
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Context
Non-alcoholic fatty liver disease (NAFLD), the most common chronic liver disease, can develop into metabolic associated fatty liver disease (MAFLD). Gypenosides (GP), the main phytochemical component of Gynostemma pentaphylla (Thunb.) Makino (Cucurbitaceae), have been applied for treatment of metabolic diseases.
Objective
We investigate how GP modulate MAFLD-related hepatic steatosis and intestinal barrier injury.
Materials and methods
In cell experiments, Caco-2 cells were treated with GP (150 or 200 μmol/L, 24 h), following lipopolysaccharide (LPS) exposure (10 μg/mL, 24 h) to mimic MAFLD in vitro. In in vivo experiments, control, model and model + GP groups were set. High fructose diet/high fat (HFD/HF)-fed (12 weeks) MAFLD rats received GP treatment (300 mg/kg, 6 weeks), followed by intra-peritoneal glucose tolerance test and histopathological examination of rat liver and intestinal mucosa using haematoxylin–eosin staining.
Results
GP at 200 μM significantly reversed LPS-induced decreases in transepithelial electrical resistance (TER) value (25%), protein expression of occludin (two fold) and ZO-1 (four fold), and the ratio of p-AMPK to AMPK (five fold), while partially repressing LPS-induced leakage of FD4 (50%) and LPS-induced increases in the Toll-like receptor 4 (TLR4) level (50%) and the ratio of p-p65 to p65 (55%). Compared with the model rats, rats with GP treatment presented a reduction in gain of weight and glucose tolerance. In addition, GP alleviated HFD/HF-induced histopathological abnormalities in rat liver and intestinal mucosa.
Conclusions
GP attenuates hepatic steatosis and intestinal barrier injury in MAFLD rats via the AMPK and TLR4/nuclear factor kappa B (NF-κB) pathways, providing a potential treatment for MAFLD patients.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease characterized by excessive fat accumulation (referring to an over-5% ratio of fat content to the whole liver), which is closely associated with metabolic syndrome (MS) and has manifestations including obesity, dyslipidaemia, insulin resistance and hypertension (Brunt et al. Citation2015; Dabke et al. Citation2019). NAFLD can progress from simple fatty liver to steatohepatitis (a risk factor reflecting MS). With the emergence of hepatocyte injury, death and inflammation in the lobules and porta, NAFLD may further develop into metabolic associated fatty liver disease (MAFLD) (Brunt et al. Citation2015). In contrast with NAFLD, MAFLD has a slow onset with no distinct clinical manifestation. MAFLD patients usually do not consume excess alcohol, but may occur as right upper abdomen discomfort or upper abdomen pain and appear to be intolerant to glucose, obese and insulin-resistant (Milić et al. Citation2014; Yki-Järvinen Citation2014), which is a cluster of symptoms indicative of MS (O'Neill and O'Driscoll Citation2015). Dietrich’s study regarding MAFLD has considered NAFLD as the hepatic manifestation of MS and has maintained that the liver is not only a passive target but affects the pathogenesis of MS and its complications (Dietrich and Hellerbrand Citation2014). Moreover, MS is associated with an increased risk for steatohepatitis and fibrosis, and the incidence of the forthcoming liver failure (Marchesini et al. Citation2003; Ryan et al. Citation2005), highlighting its significance in MAFLD.
Accumulation of hepatic lipids is the first hit to the liver in the development of MAFLD. Furthermore, prominent enlargement of adipose tissue caused by excessive weight gain leads to adipocyte dysfunction and even death, which further induces local inflammation and releases of inflammatory cytokines (Maher et al. Citation2008). These cytokines contribute to the development of insulin resistance that would impair the process of adipocyte-mediated fat storage in turn (Maher et al. Citation2008). The impaired fat storage results in accumulation of free fatty acid, and once free fatty acid enters the circulation, the progression of simple fatty liver into non-alcoholic steatohepatitis and cirrhosis will be accelerated, which eventually induces MAFLD (Dietrich and Hellerbrand Citation2014; Xu et al. Citation2015).
Interactions between the intestine and the liver are considered to deliver predominant influence on the progression of MAFLD. Altered intestinal microbiota, resulting from damaged intestinal barrier-induced diffusion of bacterial endotoxins into the blood circulation, can lead to liver diseases including MAFLD (Lambertz et al. Citation2017). More importantly, transferring the microbiota from conventional mice into germ-free mice elicited the occurrence of MS-related symptoms such as increased body fat content and insulin resistance in the germ-free mice (Dabke et al. Citation2019). Conversely, intervention with probiotics can help rebuild intestinal microbiota structure and alleviate liver pathologies in MAFLD (Xue et al. Citation2017).
Gypenosides (GP) are the main phytochemical component of Gynostemma pentaphylla (Thunb.) Makino (Cucurbitaceae) which is reported to have diverse pharmacological effects including antioxidation, immunomodulation, antitumor, antifatigue, neuroprotection and hepatoprotection (Wang et al. Citation2017; Ji et al. Citation2018). Gynostemma pentaphylla is an herbaceous climbing vine widely grown in South and East Asia (Lee et al. Citation2019). The aforementioned effects of Gynostemma pentaphylla have been reported to be associated with the molecular weight, monosaccharide composition and chemical structure of the extracted GP (Ji et al. Citation2018). Moreover, Lee et al. (Citation2019) demonstrated that GP can strengthen adenosine monophosphate activated protein kinase (AMPK) activation to reduce the high fat-induced obesity in mice (Neuschwander-Tetri Citation2017). In addition, it has been reported that AMPK and NF-κB signalling pathways play a regulatory role in the pathological process of liver injury, hepatic steatosis and hepatic fibrosis (Yang et al. 2020; Li et al. Citation2021). However, whether GP can regulate AMPK activation to affect the progression of MAFLD is undetermined yet.
