Abstract
A micellar electrokinetic chromatographic (MEKC) method has been established for the identification and determination of acid-catalyzed proanthocyanidin cleavage products in the presence of phloroglucinol. Using fused-silica capillaries (50/57 cm, 22 kV) with phosphate buffer containing sodium dodecyl sulfate as the surfactant and β-cyclodextrin as modifier, structurally similar phloroglucinol adducts and flavan-3-ols were fully separated.
Introduction
Proanthocyanidins (PAs), or condensed tannins, are a class of natural polyphenolic compounds, the occurrence of which is widespread in higher plants. PAs are claimed to be associated with a wide range of health benefits, and therefore they are attracting increasing attention (Cos et al., Citation2004). PAs consist of flavan-3-ols units usually linked by a C4–C6 or a C4–C8 bond (so-called B-type) to form compounds of different degree of polymerization. The structure of the flavan-3-ol varies with respect to its hydroxylation pattern and the presence of gallic acid ester on the C-3 position. Some PAs have two linkages (C–O–C and C–C) between of flavan-3-ol units (the so-called A-type), but these are not frequently encountered naturally in comparison with the B-type oligomers. Analyzing intact PAs provides information on their number, average molecular weight (M.n.), and weight average molecular weight (M.w.) (Williams et al., Citation1983; Bae et al., Citation1994). Analyzing PAs after acid-catalyzed cleavage provides information on their composition as well as the interflavonoid bond location (Foo & Karchesy, Citation1989; Kennedy & Jones, Citation2001); such acid catalysis need a nucleophilic cleavage reagent such as phloroglucinol or benzyl mercaptan (Geissman & Yoshimura, Citation1966; Jurd & Lundin, Citation1968; Thompson et al., Citation1972).
Capillary electrophoresis (CE) appears likely to become an indispensable tool in phytochemical laboratories to solve separation problems in cases that are difficult or time consuming to be solved by HPLC (Thomás-Barberán, Citation1995). CE has been successfully applied to separate coumarins, flavonoids, phenolic acids, flavan-3-ols, and PAs (Kreimeyer et al., Citation1998; Lee & Ong, Citation2000; Cifuentes et al., Citation2001; Spilkova et al., Citation2001; Cao et al., Citation2002; Marchart et al., Citation2003; Hamoudova et al., Citation2004). A new modified method utilizing CE, microemulsion electrokinetic chromatography (MEEKC), was developed for the separation of six catechins in several Cistus. species (Pomponio et al., Citation2003). This MEEKC method, however, uses sodium dodecyl sulfate (SDS) as surfactant, heptane as organic solvent, and butan-1-ol as cosolvent. Herrero-Martínez et al. (Citation2003) described another micellar electrokinetic chromatographic (MEKC) method for the separation of the major flavan-3-ols and their aminoethylthio-derivatives obtained after thiolysis of the grape samples with cysteamine.
Several HPLC methods for separation of phloroglucinol cleavage products and flavan-3-ols derived from the acid hydrolysis of proanthocyanidins were published. Silica gel or RP-18 phases were commonly used for the separation in about 30 min (Koupai-Abyazani & Bruce, Citation1993; Kennedy & Jones, Citation2001). Thus, we decided to develop a less time consuming, rapid CE assay (MEKC), as an alternative method for the qualitative detection of the phloroglucinol cleavage products.
Materials and Methods
Plant material
Cistus albidus. L. (Cistaceae) was collected at Massiv d' Estérel/France and identified in comparison with authentic Cistus albidus. obtained from the Botanical Institute, University Cologne. A voucher specimen is deposited at the herbarium of the Institut für Pharmazeutische Biologie, Muenster, under PBMS 188.
