
ABSTRACT
Background
BCL6 co-repressor (BCOR) gene variants are involved in oculofaciocardiodental (OFCD) syndrome, acute myeloid leukaemia, renal tumours, and photoreceptor degenerative diseases. Here, we describe a British family with a pathogenic heterozygous variant in the BCOR gene causing congenital nuclear cataract.
Methods
Whole-exome sequencing was conducted on an individual affected by X-linked dominant congenital cataract in a three-generation family to establish the underlying genetic basis. Bioinformatics analysis confirmed the variants with damaging pathogenicity scores.
Results
A novel likely pathogenic frameshift variant BCOR NM_001123385.1: c.3621del; p.Lys1207AsnfsTer31, was identified and found to co-segregate with the disease in this family.
Conclusions
This is apparently the first report of a variant in BCOR causing X-linked dominant congenital cataract which is potentially isolated or presenting with a remarkably mild systemic phenotype. Our findings extend the genetic basis for congenital cataract and add to the phenotypic spectrum of BCOR variants.
1. Introduction
The BCL6 corepressor (BCOR) is a transcription factor which plays a critical role in early embryogenesis, mesenchymal stem cell function, haematopoiesis, and lymphoid development. It is an X-linked gene (Xp11.4), with 15 exons and 17 splice variants. The most well-characterized phenotype for pathogenic BCOR variants is OFCD syndrome, a dominant disorder with wide-ranging systemic effects. These include facial abnormalities, such as cleft palate, cardiac septal, and valvular defects, various dental abnormalities, a spectrum of ocular defects, such as microphthalmia, anophthalmia, coloboma, and the focus of this paper is congenital cataract (Citation1, Citation2).
Cataract, the opacification of the eye lens, can be a stand-alone phenotype or a part of a constellation of ocular defects. It is also associated with more than 200 syndromes (Citation3–5). Congenital cataracts are clinically and genetically heterogeneous, displaying various phenotypes, such as nuclear, lamellar, posterior sub-capsule, cortical, and more. Congenital cataracts are usually detected at birth or during the first decade of life. They are mostly inherited as autosomal dominant traits, followed by recessive and X-linked inheritance (Citation6, Citation7).
Around 57 congenital cataract-causing genes have been found to date, most of which are isolated cases. This group includes genes encoding lens soluble proteins including abg-crystallins; lens membrane proteins including connexins, aquaporins, and the receptor tyrosine kinase gene EPH receptor A2; an endoplasmic reticulum membrane-embedded protein, Wolframin; chromatin modifying protein-4B; cytoskeletal proteins including BFSP1 (filensin), BFSP2 (CP49, phakinin), vimentin; and finally, genes encoding transcription or developmental factors such as EYA1, MAF, FOXE3, VSX2, PAX6, PITX3, and HSF4 (https://cat-map.wustl.edu/) (Citation8, Citation9). Here, we report a likely pathogenic variant in BCOR that causes nuclear cataract in a three-generation family pedigree.
2. Material and methods
2.1. Phenotyping
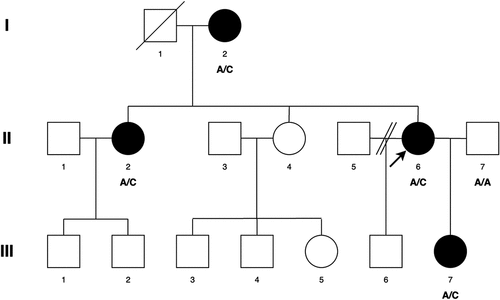
The patients in this pedigree were identified via the proband attending the Genetic Service at Moorfields Eye Hospital, London, UK. The study protocol adhered to the Tenets of the Declaration of Helsinki and was approved by the UCL research ethics committee (project ID − 4817/002). All of the family members, including the male members participating in this study gave written informed consent and underwent full ophthalmic examination, and all affected individuals were diagnosed as having bilateral congenital cataract (). No dental examination was undertaken to complement the presenting cataract phenotype, as contact with the family was lost by the time the genetic characterization was available.