Following the establishment of lipopolysaccharide (LPS)-induced Caco-2 cell models and high fat and high fructose diet (HFD/HF)-induced rat models, we investigated the effects of GP on MAFLD-related intestinal barrier dysfunction and hepatic steatosis, and explored the involvement of AMPK activation in these GP-induced effects.
Materials and methods
Cell culture and treatment
Human colorectal adenocarcinoma epithelial cells, Caco-2 cells (HTB-37), were purchased from American Type Culture Collection (ATCC, Manassas, VA), and cultured in ATCC-formulated Eagle’s Minimum Essential Media (30-2003, ATCC, Manassas, VA) supplemented with 20% foetal bovine serum (FBS, F2442, Sigma-Aldrich, St. Louis, MO) at 37 °C with 5% CO2. GP (HY-N6881, C41H70O12, purity: ≥90.0%, CAS no. 80325-22-0, MedChemExpress, Monmouth Junction, NJ) were solubilized in dimethyl sulphoxide (D2650, DMSO, Sigma-Aldrich, St. Louis, MO), and diluted into the gradient concentrations of 0, 50, 100, 150 or 200 μM for the use in drug treatment. Notably, DMSO was set at the 0.1% final concentration in in vitro experiments. Also, gradient GP were applied in the measurement of cell viability. Then, Caco-2 cells were assigned into four groups (control group, LPS group, LPS + GP150 group and LPS + GP200 group). Caco-2 cells in the LPS + GP150 group or the LPS + GP200 group were treated with 150 μM or 200 μM of GP at 37 °C for 24 h with 5% CO2. After being washed with phosphate-buffered saline (PBS, P5493, Sigma-Aldrich, St. Louis, MO), cells were exposed to 10 μg/mL LPS (HY-D1056, LPS, MedChemExpress, Monmouth Junction, NJ) at 37 °C for 24 h with 5% CO2 (He et al. Citation2020). Cells in the LPS group only underwent 10 μg/mL LPS exposure for 24 h, and cells in the Control group remained untreated.
Cell counting kit (CCK)-8 assay
The viability of Caco-2 cells was measured using CCK-8 reagent (C0037, Beyotime, Beijing, China). In a nutshell, Caco-2 cells (1 × 103 cells/well) were seeded in 96-well plates and incubated at 37 °C for 24 h. Then, the cells were washed by PBS (P5493, Sigma-Aldrich, St. Louis, MO) twice, treated with gradient concentrations of GP (0, 50, 100, 150 or 200 μM), and exposed to LPS for 24 h. Afterwards, CCK-8 reagent (10 µL) was added into the cells and incubated with the cells at 37 °C for 2 h. Ultimately, the cell absorbance at 450 nm was recorded by a microplate reader (Synergy Neo2, BioTek, Winooski, VT).
Transepithelial electrical resistance (TER) determination
The TER value of Caco-2 monolayers was determined by a Millicell-ERS system (MERS00002, Millipore, Bedford, MA) to evaluate the intestinal epithelial barrier functions. Simply put, Caco-2 cells were inoculated on a Transwell support (3396, Corning, Corning, NY) at a density of 1 × 106 cells/well, and were allowed to grow into monolayers. Once cell monolayers were formed and completely differentiated, GP (150 μM or 200 μM) were poured onto the apex of the monolayers, followed by 24-h incubation. Later, the monolayers were subjected to GP treatment or not, followed by LPS exposure for 24 h. Finally, TER value change was presented in a percentage against the corresponding basal values.
Fluorescein isothiocyanate (FITC)-labelled dextran assay
The leakage of FITC-labelled dextran (F121152, FD4, 4 kDa, Aladdin, Shanghai, China) was measured to assess the permeability of Caco-2 cell monolayers. In brief, after GP treatment and/or LPS exposure, the apical chamber of Caco-2 cell monolayers was added with FD4 (1 mg/mL). 2 h later, the medium from the basolateral chamber was collected. The fluorescence intensity at a wavelength of 520 nm was measured by a microplate reader (Synergy Neo2, BioTek, Winooski, VT).
Establishment of in vivo MAFLD models
This study was approved by the Institutional Animal Care and Use Committee (IACUC), ZJCLA (approval number: ZJCLA-IACUC-20100010). Every effort was made to minimize pain and discomfort to the animals. The animal experiments were performed in Zhejiang Animal Center.
Eighteen 12-week-old male Wistar rats weighing 220–250 g were purchased from SLAC (Shanghai, China). All the rats were housed in a controlled environment with a temperature of 25 ± 1 °C and humidity of 50–60% as well as a 12 h circadian cycle, and acclimated to the environment for one week. During this one-week habituation, the rats were fed with regular chow diet. Afterwards, they were randomized into three groups (n = 6) (control, model and GP groups).
MAFLD models were established and drug dosage was determined based on previously proposed methods (Huang et al. Citation2019; Radwan et al. Citation2020). Rats in the model group and model + GP group were fed with HFD/HF (20 kcal% protein, 20 kcal% carbohydrate and 60 kcal% fat in drinking water containing 25% d-fructose) for 18 weeks, and orally administrated with 300 mg/kg GP-containing water or distilled water from the 13th to 18th week. Rats in the control group were fed with regular chow diet for 18 weeks, followed by being administrated with the equal volume of distilled water as GP from the 13th to 18th week. The body weight of rats was measured every two weeks starting from day 0. All the rats began a 12 h fasting without water deprivation at the end of week 18. For obtainment of the serum, rat blood was centrifuged at 3000×g for 10 min. The confirmation of MAFLD in the rats was carried out according to liver fat accumulation status tested by histopathological examination. Besides, the total cholesterol (TC), triglycerides (TGs), low density lipoprotein cholesterol (LDL-C) and LPS levels in serum were assessed by biochemical analysis or enzyme-linked immunosorbent assay (ELISA).
