Abstract
A general multiresidue method cannot be applied unconditionally to all medicinal plants to extract the residues of interest. The main objective here was to evaluate the Association of Official Analytical Chemists (AOAC) official method 985.22 with Mentha piperita. L. samples. This AOAC method is one of the methods used in pesticide residue labs worldwide. The proposed method proved to show adequate recovery, repeatability, and reproducibility for most of the studied pesticides, and no cleanup step was needed. More than one pesticide residue was contaminating the tested M. piperita. samples, and the number of pesticides found reflects the diversity of compounds used.
Introduction
Medicinal plants are liable to contain pesticide residues, which accumulate from agricultural practices, such as spraying, treatment of soils during cultivation, and administration of pesticides during storage (De Smet et al., Citation1992). Although many countries have issued restricting regulations concerning the use of pesticides, pesticide residues may still contaminate crude botanical drugs. Among the reasons for this type of contamination are (1) herbs can be imported from countries where restrictive rules are still absent or disregarded, (2) many herbs continue to be picked up from natural stands, where interventions to protect the general vegetation can lead to accidental contamination, (3) large-scale cultivation of medicinal plants is not economically feasible without the use of pesticides, and (4) pesticides can persist for years in the environment and can thus lead to contamination of plants, which have not been treated accidentally or intentionally (De Smet et al., Citation1992).
Medicinal plants are commonly used worldwide for a variety of purposes; as food, spice, and flavoring agents and/or to prepare herbal teas, which are consumed as beverages or in some cases to treat minor ailments. It is therefore recommended that every country producing medicinal plant materials (naturally grown or cultivated) should have at least one control laboratory capable of performing the determination of pesticides in accordance with the guidelines of the World Health Organization (WHO) and the Food and Agriculture Organization of the United Nations (FAO) (WHO, Citation1998a).
Mentha piperita. L. (Labiatae), known as Peppermint, is a perennial herb, indigenous to the Middle East, Europe, and naturalized in the northern United States and Canada. The used parts are the leaves and stems. The plant contains volatile oils (about 1%), resin, and tannin. Peppermint is one of the most popular tonic herbs known to modern man. It has been used medicinally as a carminative, antispasmodic, choleretic, stimulant, and counterirritant. It is also used to treat indigestion, intestinal colic, as well as colds, fever, and headache. Because of its anti-inflammatory properties, peppermint is used in the treatment of rheumatism. Laboratory research studies with peppermint have found antiviral, antifungal, and antibacterial activity (Tyler et al., Citation1996). Peppermint oil has long been an extremely popular flavoring agent. Its chief commercial importance is as a pharmaceutical aid for confections, especially for chewing gum (Tyler et al., Citation1996).
The current study was performed to investigate the Association of Official Analytical Chemists (AOAC) official method 985.22 (AOAC, Citation1995) known as Luke's method for nonfatty samples, for the simultaneous determination of multiresidue pesticides in M. piperita.. This method is suitable for nonfatty samples, defined as samples with a fat percentage lower than 10% (FDA, Citation1994). Although numerous reports describe the analysis of pesticide residues in medicinal plant materials, most of the methods described are either so complicated or relatively new that they have not been validated sufficiently. Luke's method is a rapid and reproducible method whose broad utility has lead to its dominant use for the majority of Food and Drug Administration (FDA) enforcement analyses in the 1980s and 1990s (Fong et al., Citation1999). It is also an official method of the AOAC, whose methods are used widely in pesticide residue analysis, being subjected to a rigorous level of validation. A drawback of this method is the application of dichloromethane (DCM), which is less desirable in view of the environmental impact of chlorinated solvents. In Germany, DCM is gradually replaced by extraction with ethyl acetate-cyclohexane (1:1; v/vv) (Hoff & Zoonen, Citation1999).