Figure 1. Abridged pedigree presenting with congenital cataract. Squares and circles symbolise males and females respectively. Diagonal lines through these symbols indicate deceased members of the pedigree. Open and filled symbols indicate unaffected and affected individuals respectively. Arrow indicates the proband (II-6). Affected individuals with the variant change at c.3621delA are labelled A/C (I-2, II-2, II-6, III-7). A/A is normal (II-7).
2.2. Whole exome sequencing (WES) and bioinformatic analyses
Genomic DNA was extracted from EDTA treated blood samples using the Nucleon II DNA Extraction Kit (Scotlab Bioscience, Strathclyde, Scotland, UK). The DNA samples were sequenced at Macrogen Europe. Exon capture and target enrichment were performed using the SureSelectXT Human All Exon V6 (Agilent, Santa Rosa, CA, USA). Paired-end sequencing was performed on an Illumina HiSeq 2500 high-throughput sequencer, generating mean exome coverage of 50x. Raw data in FASTQ format was aligned to the UCSC Genome Browser GRCh37/hg19 human reference sequence and analysed using the Phenopolis bioinformatics platform (Citation10). We used a tiered approach to prioritise rare coding variants using Kaviar (http://db.systemsbiology.net/kaviar/.) (Citation11) and Genome Aggregation Database (GnomAD v2.1 http://gnomad.broadinstitute.org/.) or rare variants (GnomAD allele frequency <0.0001) (Citation12) in all the known cataract genes (https://cat-map.wustl.edu/). The variants were then filtered using CADD score, predicted to be moderately or highly damaging (CADD > 15) with the highest at the top for both known and unknown genes for cataracts. Further, bioinformatic validations were done on the Varsome platform (https://varsome.com; version 11.9). Direct Sanger sequencing was performed to validate the variant identified by WES using BCOR specific primers forward (caccccattagtgatgcgta) and reverse primer (tcttccgaccagcttctgtt) designed with http://bioinfo.ut.ee/primer3–0.4.0/.
3. Results
A three-generation British pedigree with 4 affected, 7 unaffected, and 4 unrelated spouses presented with bilateral congenital nuclear cataract (). All family members were examined at the slit lamp and those who were affected had bilateral nuclear cataracts. An affected DNA sample III-7 was sent for WES. After the Phenopolis genetic variant analysis pipeline, variants were filtered by allele frequency and from a total of 3036 rare coding variants, 58 rare variants remained. The top scoring variant for CADD was a rare heterozygous variant with high impact c.3621delA; p.K1207Nfs *31 in exon 8 of 15 coding the BCOR gene with a score of 28.9 (). Direct Sanger sequencing confirmed the variant sequence (), which co-segregated in all the available affected family members. The conserved lysine p.1207 is mutated to asparagine, and it is followed by a 31-residue frameshifted sequence ().
Figure 2. Sequence analysis - (a) BCOR–frameshift variant c.3621delA in affected member, (b) BCOR wild type in normal individual.
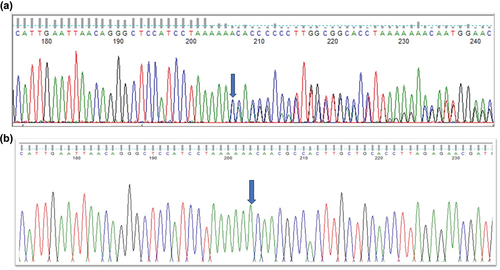
Figure 3. The multiple-sequence alignments from different vertebrate species. Arrows show conserved lysine at p.1207 (https://www.ncbi.nlm.nih.gov/nuccore/?term=BCOR).

Table 1. Pathogenicity scores of BCOR variant.