Once the fasting was finished, intra-peritoneal glucose tolerance test (IPGTT) was performed. Following the above tests, the rats were anaesthetized by 2% pentobarbital sodium (P-010, Sigma-Aldrich, St. Louis, MO), and then sacrificed by spinal dislocation. Rat liver and intestinal mucosa were harvested and fixed in 4% paraformaldehyde (P6148, Sigma-Aldrich, St. Louis, MO) for histopathological examination, or quick-frozen in liquid nitrogen at −80 °C for Western blot analysis. The subsequent in vivo experiments were repeated three times to obtain average values.
Biochemical analysis
For the assessment of the hepatic function impairment in HFD/HF Wistar rats with or without GP treatment (300 mg/kg), the serum levels of glucose, alanine transaminase (ALT) and aspartate transaminase (AST) were measured by an automatic biochemical analyser (AU5400, Olympus, Tokyo, Japan). For IPGTT, the rats were intraperitoneally injected with glucose (2.0 g/kg) following the 12 h fasting and their glucose levels were determined at 0, 30, 60, 90 and 120 min after the injection.
ELISA
The serum levels of insulin (kt30476, MSKbio, Wuhan, China), TC (kt30156, MSKbio, Wuhan, China), TGs (kt50023, MSKbio, Wuhan, China), LDL-C (kt25323, MSKbio, Wuhan, China) and LPS (E-EL-R0589c, Elabscience, Wuhan, China) as well as tumour necrosis factor-alpha (TNF-α, ml002859, mlbio, Shanghai, China), interleukin 6 (IL-6, ml064292, mlbio, Shanghai, China) and IL-1β (PI303, Beyotime, Beijing, China) levels in liver tissue were detected by specific ELISA kits. Briefly, rat serum was added into the enzyme-labelled plates and incubated at 37 °C for 90 min. Then, the fluid in the plates was discarded, and 100 μL biotinylated antibody was added into the plates. After 60 min incubation at 37 °C, the plates were washed by the buffer solution thrice, added with the enzyme-conjugated working solution and incubated at 37 °C for 30 min. Subsequently, the plates were supplemented with the substrate Reagent and incubated at 37 °C for 15 min. Following these, H2SO4 (2 M, 50 μL) was added to each well to terminate the reaction. Ultimately, the optical density at 450 nm was recorded under a microplate reader (ELx808, BioTek, Winooski, VT).
Later, homeostasis model assessment-insulin resistance (HOMA-IR) was calculated according to this formula (Jia N et al. Citation2018):
Haematoxylin–eosin staining
After rat liver and intestinal mucosa were fixed in 4% paraformaldehyde (16005, Sigma-Aldrich, St. Louis, MO) for 24 h, they were dehydrated by gradient alcohol, transparentized by xylene (95682, Sigma-Aldrich, St. Louis, MO), and embedded in paraffin (1496904, Sigma-Aldrich, St. Louis, MO). Subsequently, the paraffinized tissues were cut into 5 μm thick sections, dewaxed by xylene and rehydrated by gradient alcohol. Then, the sections were stained with haematoxylin (H3136, Sigma-Aldrich, St. Louis, MO) for 12 min. After being differentiated by hydrochloric alcohol, the sections were stained with eosin (E4009, Sigma-Aldrich, St. Louis, MO) for 5 min. Thereafter, the stained sections were sealed by neutral balsam (N861409, Macklin, Shanghai, China) and dried at 37 °C for 4 h. Afterwards, the histopathological changes in the liver tissues and intestinal mucosa tissues were observed by an optical microscope (ZEISS Primotech, Carl Zeiss, Oberkochen, Germany) under magnifications of ×200 and ×40, respectively.
The morphological changes in liver were determined by NAFLD activity score, which includes histological features and has been defined as unweighted sum of scores for steatosis (0–3), lobular inflammation (0–3) and ballooning (0–2).
Western blot
Caco-2 cells, intestinal mucosa tissues and liver tissues were lysed in radio-immunoprecipitation assay (RIPA) Buffer (89900, ThermoFisher, Waltham, MA) cocktailed with a protease-phosphatase inhibitor (A32959, ThermoFisher, Waltham, MA) for extraction of total protein. Next, protein concentration was determined by bicinchoninic acid (BCA) kits (A53227, ThermoFisher, Waltham, MA). Subsequently, total protein (45 μg) and marker (4 μL) (PR1910, Solarbio, Beijing, China) were separately loaded and electrophoresed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gel (12% (P0672), 8% (P0678), Beyotime, Shanghai, China). Then, the proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (P2438, Sigma-Aldrich, St. Louis, MO) and blocked using 5% skim milk in Tris-buffered saline with 1% Tween 20 (TBST, TA-125-TT, ThermoFisher, Waltham, MA) at room temperature for 1 h. Afterwards, the membranes were blotted with primary antibodies against occludin (ab216327, 65 kDa, 1:1000, Abcam, Cambridge, MA), tight junction protein 1 (ZO-1, ab96587, 187 kDa, 1:500, Abcam, Cambridge, MA), peroxisome proliferator-activated receptor α (PPARα, ab3484, 52 kDa, 1:100, Abcam, Cambridge, MA), sterol regulatory element-binding protein 1c (SREBP-1c, ab28481, 68 kDa, 1:500, Abcam, Cambridge, MA), Toll-like receptor 4 (TLR4; ab13867, 90 kDa, 1:1000, Abcam, Cambridge, MA), phosphorylated (p)-p65 (ab76302, 65 kDa, 1:1000, Abcam, Cambridge, MA), p65 (ab16502, 64 kDa, 1:1000, Abcam, Cambridge, MA), AMPK (ab32047, 63 kDa, 1:1000, Abcam, Cambridge, MA), p-AMPK (ab131357, 64 kDa, 1:500, Abcam, Cambridge, MA) and GAPDH (ab8245, 36 kDa, 1:10,000, Abcam, Cambridge, MA) at 4 °C overnight. After being washed with TBST, the membranes were incubated with a secondary antibody Goat anti-Rabbit IgG (A32731, 1:10,000, ThermoFisher, Waltham, MA) or Goat anti-Mouse IgG (A32733, 1:1000, ThermoFisher, Waltham, MA). Later, the protein bands were visualized by Novex™ enhanced chemiluminescence reagent kit (WP20005, ThermoFisher, Waltham, MA), visualized using Tanon 5200 chemiluminescence instrument (Tanon Inc., Shanghai, China), and analysed by ImageJ software (1.52s version, National Institutes of Health, Bethesda, MA).