Materials and Methods
Reagents and equipment
Acetone, DCM, hexane, and petroleum ether (PE) were all pesticide residue (PR) grade (Scharlau, Barcelona, Spain). Pesticide standards were purchased from Ehrenstorfer (Augusburg, Germany). Standards, their purity levels, and groups to which they belong are listed in . Stock solutions of the individual pesticides (1000 µg/ml) were prepared and stored at −20°C, except trans.-chlordane, which was purchased as a 10 µg/ml standard solution. Individual dilutions were prepared as needed and stored at 4°C. Seven mixed standard solutions of the chlorinated hydrocarbons (CH), pyrethroids and miscellaneous pesticides, and one mixed solution of the organophosphorus pesticides (OPP) and primicarb (a carbamate) were prepared with concentrations ranging from 0.01 to 0.5 µg/ml (), which were selected to suite the sensitivity of the detectors used. An internal standard solution of endrin (0.03 µg/ml) and bromophos (0.2 µg/ml) was also prepared and added to all pesticide standard mixed solutions and to all extracts in the final step before gas chromatography (GC) analysis. All pesticide standard solutions and dilutions were prepared in acetone-hexane (10:90% v/v). Anhydrous sodium sulfate analytical reagent (AR) grade (Merck, Darmstadt, Germany) was heated in Memmert's oven (Schawbach, Bavaria, Germany) at 130°C for 5 h and then was stored in 500-ml glass jars with glass stoppers in desicator (Pragati, Maharashtra, India). Sodium chloride AR grade (Nottingham, UK), Whatman filter paper (cat. no. 1002 110) (Medicell International Ltd., UK).
Table 1 Groups of the pesticides and the % purity and concentrations of their standards.
The equipment used included a high-speed blender with a stainless steel jar (Moulinex, France), a Rotavapor, R110 (Büchi, Switzerland), a cooler circulator (Julabo, Seelbach, Germany), and microliter syringes (Hamilton Bonadus AG, Switzerland). All glassware was rinsed thoroughly using soap and deionized water, then washed with acetone and dried in oven (100–130°C) overnight.
Plant materials
Eight different fresh samples—leaves and stems (M1–M8)—were purchased through the year 2001–2002 from the local market in Jordan. The plants were authenticated by Dr. Sawsan Oran, Department of Biological Sciences, Faculty of Science, University of Jordan. A voucher sample from all plant materials was stored at the Department of Pharmaceutical Sciences, Faculty of Pharmacy, University of Jordan. The purchased samples were processed without washing. The plant material was analyzed as fresh samples and as dried samples. After analyzing the fresh samples, the plant material was dried at room temperature (23°C) for 3 days, then powdered and sieved through a no. 60 mesh sieve. M. piperita. samples were extracted according to the AOAC method, and no additional cleanup was undertaken. Sample size was 10 g for fresh samples and 3–5 g for dry samples; half quantities of solvents were used for the extraction of pesticide residues.
Chromatographic instrumentation for determination of CH, pyrethroids, and miscellaneous pesticides
An HP-5890 series II gas chromatograph (GC) equipped with an HP-608 capillary column (30 m, 0.53 id, 0.5-µm film thickness) with the stationary phase (50%-phenyl)–methylpolysiloxane and 63Ni electron capture detector (ECD) was used. The GC instrument was operated under the following conditions: injector in the split mode (split ratio 1:17), injector temperature 250°C, detector temperature 300°C, argon-methane (5:95% v/v) as the carrier gas at flow rate of 1 ml/min, which was also used as the make-up gas at a flow rate of 24 ml/min. Column temperature was initially held at 80°C for 1 min, then programmed at 30°C/min to 180°C, followed by 5°C/minto 200°C, and 10°C/min to 280°C and held for 14 min.
Chromatographic instrumentation for determination of OPP
An HP-5890 series II GC, equipped with an HP-1 capillary column (25 m, 0.2 mm id, 0.5-µm film thickness) with the stationary phase 100% dimethyl polysiloxane and nitrogen phosphorus detector (NPD) was used. The instrument was operated under the following conditions: injector in the split mode (split ratio 1:10), injector temperature 225°C, detector temperature 280°C, helium was the carrier gas with flow rate of 1 ml/min, detector gas flow rates were 3–3.5 ml/min for hydrogen and 100 ml/min for air. Column temperature was initially held at 90°C for 2 min, then programmed at 20°C/min to 150°C, followed by 6°C/min to 270°C and held for 15 min.