4. Discussion
We report a truncating and frameshift variant in BCOR that causes nuclear cataract in females, suggesting an X-linked heterozygous inheritance in a three-generation British pedigree. BCOR is a ubiquitously expressed protein, but two dramatically different syndromes arise from pathogenic variants in BCOR (BCL-6 corepressor): OFCD syndrome (Citation1, Citation2), which affects females, and a severe microphthalmia (“Lenz”-type) syndrome affecting primarily males. OFCD syndrome is an X-linked dominant syndrome caused by a variety of BCOR null mutations. As it manifests only in females, it is presumed to be lethal in males. OFCD syndrome patients have a much broader spectrum of phenotypes associated with ocular abnormalities including congenital cataracts, microphthalmia, and microcornea; dysmorphic facial features; cleft palate; cardiac abnormalities and dental anomalies (Citation13–16). The severe male X-linked recessive microphthalmia syndrome (“Lenz”) usually includes developmental delay and is caused by hypomorphic BCOR variants, mainly by a specific missense variant c.254C > T, p.Pro85Leu (Citation1, Citation17, Citation18). Given that the severe and apparent phenotype of OFCD syndrome is not included in the clinical notes of any participating family members, and no males in the family are affected, we hypothesise that this is a case of isolated congenital cataract. It is therefore clear that BCOR is likely involved in many different transcriptional networks, and the study here suggests this includes those important in eye and lens development. However, this conclusion should be taken cautiously, as dental x-ray and echocardiogram records are necessary for validation, but are unavailable for this study.
The c.3621delA variant reported here impacts the C-terminal region of BCOR, just upstream of the ankyrin repeat region (Pfam: PF15808 https://www.ebi.ac.uk/interpro/entry/pfam/PF15808/). BCOR is a 1755 amino-acid protein (https://www.uniprot.org/uniprotkb/Q6W2J9/entry.) identified as a POZ/zinc finger transcriptional repressor (Citation19). Within this C-terminal domain that would be removed in the c.3621delA variant, there is a PCGF1 binding site, called the PCGF Ub-like fold discriminator (PUFD) domain (). A complex between BCOR and PCGF1 along with the RING-domain protein RING1B is required for the binding of KDM2B along with the F-box binding protein SKP1 that constitutes the Polycomb Repressor Complex PRC1 () (Citation20).
Figure 4. Protein interactions between BCOR1, PCGF1 and KDM2B in the PRC1.1 complex. The schematic highlights the important interactions between BCOR1, PCGF1 and KDM2B involved in the formation of the complex PRC1.1. Both the BCL2 binding site and the p.1207 frameshift mutation in BCOR1 are indicated relative to the ankyrin repeats and the important PUFD domain. (b) The work of Wong et al. 2016 showed that BCOR1-PCGF1 dimers were required to form a hetero-tetrameric complex with the KDM2B-SKP1 dimer. PCGF1 binds RING1B to form the subcomplex 1 via the RING domains. KDM2B binds SKP1 via its F-box motif. These two subdomains combine to provide the core PRC1.1 which can then bind a wide range of accessory proteins. The p1207 frameshift mutation reported here will truncate BCOR1 before the ankyrin repeats and will therefore remove the PUFD domain needed for dimerization with PCGF1 with the result that KDMK2 will also not bind. Note the proteins and their domains are not drawn to scale.
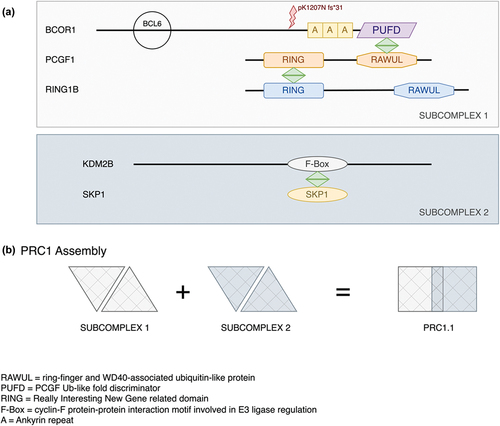
The PRC1 complex is involved in chromatin remodelling during mammalian development, and it is necessary for the correct differentiation of lens fibre cells (Citation21–23). KDM2B, a lysine demethylase () is highly expressed during lens development (https://research.bioinformatics.udel.edu/iSyTE/ppi/expression.php). When KDM2B is knocked out in the developing retina, micro-ophthalmia results (Citation24). The C-terminal-affecting mutation reported here in BCOR would be expected to abolish the interaction of BCOR with both KDM2B and PCGF1. This, we believe, resulted in the lens cataract phenotype in the pedigree reported here.