Statistical analysis
Collected data were expressed as mean ± standard deviation. Statistical analyses were performed using GraphPad prism (version, 8.0, GraphPad Software Inc., San Diego, CA). Comparisons among multiple groups were conducted by one-way analysis of variance, followed by a Tukey post hoc test. p < 0.05 was considered as statistically significance. All experiments were performed in triplicate.
Results
GP reversed LPS-induced effects on TER value, extracellular permeability and expressions of occludin and ZO-1 in Caco-2 cells possibly via inhibiting the TLR4/NF-κB pathway and activating the AMPK pathway
A recent in vitro experiment was conducted to reveal the effects of GP on H2O2-induced retinal ganglion cells injury, the results of which showed that treatment with GP at 50, 100 and 200 μg/mL reversed the decrease in viability of H2O2-induced retinal ganglion cells (Zhang et al. Citation2020). In contrast, GP treatment (50, 100, 150 and 200 μM) failed to demonstrate any alterations in Caco-2 cell viability (). In order to better show the effect of GP on LPS-treated cells, we selected high concentrations of GP (150 and 200 μmol/L) for follow-up cell experiments. As depicted in , LPS (10 μg/mL) treatment decreased TER value by 45% in Caco-2 cells, but increased the leakage of FD4 from the apical chamber of a Caco-2 cell monolayer to the basolateral chamber by approximately three fold (p < 0.001). Furthermore, the protein expression of occludin was decreased by approximately 60% and that of ZO-1 was reduced by approximately 85% after exposure to LPS (p < 0.001, ). Conversely, LPS exposure led to markedly increases in protein expression of TLR4 (2-fold change) and p-p65 (2.3-fold change) and a ratio of p-p65 to p65 (2.3-fold change) (p < 0.001, ). Additionally, LPS treatment did not cause a significant change in AMPK protein expression, but reduced p-AMPK protein expression by approximately 80%, thereby decreasing the p-AMPK/AMPK by 80% (p < 0.001, ).
Figure 1. GP reversed LPS-induced effects on TER value, extracellular permeability and expression of occludin and ZO-1 in Caco-2 cells possibly via activating the AMPK pathway. (A) The viability of Caco-2 cells treated with gradient concentrations of GP (0, 50, 100, 150 or 200 μM) was measured by CCK-8 assay. (B) The TER value of Caco-2 cell monolayers which received LPS exposure alone or in combination with GP treatment was determined by a Millicell-ERS system. (C) The flow of FD4 in Caco-2 cell monolayers which received LPS exposure alone or in combination with GP treatment was measured by FITC-labelled dextran assay. The protein expression of occludin, ZO-1 (D–F), TLR4, p-p65, p65 (G–J), AMPK and p-AMPK (L–N) in Caco-2 cells which received LPS exposure alone or in combination with GP treatment was analysed by Western blots, with GAPDH serving as a reference gene. The ratios of p-p65 to p65 (K) and p-AMPK to AMPK (O) in Caco-2 cells which received LPS exposure alone or in combination with GP treatment. GP: gypenosides; LPS: lipopolysaccharides; TER: transepithelial electrical resistance; FD4: FITC-labelled dextran of 4 kDa; CCK-8: cell counting kit-8; ZO-1: tight junction protein 1; TLR4: Toll-like receptor 4; AMPK: adenosine monophosphate activated protein kinase; p: phosphorylated. ***p< 0.001 vs. Control; #p< 0.05, ##p< 0.01, ###p< 0.001 vs. LPS.
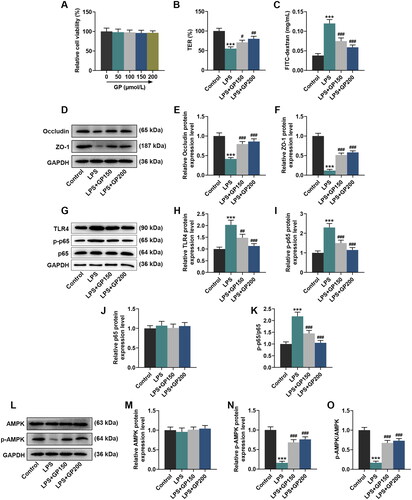
Treatment with GP at 150 μM or 200 μM partially reversed LPS-induced decreases in TER value, protein expression of occludin, ZO-1 and p-AMPK, and the ratio of p-AMPK to AMPK (p < 0.05), while partially repressing LPS-induced leakage of FD4 and LPS-induced increases in the protein expression of TLR4 and p-p65 as well as the ratio of p-p65 to p65 (p < 0.01, ). Concretely, the TER value (GP150: 15% change; GP200: 25% change), protein expression of occludin (GP150: 1.8-fold change; GP200: 2-fold change), ZO-1 (GP150: 3.5-fold change; GP200: 4-fold change) and p-AMPK (GP150: 4-fold change; GP200: 5-fold change) and the ratio of p-AMPK to AMPK (GP150: 4-fold change; GP200: 5-fold change) in the LPS + GP150 group and LPS + GP200 group was increased to various degrees, as compared with those in LPS group (p < 0.05, ). Also, various changes were observed in the leakage of FD4 (GP150: 38% decrease; GP200: 50% decrease), protein levels of TLR4 (GP150: 25% decrease; GP200: 50% decrease) and p-p65 (GP150: 40% decrease; GP200: 55% decrease) as well as the ratio of p-p65 to p65 (GP150: 40% decrease; GP200: 55% decrease) between LPS + GP150 group or LPS + GP200 group and LPS group (p < 0.01, ). These results suggested that GP may attenuate intestinal barrier dysfunction possibly through activating the AMPK pathway.