Extraction and preparation of the oligomeric proanthocyanidin fraction
Air-dried herb material of C. albidus. (2 kg) was exhaustively extracted with Me2CO:H2O (7:3, 12 l) and the combined extracts evaporated in vacuo. to 1.5 l, filtered to remove the precipitated chlorophyll, concentrated, and defatted with petroleum benzene (30–70°C). Successive extractions with EtOAc (7.5 l) of the remaining water-extract gave, on evaporation of solvent, a solid of 32.5 g EtOAc extract and 200 g of a remaining H2O extract. A portion (2 × 50 g) of the H2O extract was successively applied to column chromatography on Sephadex LH-20 (55 × 900 mm) with 3 l EtOH:H2O (1:1) and 6.5 l EtOH:H2O:Me2CO (1:1:2) until the eluent was colorless; then 2.5 l Me2CO:H2O (3:7) as eluents to give 22.5 g of a fraction containing the oligomeric proanthocyanidins (Qa'dan et al., Citation2003).
Degradation with phloroglucinol and isolation of proanthocyanidin cleavage products
An aliquot of the oligomeric fraction of Cistus albidus. L. obtained as described above (6 g) was treated for 30 min at room temperature with phloroglucinol (4.5 g) in 1% HCl in EtOH (100 ml) under shaking (Foo & Karchesy, Citation1989). The solution was concentrated under reduced pressure to give 10.5 g (PA degradation fraction). With the exception of catechin-(4α → 2)-phloroglucinol (7), all flavan-3-ols and phloroglucinol adducts (1–6) were isolated from PA degradation fraction of Cistus albidus. ().
Figure 1 Flavan-3-ols and phloroglucinol cleavage products
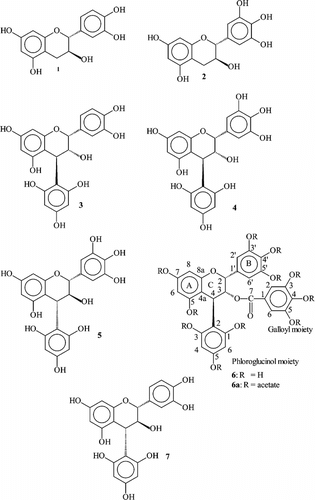
A portion (8.5 g) of the PA degradation fraction was fractionated by CC on Sephadex LH-20 (55 × 900 mm) using EtOH (96%) (17 l), EtOH:MeOH 1:1 (3 l), and Me2CO:H2O 3:7 (3 l) as eluents to give 8 fractions. Fraction 2 (3550–3900 ml; 430 mg) was subjected to chromatography on MCI-gel CHP 20 P (2.5 × 25 cm, 75–100 µm; Mitsubishi Kasei Corporation, Tokyo, Japan) with a 10–50% MeOH linear gradient (17 ml/frs.) to afford catechin 1 (subfrs. 89–112; 61 mg) and gallocatechin 2 (subfrs. 49–71; 143 mg). Fraction 3 (3900–4600 ml; 1.6 g) was separated on MCI-gel with the same gradient as above to afford epicatechin-(4β → 2)-phloroglucinol (3) (subfrs. 71–80; 53 mg). Fraction 4 from CC on Sephadex LH-20 (4600–4800 ml; 0.8 g) was separated on MCI-gel to afford epigallocatechin-(4β → 2)-phloroglucinol (4) (subfrs. 33–50; 447 mg). Gallocatechin-(4α → 2)-phloroglucinol (5) was isolated from fraction 5 (4800–5200 ml; 0.7 g) and after MCI-gel chromatography (subfrs. 42–60; 235 mg). Epigallocatechin-3-O.-gallat-(4β → 2)-phloroglucinol (6) was achieved from fraction 6 (5200–5700 ml, 0.4 g) followed by MCI-gel chromatography as described above (subfrs. 69–81, 133 mg).