Author’s contribution
V.B. conceived, analysed the data, wrote, and provided critical revision of the manuscript. M.P. created figures, provided analysis and critical revision. N.P. provided bioinformatic information, R.Q. and M.M. provided critical revision of the manuscript.
Ethics approval
This study involves human participants. This study was approved by the UCL research ethics committee (project ID: 4817/002). The study protocol adhered to the Tenets of the Declaration of Helsinki. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank the family members for their cooperation in this study. For this work, VB and MM were supported by grants from the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology (BRC-D-CON 546795), Moorfields Eye Hospital Special Trustees.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Ng D, Thakker N, Corcoran CM, Donnai D, Perveen R, Schneider A, Hadley DW, Tifft C, Zhang L, Wilkie AOM, et al. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet. 2004;36(4):411–416. doi:10.1038/ng1321.
- Traboulsi EI, Lenz W, Gonzales-Ramos M, Siegel J, Macrae WG, Maumenee IH. The Lenz microphthalmia syndrome. Am J Ophthalmol. 1988;105:40–45. doi:10.1016/0002-9394(88)90119-5.
- François J. Genetics of cataract. Ophthalmologica. 1982;184(2):61–71. doi:10.1159/000309186.
- Ding X, Patel M, Herzlich AA, Sieving PC, Chan C-C. Ophthalmic pathology of nance-horan syndrome: case report and review of the literature. Ophthalmic Genet. 2009;30(3):127–135. doi:10.1080/13816810902822021.
- Loi M. Lowe syndrome. Orphanet J Rare Dis. 2006;1(1):16. doi:10.1186/1750-1172-1-16.
- Ionides A, Francis P, Berry V, Mackay D, Bhattacharya S, Shiels A, Moore A. Clinical and genetic heterogeneity in autosomal dominant cataract. Br J Ophthalmol. 1999;83:802–808. doi:10.1136/bjo.83.7.802.
- Francis PJ, Berry V, Bhattacharya SS, Moore AT. The genetics of childhood cataract. J Med Genet. 2000;37:481–488. doi:10.1136/jmg.37.7.481.
- Shiels A, Hejtmancik JF. Mutations and mechanisms in congenital and age-related cataracts. Exp Eye Res. 2017;156:95–102. doi:10.1016/j.exer.2016.06.011.
- Berry V, Georgiou M, Fujinami K, Quinlan R, Moore A, Michaelides M. Inherited cataracts: molecular genetics, clinical features, disease mechanisms and novel therapeutic approaches. Br J Ophthalmol. 2020;104:1331–1337. doi:10.1136/bjophthalmol-2019-315282.
- Pontikos N, Yu J, Moghul I, Withington L, Blanco-Kelly F, Vulliamy T, Wong TLE, Murphy C, Cipriani V, Fiorentino A, et al. Phenopolis: an open platform for harmonization and analysis of genetic and phenotypic data. Bioinformatics. 2017;33(15):2421–2423. doi:10.1093/bioinformatics/btx147.
- Glusman G, Caballero J, Mauldin DE, Hood L, Roach JC. Kaviar: an accessible system for testing SNV novelty. Bioinformatics. 2011;27:3216–3217. doi:10.1093/bioinformatics/btr540.
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi:10.1186/s13059-016-0974-4.