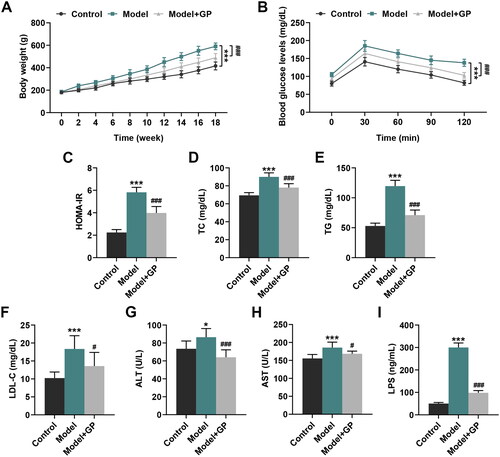
GP counteracted HFD/HF-induced effects on body weight, glucose tolerance, insulin resistance and the levels of hepatic steatosis and damage-related indices in rats
Given that obesity is a common phenomenon in patients with metabolic MS, the body weight of MS model rats was assessed. Rats fed with HFD/HF started to show an excessive gain of weight at the 10th week after the first feeding, compared to the control rats (p < 0.001, ). However, rats with GP treatment (300 mg/kg) presented a reduction in gain of weight, as compared with the model rats (p < 0.001, ). Moreover, glucose intolerance, insulin resistance and hepatic steatosis are thought to be prominent features in MAFLD and meanwhile can accelerate MAFLD progression (Pagano et al. 2002; Yki-Järvinen Citation2005). As shown in , rats with HFD/HF feeding exhibited a significantly impaired glucose tolerance relative to the control rats (p < 0.001). However, this HFD/HF-induced impairment on the glucose tolerance was attenuated by GP treatment (p < 0.001, ). Meanwhile, HOMA-IR (2.6-fold change), levels of TC (1.3-fold change), TG (2.3-fold change) and LDL-C (1.8-fold change) as well as ALT (1.1-fold change), AST (1.2-fold change) and LPS (6-fold change) were promoted in rats with HFD/HF feeding, when compared with those in the control rats (p < 0.05, ). Yet, the aforementioned indices were evidently decreased in rats after GP treatment (HOMA-IR: 32% decrease; TC: 14% decrease; TG: 40% decrease; LDL-C: 27% decrease; ALT: 26% decrease; AST: 10% decrease; LPS: 68% decrease), relative to those in the Model rats (p < 0.05, ). These results uncovered that GP could counteract HFD/HF-induced glucose tolerance, insulin resistance, hepatic steatosis and damage in vivo.
Figure 2. GP reversed HFD/HF-induced effects on body weight, glucose tolerance, insulin resistance and levels of hepatic steatosis and damage-related indices in rats. (A) Changes in the body weight of HFD/HF-fed Wistar rats receiving GP treatment (300 mg/kg) or not. (B) The blood glucose tolerance of HFD/HF-fed Wistar rats receiving GP treatment (300 mg/kg) or not was assessed by intra-peritoneal glucose tolerance test. (C–F, I) The serum levels of insulin, TC, TG, LDL-C and LPS in HFD/HF-fed Wistar rats receiving GP treatment (300 mg/kg) or not were determined by specific ELISA kits. (G, H) The serum levels of ALT and AST in HFD/HF-fed Wistar rats receiving GP treatment (300 mg/kg) or not were measured by an automatic biochemical analyser. GP: gypenosides; HFD/HF: high-fat/high fructose diet; HOMA-IR: homeostasis model assessment-insulin resistance; TC: total cholesterol; TG: triglycerides, LDL-C: high density lipoprotein cholesterol; LPS: lipopolysaccharides; ALT: alanine transaminase; AST: aspartate transaminase; ELISA: enzyme-linked immunosorbent assay. *p< 0.05, ***p< 0.001 vs. Control; #p<0.05, ###p< 0.001 vs. Model.
GP alleviated HFD/HF-induced histopathological abnormalities in rat liver and intestinal mucosa
Haematoxylin–eosin staining assay was carried out in rat liver. The results revealed that the control rats had normal liver lobule structures, where their outlines were clear with neatly arranged cell cords as well as radially arranged hepatic sinusoids and hepatic cords around central vein, and they showed normal liver cell structures without fat droplets, vacuoles and inflammatory cell infiltration, with normal nucleus and rich cytoplasm (). In contrast, the model rats exhibited disordered liver lobule structures without hepatocyte cords, the infiltration of inflammatory cells and enlarged hepatocytes which were presented as mild to moderate bullous steatosis with loose cytoplasm, squeezed nucleus and balloon-shaped deformation (). Moreover, all of these histopathological abnormalities above in rat liver were alleviated by GP treatment (). In the meantime, the NAFLD activity score showed that the levels of steatosis, lobular inflammation and ballooning degeneration in model rats were reduced after treatment with GP (). According to the results of ELISA, the increased levels of TNF-α, IL-6 and IL-1β in model rat liver tissue were remarkably decreased by GP treatment (p < 0.001, ). In addition, haematoxylin–eosin staining assay was also conducted in rat intestinal mucosa. It turned out that the intestinal villi were sparse in the model rats. In contrast, the control rats showed abundant intestinal villi (). Compared with the model group, the rats in the GP group had more regular microvilli, improved tight junctions and desmosomes, and smaller gaps ().