Catechin-(4α → 2)-phloroglucinol (7) was prepared as follows: Catechin-(4α → 8)-catechin [isolated from Cistus albidus. (Qa'dan et al. Citation2003), 50 mg] and 30 mg phloroglucinol were treated for 30 min at room temperature in 1% HCl in EtOH (10 ml) under shaking (Foo & Karchesy, Citation1989). The solution was concentrated under reduced pressure to give a brown mixture. The mixture was separated on MCI-gel with a 10–50% MeOH linear gradient (17 ml/frs.) to afford 7 (33 mg). Flavan-3-ols (1–2) and phloroglucinol adducts (3–7) were characterized by 1D-, 2D-NMR, circular dicroism (CD), [α] and MALDI-TOF-MS of the corresponding derivatives obtained as peracetates in comparison with authentic samples and published values (Fletcher et al., Citation1977; Foo & Porter, Citation1978Citation1983; Ploss et al., Citation2001). The new compound 6 was isolated as: weak violet amorphous powder: [α]D20 + 113° (c 0.10, MeOH). MALDI-TOF-MS of its peracetate (6a): 1066 [M + Na]+. Further characterization by 1H NMR (600 MHz, Brucker AM 600), 13C NMR (150 MHz), 2D NMR (1H-1H COSY, HMBC, HSQC) and CD spectroscopy provided structural confirmation of 6a ().
Table 1 1H (600 MHz) and 13C (150 MHz) NMR data of 6a in chloroform-d.1 relative to CHCl3
CE apparatus
MEKC was performed on a P/ACE System 5510 (Beckman Coulter Instruments, Palo Alto, CA, USA) with the following operating conditions: uncoated fused-silica capillary, length 57 cm, 50 cm to the detector (DAD), 50 µm i.d.; analysis were carried out at temperature of 27°C. After testing different electrolytes, the optimized composition was as follows: 80 mM phosphate, pH 7.0, 120 mM sodium dodecyl sulfate (SDS), and 10 mM β-cyclodextrin. The applied voltage was 22 kV, the injection time was 3 s at 0.5 psi; detection was effected at 280 nm.
Determination of capacity factor
In MEKC, “apparent” capacity factors are related to the migration time of an unincorporated solute (t.0), the migration time of the analyte (t.a) and that of the micelle (t.mc) by the formula k.′ = (t.a − t.0)/t.0 (1 − t.a/t.mc)(Engelhard, Citation1994). Methanol, almost totally excluded from the micelles, served as a marker to measure t.0, and quinine hydrochloride was employed as a marker for t.mc, as it is almost completely incorporated into the micelle (Terabe, Citation1992). t.0 and t.m, respectively, were determinated by co-injection of methanol (migration time: 1.90 min) and quinine hydrochloride (migration time 12.70 min).
Sample preparation
Mixtures of ca. 1.0 mg from each compound (1–7) were dissolved in 1 ml sodium phosphate buffer (80 mM, pH 5.0) and analyzed by capillary electrophoresis.
Results and Discussion
Compounds 1–7 () are uncharged molecules under neutral and acid conditions. Basic pH should be avoided because phenolates are not stable under such conditions. As expected, without adding a charged surfactant, a separation could not be achieved. Thus, a selective MEKC method was developed with combined advantages of reversed-phase chromatography and capillary electrophoresis. In preliminary experiments, anionic surfactant additives in phosphate and borate buffer systems were tested. Only SDS in phosphate buffer solution showed a promising separation. In order to optimize the buffer concentration, five phosphate concentrations (20, 40, 60, 80, 100 mM) were tested. Increasing phosphate caused an improved separation but prolonged the time of analysis. Best results were obtained at a buffer concentration of 80 mM and pH = 7.0.
The influence of the SDS concentration in the range of 20 to 140 mM on the separation of these compounds was studied. As expected, migration times and retention of the solutes appeared to increase proportionally with the increase in the SDS concentration. The concentration of 120 mM SDS provides satisfactory resolution of all compounds. Higher SDS (>120 mM) concentrations caused peak broadening, unacceptably high Joule heating, and longer analysis time. Therefore, the best separation was obtained with 120 mM SDS in the phosphate buffer 80 mM. In spite of these results obtained, the analysis time was similar to the one obtained with classic HPLC methods (Koupai-Abyazani & Bruce, Citation1993; Kennedy & Jones, Citation2001). To shorten time of analysis and modify selectivity, β-cyclodextrin (β-CD) as a neutral pseudostationary phase was added. Of all tested concentrations, 10 mM β-CD showed the best CD-modified-MEKC separation with a total migration time below 5 min. ( and ).