- Aalfs CM, Oosterwijk JC, Van Schooneveld MJ, Begeman CJ, Wabeke KB, Hennekam RCM. Cataracts, radiculomegaly, septal heart defects and hearing loss in two unrelated adult females with normal intelligence and similar facial appearance: confirmation of a syndrome? Clin Dysmorphol. 1996;5:93. doi:10.1097/00019605-199604000-00001.
- Davoody A, Chen I-P, Nanda R, Uribe F, Reichenberger EJ. Oculofaciocardiodental syndrome: a rare case and review of the literature. Cleft Palate Craniofac J. 2012;49:55–60. doi:10.1597/10-256.
- Feberwee HE, Feenstra I, Oberoi S, Sama IE, Ockeloen CW, Clum F, Slavotinek A, Kuijpers MAR, Dooijes D, Kuijpers‐Jagtman AM, et al. Novel BCOR mutations in patients with oculofaciocardiodental (OFCD) syndrome. Clin Genet. 2014;85(2):194–197. doi:10.1111/cge.12125.
- Gorlin RJ, Marashi AH, Obwegeser HL. Oculo-facio-cardio-dental (OFCD) syndrome. Am J Med Genet. 1996;63(1):290–292. doi:10.1002/(SICI)1096-8628(19960503)63:1<290:AID-AJMG47>3.0.CO;2-G.
- Hilton E, Johnston J, Whalen S, Okamoto N, Hatsukawa Y, Nishio J, Kohara H, Hirano Y, Mizuno S, Torii C, et al. BCOR analysis in patients with OFCD and Lenz microphthalmia syndromes, mental retardation with ocular anomalies, and cardiac laterality defects. Eur J Hum Genet. 2009;17(10):1325–1335. doi:10.1038/ejhg.2009.52.
- Suzumori N, Kaname T, Muramatsu Y, Yanagi K, Kumagai K, Mizuno S, Naritomi K, Saitoh S, Sugiura‐Ogasawara M. Prenatal diagnosis of X-linked recessive Lenz microphthalmia syndrome. J Obstet Gynaecol. 2013;39:1545–1547. doi:10.1111/jog.12081.
- Huynh KD, Fischle W, Verdin E, Bardwell VJ. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 2000;14:1810–1823. doi:10.1101/gad.14.14.1810.
- Wong SJ, Gearhart MD, Taylor AB, Nanyes D, Ha D, Robinson A, Artigas J, Lee O, Demeler B, Hart P, et al. KDM2B recruitment of the polycomb group complex, PRC1.1, requires cooperation between PCGF1 and BCORL1. Structure. 2016;24(10):1795–1801. doi:10.1016/j.str.2016.07.011.
- Macrae TA, Fothergill-Robinson J, Ramalho-Santos M. Regulation, functions and transmission of bivalent chromatin during mammalian development. Nat Rev Mol Cell Biol. 2023;24(1):6–26. doi:10.1038/s41580-022-00518-2.
- Limi S, Senecal A, Coleman R, Lopez-Jones M, Guo P, Polumbo C, Singer RH, Skoultchi AI, Cvekl A. Transcriptional burst fraction and size dynamics during lens fiber cell differentiation and detailed insights into the denucleation process. J Biol Chem. 2018;293:13176–13190. doi:10.1074/jbc.RA118.001927.
- He S, Limi S, McGreal RS, Xie Q, Brennan LA, Kantorow WL, Kokavec J, Majumdar R, Hou H, Edelmann W, et al. Chromatin remodeling enzyme Snf2h regulates embryonic lens differentiation and denucleation. Development. 2016;143(11):1937–1947. doi:10.1242/dev.135285.
- Iwagawa T, Fukushima M, Takeuchi S, Kawamura Y, Aihara Y, Ozawa M, Yakushiji‐Kaminatsui N, Aihara M, Koseki H, Suzuki Y, et al. The histone H3K36 demethylase Fbxl11 plays pivotal roles in the development of retinal late-born cell types. Genes Cells. 2023;28(7):482–495. doi:10.1111/gtc.13028.