Figure 3. GP alleviated HFD/HF-induced histopathological abnormalities in rat liver and intestinal mucosa. (A) Histopathological changes in the liver (magnification: ×200, scale: 50 µm) were examined by haematoxylin–eosin staining. The levels of TNF-α (B), IL-6 (C) and IL-1β (D) in the liver tissue of rats were determined by ELISA. (E) Histopathological changes in the intestinal mucosa (magnification: ×200, scale: 200 µm) were detected by haematoxylin–eosin staining. GP: gypenosides; TNF-α: tumour necrosis factor-alpha; IL-6: interleukin 6; ELISA: enzyme-linked immunosorbent assay. ***p< 0.001 vs. Control; ###p< 0.001 vs. Model.
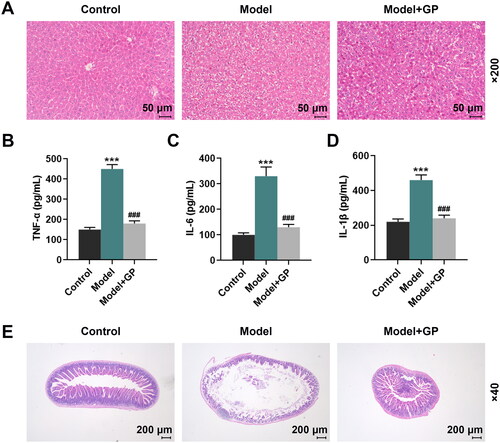
Table 1. NAFLD activity score in each group.
GP attenuated HFD/HF-induced intestinal mucosa damage and hepatic steatosis in rats possibly via inhibiting the TLR4/NF-κB pathway and activating the AMPK pathway
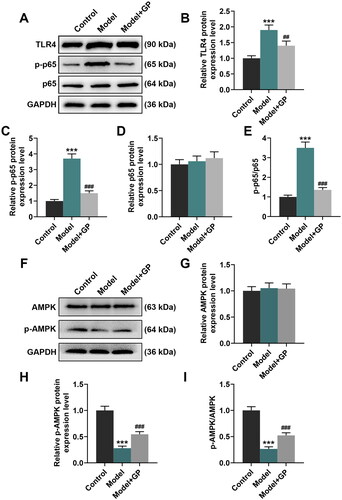
PPARα exerts an essential effect on fatty acid metabolism in liver (Pawlak et al. Citation2015), and SREBP-1c is a mediator of the activation of lipogenesis (Moon Citation2017). Thus, their expression in the livers of HFD/HF-fed rats were both assessed. HFD/HF feeding led to decreases in the protein expression of PPARα, occludin, ZO-1 and p-AMPK as well as the ratio of p-AMPK to AMPK (p < 0.001, and ). Besides, it also elicited increases in the protein expression of SREBP-1c, TLR4 and p-p65 and the ratio of p-p65 to p65 (p < 0.01, p < 0.001, and ). Rats with GP treatment showed notable increases in the protein expression of occludin, ZO-1, PPARα and p-AMPK, and visible decreases in SREBP-1c, TLR4 and p-p65 expression, when compared with the model rats (p < 0.001, and ). These results mirrored that GP could regulate metabolic disorders in NAFLD through inhibiting the TLR4/NF-κB pathway and activating the AMPK pathway.
Figure 4. GP attenuated HFD/HF-induced intestinal mucosa damage and hepatic steatosis in rats. The protein expression of occludin and ZO-1 (A–C) in the intestinal mucosa, and the protein expression of PPARα and SREBP-1c (A, D, E) in the livers of HFD/HF-fed Wistar rats receiving GP treatment (300 mg/kg) or not was analysed by Western blots, with GAPDH serving as a reference gene. GP: gypenosides; ZO-1; tight junction protein 1; PPARα: peroxisome proliferator-activated receptor α; SREBP-1c: sterol regulatory element-binding protein 1c; AMPK: adenosine monophosphate activated protein kinase; p: phosphorylated. ***p< 0.001 vs. Control; ###p< 0.001 vs. Model.
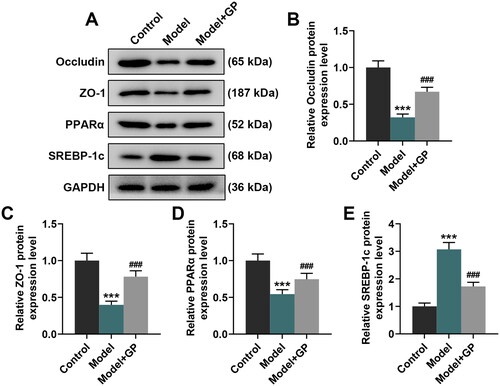
Figure 5. GP counteracted HFD/HF-induced TLR4/NF-κB activation and AMPK inhibition in rats. The protein expression of TLR4, p-p65, p65 (A–D), AMPK and p-AMPK (F–H) in the livers of HFD/HF-fed Wistar rats receiving GP treatment (300 mg/kg) or not was analysed by Western blots, with GAPDH serving as a reference gene. Ratios of p-p65 to p65 (E) and p-AMPK to AMPK (I) in the livers of HFD/HF-fed Wistar rats receiving GP treatment (300 mg/kg) or not. GP: gypenosides; TLR4: Toll-like receptor 4; AMPK: adenosine monophosphate activated protein kinase; NF-κB: nuclear factor kappa B; p: phosphorylated. ***p< 0.001 vs. Control;##p< 0.01, ###p< 0.001 vs. Model.