Figure 2 Electropherogram, registered at 280 nm, shows the separation of the compounds 1–7
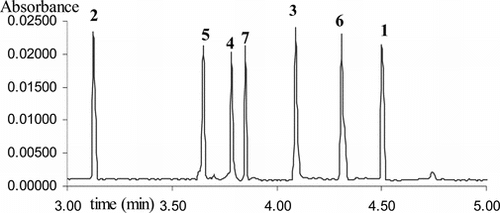
Table 2 Migration times and corresponding capacity factors (k´) of reference substances
The relationship between structural features of the phloroglucinol adducts 3–7 and their electrophoretic behavior was similar to the rules established by Kreimeyer et al. (1997) for the flavan-3-ols and dimeric proanthocyanidins: the relative cis./trans. stereoisomerism at C2–C3 carbon axis influences migration time. The trans.-isomers (gallo)-catechin-(4α → 2)-phloroglucinol migrate faster than the corresponding cis.-isomer epi-(gallo)-catechin-(4β → 2)-phloroglucinol. An increase in the size of the phloroglucinol adduct increases the affinity to the SDS micelles and to β-cyclodextrine leading to an increase in the migration time. Because epigallocatechin-3-O.-gallat-(4β → 2)-phloroglucinol is bulkier than epigallocatechin-(4β → 2)-phloroglucinol, the gallate group at C3 carbon makes compound 6 migrate slower than compound 4.
In conclusion, the developed method provides a rapid CD-modified-MEKC determination for analysis of five major monomeric proanthocyanidin cleavage products after acid-catalyzed hydrolysis in the presence of excess phloroglucinol. According to our knowledge, this is the first qualitative analysis of these compounds using MEKC technique.
Acknowledgments
Fadi Qa'dan would like to acknowledge gratefully the DAAD for funds and grants. We thank Mrs. Bettina Quandt for her valuable technical assistance. We acknowledge also the help of Dr. H. Lahl (Inst. f. Pharmazeutische Chemie, Muenster) and Ms. M. Heim for runing the NMR-spectra. The help of Dr. H. Luftmann (Inst. f. Organische Chemie, Muenster) for the MALDI-MS-spectra is gratefully acknowledged.
References
- Bae YS, Foo LY, Karchesy JJ (1994): GPC of natural procyanidin oligomers and polymers. Holzforschung 48: 4–6. [CSA]
- Cao Y, Chu Q, Fang Y, Ye J (2002): Analysis of flavonoids in Ginkgo biloba. L. and its phytopharmaceuticals by capillary electrophoresis with electrochemical detection. Anal Bioanal Chem 374: 294–299. [INFOTRIEVE], [CROSSREF], [CSA]
- Cifuentes A, Bartolome B, Gomez-Cordoves C (2001): Fast determination of procyanidins and other phenolic compounds in food samples by micellar electrokinetic chromatography using acidic buffers. Electrophoresis 22: 1561–1567. [INFOTRIEVE], [CROSSREF], [CSA]
- Cos P, De Bruyne T, Hermans N, Apers S, Berghe DV, Vlietinck AJ (2004): Proanthocyanidins in health care: Current and new trends. Curr Med Chem 11: 1345–1359. [INFOTRIEVE], [CSA]
- Engelhard H (1994): Kapillarelektrophorese. Braunschweig/Wiesbaden, Friedrich Vieweg und Sohn Verlagsgesellschaft mbH.