Chromatographic instrumentation for confirmation of identity
An HP-5890 Series II GC, equipped with an HP-5 capillary column (30 m, 0.25 mm id, 0.25-µm film thickness) with the stationary phase 5% diphenyl and 95% dimethyl polysiloxane and 63NI ECD was used. The instrument was operated under the following conditions: injector was operated in the split mode (split ratio 1:17), injector temperature 280°C, detector temperature 300°C, carrier gas was helium with a flow rate of 2 ml/min, make-up gas was argon-methane (5:95% v/v) with a flow rate of 30 ml/min, column temperature was initially held at 80°C for 2 min, then followed by 30°C/min to 175°C, and at 10°C/min to 225°C and held for 2 min, then at 20°C/min to 280°C and held for 10 min.
Extraction and partitioning
Plant material was extracted with water/acetone, liquid-liquid partitioning in petroleum ether, dichloromethane, known as AOAC method 985.22 and PAM method 302 was used (AOAC, Citation1995; FDA, Citation1994).
Determination of pesticides retention times (tR) and relative retention times (RRTs)
For the determination of the tR for each individual pesticide, 1 µl of the 1.0 µg/ml pesticide solution was injected into the GC-column. Standard mixtures of the pesticides were prepared in the concentrations as listed in . One microliter of each standard mixture was also injected into the GC-column. Pesticides were identified by comparing their tRs and RRTs ().
Table 2 Types of detectors, pesticides' retention times, relative retention times, and limits of detection.
Limits of detection (LOD)
Limits of detection of the used instruments, equipped with ECD and NPD, were determined for each pesticide by successive dilution of the standard mixed pesticide solution followed by injection into the GC-column several times. Serial dilution experiments provided the necessary information to calculate the detection limits (Boyd-Boland & Pawliszyn, Citation1995; Lehotay & Valverde-García, Citation1997).
Recovery tests
Recovery tests were evaluated with all 33 pesticides used in the current study. This was performed by spiking plant samples with a concentrated mixed pesticide solution in concentrations ranging from 0.01 to 0.5 µg/ml. The spikded plant samples were then extracted according to the proposed method. Samples used in recovery tests were chosen to be pesticide-free and tested in the same methods mentioned. In order to evaluate the recoveries of the residue analytical procedure without being affected by interferences from plant matrices, spiking water instead of plant samples was also performed. All tests were performed in triplicate. Recovery studies were performed on one concentration level, differing from one pesticide to another, that would be applicable to monitoring the maximum residue levels of these pesticides (Obana et al., Citation2001).
Reagent blank analysis
Reagent blank analysis was performed using reagents only (without plant sample) to show whether there were any detector responses that could be mistaken for the pesticide residues. These blank analyses were performed once weekly.
Control samples
Chromatograms of plant extracts, which were found to be free from contamination with pesticide residues, were used for comparison purposes with chromatograms of plant samples that were contaminated with pesticide residues. These pesticide-free plant samples (control samples) were used for fortification purposes in recovery studies.
Residue analysis
For residue analysis, the purchased samples (M1–M8) were ground mechanically and sieved through no. 60 mesh sieve. Samples were extracted according to the AOAC method.
Results
The limits of detection () for GC-ECD and GC-NPD were between 0.0008 and 0.05 pg for the CH and the miscellaneous pesticides, 0.02 and 0.1 pg for pyrethroids, and between 0.006 and 0.5 pg for OPP.
The mean recoveries of the studied pesticides, in case of fortified water samples, ranged from 83% to 120% (); no pesticide showed very high recovery (150%), but some pesticide showed recoveries >100%. In case of M. piperita. spiked samples, the mean recoveries were between 82% and 119% (>150%) () with the exception of p.,p.-DDT (>150) and phosalone (variable). A representative chromatogram for a contaminated M. piperita. sample is shown in .
Figure 1Representative GC-ECD chromatogram of Mentha piperita. (M1) extract contaminated with HCB and cypermethrin.