Discussion
At present, there are no specific agents approved for the treatment of NAFLD and its advanced forms, but several potential agents have been widely investigated, including glucose-lowering drugs, statins and other lipid lowering therapies and anti-hypertensive drugs (Xian et al. Citation2020; Mantovani and Dalbeni Citation2021). Within MAFLD, NAFLD is considered as a cause and a consequence of MS which is related to increased fasting plasma glucose and obesity (Yki-Järvinen Citation2014). Two key components of MS, glucose and TGs, are overproduced by the fatty liver. The liver is a key determinant of MS, and fat re-synthesis can be promoted by intake of high fat and high fructose which inhibits fatty acid β oxidation to induce hepatic steatosis and inflammatory responses, thus leading to the development of MAFLD (Liu et al. Citation2016). The initiation and progression of MAFLD are closely related to the altered intestinal flora-induced intestinal barrier dysfunction and abnormal lipid metabolism, which is termed as ‘dysbiosis’ (Day and James Citation1998; Festi et al. Citation2014; Dabke et al. Citation2019). Intestinal dysbiosis is thought to break down the intestinal barrier by increasing the permeability of intestinal mucosa towards inflammation inducers like bacteria which can injure the liver through the portal system and thus affect metabolic balance (Festi et al. Citation2014; Tripathi et al. Citation2018; Yan et al. Citation2019). These previous findings suggest that keeping an appropriate permeability of intestinal mucosa to maintain a normal intestinal barrier function is conducive to alleviating MAFLD.
GP treatment can increase bacterial biodiversity by reducing the value of Firmicutes/Bacteroidetes, which is positively correlated with the degree of obesity (Abdallah Ismail et al. Citation2011), and by promoting the abundances of Akkermansia, whose reduction in the intestinal mucus layer promotes MAFLD (Everard et al. Citation2013), in high fat and high cholesterol-induced MAFLD mice (Huang et al. Citation2019). 2α-OH-protopanaxadiol is a metabolite of GP, which has been reported to ameliorate MS via the intestinal FXR/GLP-1 axis through gut microbiota remodelling (Xie et al. Citation2020). These findings indicate that GP alleviate MAFLD through improving intestinal flora. LPS-induced intestinal barrier dysfunction exhibits a decreased TER value and an increased FD4 flux in the basolateral chamber of cell monolayers (He et al. Citation2020). Besides, occludin and ZO-1 are two of the major components of the apical tight junction (AJC) which regulates the selective permeability of the intestinal barrier, and decreased levels of occludin and ZO-1 are detected in intestinal barrier dysfunction (Yoseph et al. Citation2016). Our study demonstrated that GP treatment (150 μM and 200 μM) reversed the effect of LPS through reducing the FD4 leakage yet increasing TER value as well as protein levels of occludin and ZO-1 in LPS-injured Caco-2 cells, though no obvious effect of GP treatment was observed on the viability of normal Caco-2 Cells, which confirms our aforementioned surmise. Moreover, low-grade inflammation is a hallmark of metabolic disorders in MAFLD, this metabolism-associated inflammation may be fuelled by dysbiosis and intestinal barrier dysfunction (Tilg et al. Citation2020). TLR-4 signalling is found to be associated with thinner intestinal mucus layer and promote gut permeability in MAFLD (Miura and Ohnishi Citation2014), which is also identified as a stimulator in the production of inflammatory kinases (such as JNK, IKK and p38) as well as an impairer of the insulin signal transduction pathway (Jia L et al. Citation2014; Miura and Ohnishi Citation2014). Studies have confirmed that TLR-4 can drive ‘metabolic endotoxemia’-induced inflammatory reactions, and induce insulin resistance in MAFLD rats (Dowman et al. Citation2010; Henao-Mejia et al. Citation2012). Meanwhile, TLR-4 can identify the conserved pathogen-associated molecular patterns (PAMPs), which thereby triggers the activation of NF-κB during intestinal inflammatory responses, and leads to the enhanced phosphorylation of p65 (Qiao et al. Citation2020). Our study displayed that GP treatment (150 μM and 200 μM) repressed LPS-induced elevated levels of TLR-4 and p-p65, suggesting that the inhibition of TLR-4/NF-κB pathway is implicated in the protective effect of GP on the intestinal barrier.
In addition, accumulating studies have revealed that the activation of AMPK pathway is an important mechanism for counteracting LPS-induced intestinal barrier dysfunction (Sun et al. Citation2017; Wu et al. Citation2018). AMPK, an energy sensor playing a crucial role in regulating mitochondrial function (Garcia and Shaw Citation2017), can facilitate the formation of AJC via CDX2-induced epithelial differentiation (Sun et al. Citation2017). Certainly, AMPK inhibition hinders AJC assembly, triggering intestinal barrier dysfunction (Peng et al. Citation2009). Our study signified that GP treatment partially reversed LPS-induced AMPK inhibition in Caco-2 cells, indicating that GP may induce AMPK activation to protect the intestinal barrier.
