich H, Ohlenbusch A, Gärtner J. Genetic and clinical aspects of Zellweger spectrum patients with PEX1 mutations. J Med Genet. 2005;42:e58. doi:10.1136/jmg.2005.033324.
- Preuss N, Brosius U, Biermanns M, Muntau AC, Conzelmann E, Gärtner J. PEX1 mutations in complementation group 1 of Zellweger spectrum patients correlate with severity of disease. Pediatr Res. 2002;51:706–14. doi:10.1203/00006450-200206000-00008.
- Crane DI, Maxwell MA, Paton BC. PEX1 mutations in the Zellweger spectrum of the peroxisome biogenesis disorders. Hum Mutat. 2005;26:167–75. doi:10.1002/humu.20211.
- Argyriou C, Polosa A, Cecyre B, Hsieh M, Di Pietro E, Cui W, Bouchard JF, Lachapelle P, Braverman N. A longitudinal study of retinopathy in the PEX1-Gly844Asp mouse model for mild zellweger spectrum disorder. Exp Eye Res. 2019;186:107713. doi:10.1016/j.exer.2019.107713.
- Zaki MS, Heller R, Thoenes M, Nürnberg G, Stern-Schneider G, Nürnberg P, Karnati S, Swan D, Fateen E, Nagel-Wolfrum K, et al. PEX6 is expressed in photoreceptor cilia and mutated in Deafblindness with enamel dysplasia and microcephaly. Hum Mutat. 2016;37(2):170–74. doi:10.1002/humu.22934.
- Das Y, Swinkels D, Baes M. Peroxisomal disorders and their mouse models point to essential roles of peroxisomes for retinal integrity. Int J Mol Sci. 2021;22(8):4101. doi:10.3390/ijms22084101.
- Hiebler S, Masuda T, Hacia JG, Moser AB, Faust PL, Liu A, Chowdhury N, Huang N, Lauer A, Bennett J, et al. The Pex1-G844D mouse: a model for mild human Zellweger spectrum disorder. Mol Genet Metab. 2014;111(4):522–32. doi:10.1016/j.ymgme.2014.01.008.
- Boon CJF, Jeroen Klevering B, Keunen JEE, Hoyng CB, Theelen T. Fundus autofluorescence imaging of retinal dystrophies. Vision Res. 2008;48:2569–77. doi:10.1016/j.visres.2008.01.010.
- Miyamoto T, Hosoba K, Itabashi T, Iwane AH, Akutsu SN, Ochiai H, Saito Y, Yamamoto T, Matsuura S. Insufficiency of ciliary cholesterol in hereditary zellweger syndrome. EMBO J. 2020;39:e103499. doi:10.15252/embj.2019103499.