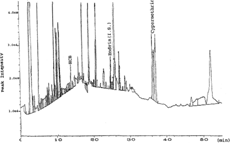
Table 3 The spiked level of each pesticide, mean recovery, RSD, relative errors, and total errors for method I.
Table 4 The spiked level of each pesticide, mean recovery, RSD, relative errors, and total errors in fortified Mentha piperita. samples.
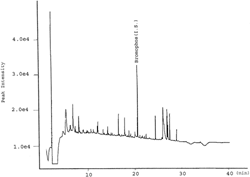
Chromatograms of the reagent blanks showed unfamiliar peaks ( and ). These peaks are most probably originating from contaminants in sodium sulfate or in sodium chloride, as these were analytical grade materials and all other used chemicals were PR-grade. Literature review revealed that sodium sulfate is one of the possible sources of background peaks in blank extracts (FDA, 1994).
Figure 2Representative GC-ECD chromatogram of a reagent blank.
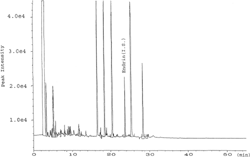
Figure 3Representative GC-NPD chromatogram of a reagent blank.
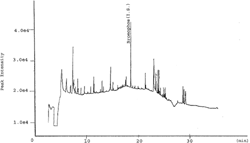
Chromatograms of control samples showed several peaks that are most probably due to the different constituents of the medicinal plant materials—detected on the detector—in addition to the peaks originating from contaminants that are already present in the reagent blanks (). Control sample analysis was also carried out in accordance with recovery tests to make sure that the spiked plant samples were originally pesticide-free.
Figure 4Representative GC-NPD chromatogram of a pesticide-free Mentha piperita. sample.
All eight analyzed M. piperita. samples were contaminated with CH, pyrethroid, and miscellaneous pesticide residue (). The pesticides identified in all samples were HCB, α.-HCH, vinclozolin, dicofol, procymidone, p.,p.-DDE, dieldrin, β.-endosulfan, fenpropathrin, and cypermethrin.
Table 5 Pesticide residues in Mentha piperita. samples (M1–M8).
Discussion
During the current study, some modifications were made on the original method: (1) The quantity of the medicinal plant employed in this study for the analysis using method I was sometimes reduced to about 1/2 or even 1/4 the amount suggested by Luke's method, accomplished by reducing the consumption of all reagents, with no decrease in method performance; (2) Rotavapor was used instead of the Kurdena-Danish Concentrator. In recovery studies form M. piperita. spiked samples, p., p.-DDT had recovery >150 and phosalone's recovery was variable. These two exceptions are most probably due to matrix interferences. When a pesticide-free sample was not readily available, recovery studies may be performed on another portion of the plant sample by fortifying it with at least twice the level of residue found (FDA, Citation1994).
Two parameters were calculated in order to determine the accuracy and precision of the used method: (1) relative standard deviation (RSD), a measure of the method's precision, RSD = SD/% Recovery × 100 (SD: standard deviation); (2) relative error (RE), a measure of the method's accuracy.
Relative errors of 20% or less are considered satisfactory. When the best method available gives less than 80% recovery, it may still be used provided the percent recovery is reproducible (Fong et al., Citation1999). It is useful sometimes to calculate the method's total error where both RSD and RE are included: Total error = RE + 2RSD. Total errors tend to run high in trace analyses. A total error of <50% is considered good, 50–100% acceptable, and occasionally methods with >100% total error can still be usable if no better method exists (McFarren et al., Citation1970; Fong et al., Citation1999). Values of RSD, relative errors, and total errors are listed in Tables and .
Pesticide residues present in the real samples were identified tentatively by comparing the RRTs of the suspected peaks with RRTs of the injected standards and then quantitated using the following equation:
where Cs is concentration of pesticide residues in sample in mg/kg dry plant material, Cst is concentration of the pesticide in the mixed pesticide standard solution, As is average peak area obtained for the pesticide found in sample, Ais is average peak area obtained for the internal standard injected with the sample, Ast is peak area obtained for the pesticide in the mixed-pesticide standard solution, Aist is peak area obtained for the internal standard found in the mixed-pesticide standard solution, R is recovery factor calculated from 100% recovery, 5 is final volume (Vfinal) of the analyzed sample in ml, and F is extraction factor.