- Fletcher AC, Porter LJ, Haslam E, Gupta RK (1977): Plant proanthocyanidins. Part 3. Conformational and configurational studies of natural procyanidins. J Chem Soc Perkin Trans I 14: 1628. [CROSSREF], [CSA]
- Foo LY, Porter LJ (1978): Prodelphinidin polymers: Definition of structural units. J Chem Soc Perkin Trans I 10: 1186–1190. [CROSSREF], [CSA]
- Foo LY, Porter LJ (1983): Synthesis and conformation of procyanidins diastereoisomers. J Chem Soc Perkin Trans I: 1535. [CROSSREF], [CSA]
- Foo LY, Karchesy JJ (1989): Procyanidin polymers of Douglas fir bark: Structure from degradation with phloroglucinol. Phytochemistry 28: 3185–3190. [CROSSREF], [CSA]
- Geissman TA, Yoshimura NN (1966): Synthetic procyanidin. Tetrahedron Lett. 7(24): 2669–2673. [CROSSREF], [CSA]
- Hamoudova R, Urbanek M, Pospisilova M, Polasek M (2004): Assay of phenolic compounds in red wine by on-line combination of capillary isotachophoresis with capillary zone electrophoresis. J Chromatogr A 1032: 281–287. [INFOTRIEVE], [CROSSREF], [CSA]
- Herrero-Martínez JM, Ràfols C, Rosés M, Torres JL, Bosch E (2003): Mixed micellar electrokinetic capillary chromatography separation of depolymerised grape procyanidins. Electrophoresis 24: 707–713. [CROSSREF], [CSA]
- Jurd L, Lundin R (1968): Anthocyanidins and related compounds-XII. Tetra methylleucocyanidin-phloroglucinol and resorcinol condensation products. Tetrahedron 24: 2653–2661. [CROSSREF], [CSA]
- Kennedy JA, Jones GP (2001): Analysis of proanthocyanidin cleavage products following acid-catalysis in the presence of excess phloroglucinol. J Agric Food Chem 49: 1740–1746. [INFOTRIEVE], [CROSSREF], [CSA]
- Koupai-Abyazani MR, Bruce AB (1993): The condensed tannins of Leptarrhena pyrolifolia.. Phytochemistry 33: 1485–1487. [CROSSREF], [CSA]
- Kreimeyer J, Petereit F, Nahrstedt A (1998): Separation of flavan-3-ols and dimeric proanthocyanidins by capillary electrophoresis. Planta Med 64: 63–67. [CSA]
- Lee BL, Ong CN (2000): Comparative analysis of tea catechins and theaflavins by high-performance liquid chromatography and capillary electrophoresis. J Chromatogr A 881: 439–447. [INFOTRIEVE], [CROSSREF], [CSA]
- Marchart E, Krenn L, Kopp B (2003): Quantification of the flavonoid glycosides in Passiflora incarnata. by capillary electrophoresis. Planta Med 69: 452–456. [INFOTRIEVE], [CROSSREF], [CSA]
- Ploss O, Petereit F, Nahrstedt A (2001): Procyanidins from the herb of Hypericum perforatum.. Pharmazie 56: 506–511. [CSA]
- Pomponio R, Gotti R, Santagati NA, Cavrini V (2003): Analysis of catechins in extracts of Cistus. species by microemulsion electrokinetic chromatography. J Chromatogr A 990: 215–223. [INFOTRIEVE], [CROSSREF], [CSA]
- Qa'dan F, Petereit F, Nahrstedt A (2003): Prodelphinidin trimers and characterization of a proanthocyanidin oligomer from Cistus albidus.. Pharmazie 58: 416–419. [INFOTRIEVE], [CSA]
- Spilkova J, Bednar P, Stroblikova R (2001): Capillary electrophoretic analysis of hydroxycinnamic acids from Ononis arvensis. L. Pharmazie 56: 424–425. [INFOTRIEVE], [CSA]
- Terabe S (1992): Micellar Electrokinetic Chromatography. Fullerton, CA, Beckman Instruments, p. 12.
- Thomás-Barberán FA (1995): Capillary electrophoresis: A new technique in the analysis of plant secondary metabolites. Phytochem Anal 6: 177–192. [CSA]
- Thompson RS, Jacques D, Haslam E, Tanner RJN (1972): Plant proanthocyanidins. Part I. Introduction; the isolation, structure, distribution in nature of plant procyanidins. J Chem Soc Perkin Trans I: 1387–1399. [CROSSREF], [CSA]
- Williams VM, Porter LJ, Hemingway RW (1983): Molecular weight profiles of proanthocyanidin polymers. Phytochemistry 22: 569–572. [CROSSREF], [CSA]