The identity of the found pesticides was confirmed using GC-ECD equipped with another column of a different polarity, namely HP-5.
The pesticide residues found in the analyzed M. piperita. samples were those that can be extracted with the proposed method. Residues of DDT, HCB, dieldrin, and endosulfan probably came from environmental contamination rather than from current spraying operations. The inability to detect pesticide residues in some plant samples does not necessarily mean that these samples were pesticide-free. Some pesticides may exist but cannot be extracted by the applied methods. It is also possible that some pesticide residues may be present in concentrations below the detection limits achieved in this study and consequently were not detected. In general, the concentrations of most pesticide residues found in the study were below tolerance levels. But the important issue here is the presence of several residues in the same sample, some of which might have similar mechanisms of toxicity.
Conclusions
The AOAC official method 985.22 was found to be efficient for the determination of multiresidue pesticides in M. piperita.. Results of residue analysis in real samples showed more than one pesticide residue contaminating most purchased M. piperita. samples, and the number of pesticides found reflects the diversity of compounds used. The absence of OPP residues in the analyzed samples could be due to (1) the generally shorter half-lives of these pesticides or (2) the applied methods of analysis are not suitable for extracting such pesticides, which are relatively more polar than the CH and pyrethroids.
Acknowledgments
The authors express their gratitude to the staff members at the Pesticide Residue Laboratory in Al-Hussein Agricultural Station, Ministry of Agriculture, especially to Mr. B. Sokker, and the staff at the Industrial Chemistry Center at the Royal Scientific Society, especially to Mr. H. Esteitieh.
References
- AOAC (1995): Official Methods of Analysis of AOAC International, Vol. 1. Arlington, Association of Official Analytical Chemists, p. 2, 7, 10.
- Boyd-Boland AA, Pawliszyn JB (1995): Solid-phase microextraction of nitrogen-containing herbicides. J Chromatogr A 704: 163–172. [CSA]
- De Smet PAGM, Keller K, Hänsel R, Chandler RF (1992): Adverse Effects of Herbal Drugs, Vol. 1. Berlin, Heidelberg, Springer-Verlag, pp. 20, 21.
- FDA (1994): Pesticide Analytical Manual of the Food and Drug Administration, Vol. I. Washington, OC, Food and Drug Administration, Department of Health and Human Services, Sec. 302, p. 1, 13, 14; Sec. 304, p. 1, 15–17; Sec. 304-a, p. 12.
- Fong GW, Moye AH, Seiber JN, Toth JP (1999): Pesticide Residues in Food. New York, John Wiley & Sons, p. 10, 39, 45.
- Hoff GR, Zoonen P (1999): Trace analysis of pesticides by gas chromatography. J Chromatogr A 843: 301–322. [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Lehotay SJ, Valverde-García A (1997): Evaluation of different solid-phase traps for automated collection and cleanup in the analysis of multiple pesticides in fruits and vegetables after supercritical fluid extraction. J Chromatogr A 765: 69–84. [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- McFarren EF, Liskka RJ, Parker JH (1970): Criterion for judging acceptability of analytical methods. Anal Chem 42: 358–365. [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Obana H, Akutsu K, Okihashi M, Hori S (2001): Multiresidue analysis of pesticides in vegetables and fruits using two-layered column with graphitized carbon and water absorbent polymer. Analyst 126: 1529–1534. [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Tyler VE, Speedie MK, Robbers JE (1996): Pharmacognosy and Pharmacobiotechnology. Baltimore, Williams and Wilkins, pp. 116–121.
- WHO (1998a): Quality Control Methods for Medicinal Plant Materials. Geneva, Switzerland, World Health Organization, pp. 1–4.
- WHO (1998b): Regulatory Situation of Herbal Medicines. A Worldwide Review. Geneva, Switzerland, World Health Organization, pp. 47–48.
- Worthing CR (1991): The Pesticide Manual. A World Compendium, 9th ed. Great Britain: British Crop Protection Council, Lavenham Press, pp. 208, 232.