Notably, AMPK is recorded to initiate catabolic processes including fatty acid oxidation promotion and glycolysis, and inhibit anabolic processes such as fatty acid synthesis and glucose storage, thereby achieving the restoration of metabolic balance (Garcia and Shaw Citation2017). MS generally occurs in MAFLD and comprises insulin resistance, excessive accumulation of adipose tissue, and altered intestinal flora (Buzzetti et al. Citation2016). Therefore, we hypothesized that GP may dispel these metabolic disorders possibly via AMPK activation. Our in vivo experiment first probed into how GP improved MAFLD-related metabolic disorders and then investigated whether the mechanism was associated with AMPK activation. Similar to the findings of Huang et al. (Citation2019), our study found that HFD/HF-induced rats gained excessive body weight and showed high blood glucose level, but these abnormal manifestations were attenuated by GP treatment. Moreover, the fatty liver accounts for the overproduction of two key components in MS, glucose and TGs (Yki-Järvinen Citation2014). As the study of Jia N et al. (Citation2018) demonstrated, GP treatment exerts inhibitory effects on high fat-induced upregulation of HOMA-IR, TC, TG, LDL-C, AST and ALT, of which AST and ALT are both indicative of the hepatocyte impairment. Meanwhile, LPS level is lifted with high fat-induced intestinal microbiota alteration, which can induce intestinal barrier dysfunction (de Faria Ghetti et al. Citation2018). In line with these findings, our study also detected that levels of above-mentioned indices were upregulated in rats after HFD/HF feeding, but GP treatment reversed these upward trends. Our findings suggested that GP can counteract HFD/HF intake-induced insulin resistance, hepatic steatosis, impaired hepatic function and intestinal barrier dysfunction. Besides, our histopathological examinations on the liver tissues and intestinal mucosa tissues of HFD/HF-fed rats showed evidently excessive lipogenesis in the liver and exorbitant permeability of intestine barrier, accompanied by the downregulation of occludin and ZO-1 in the intestine. Moreover, PPARα activation lowers TG via increasing lipolysis through induction of lipoprotein lipase (Pawlak et al. Citation2015). SREBP-1c can activate the transcription of genes participating in the biosynthesis of fatty acid and TG (Kobayashi et al. Citation2018). In our study, HFD/HF feeding led to downregulated PPARα and upregulated SREBP-1c in the liver. Apart from these, we found that GP treatment reversed HFD/HF-induced changes in levels of all those indices, indicating that GP alleviated HFD/HF-induced histological abnormalities, excessive lipogenesis in the liver and exorbitant permeability of intestine barrier (intestinal villi were sparse), which further confirmed that GP regulated HFD/HF-induced metabolic disorders. Meanwhile, activated TLR4/nuclear factor kappa B (NF-κB) pathway has been reported to be associated with metabolic disorders including hepatic steatosis and insulin resistance, as well as dysbiosis and gut-liver axis alteration in HFD-induced mice (Porras et al. Citation2017). Additionally, Lee et al. (Citation2019) verified the involvement of AMPK activation in GP-reduced and high fat-induced obesity (Neuschwander-Tetri Citation2017). On the basis of these results, we further authenticated that HFD/HF-induced AMPK inhibition and TLR4/NF-κB activation in rats with MS were reversed by GP treatment, signifying that GP improved MAFLD by activating AMPK and inhibiting the TLR4/NF-κB pathway. However, there are some limitations in this study. For example, only one intestinal cell line was used in this study to analyse intestinal barrier function. A co-culture model of Caco-2 cells and hepatocytes or a model of Kupffer cells is required in our future experiments. In addition, the effectiveness of GP, the TG level in the liver and the steatosis score in MAFLD model need to be further detected.
Conclusions
We reveal that GP treatment may help restore the metabolic balance in MAFLD rats through inhibiting HFD/HF diet-caused insulin resistance, hepatic steatosis, impaired hepatic function and intestinal barrier dysfunction possibly via AMPK activation and TLR4/NF-κB inhibition. Therefore, GP may become new regimens for the treatment of NAFLD, providing more options for patients with NAFLD.
Disclosure statement
The authors declare no conflicts of interest.
Data availability statement
The analysed data sets generated during the study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Abdallah Ismail N, Ragab SH, Abd Elbaky A, Shoeib AR, Alhosary Y, Fekry D. 2011. Frequency of Firmicutes and Bacteroidetes in gut microbiota in obese and normal weight Egyptian children and adults. Arch Med Sci. 3:501–507.
- Brunt EM, Wong VW, Nobili V, Day CP, Sookoian S, Maher JJ, Bugianesi E, Sirlin CB, Neuschwander-Tetri BA, Rinella ME. 2015. Nonalcoholic fatty liver disease. Nat Rev Dis Primers. 1:15080.
- Buzzetti E, Pinzani M, Tsochatzis EA. 2016. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 65(8):1038–1048.
- Dabke K, Hendrick G, Devkota S. 2019. The gut microbiome and metabolic syndrome. J Clin Invest. 129(10):4050–4057.
- Day CP, James OF. 1998. Steatohepatitis: a tale of two "hits"? Gastroenterology. 114(4):842–845.
- de Faria Ghetti F, Oliveira DG, de Oliveira JM, de Castro Ferreira L, Cesar DE, Moreira APB. 2018. Influence of gut microbiota on the development and progression of nonalcoholic steatohepatitis. Eur J Nutr. 57(3):861–876.
- Dietrich P, Hellerbrand C. 2014. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract Res Clin Gastroenterol. 28(4):637–653.
- Dowman JK, Tomlinson JW, Newsome PN. 2010. Pathogenesis of non-alcoholic fatty liver disease. QJM. 103(2):71–83.
- Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, et al. 2013. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 110(22):9066–9071.
- Festi D, Schiumerini R, Eusebi LH, Marasco G, Taddia M, Colecchia A. 2014. Gut microbiota and metabolic syndrome. World J Gastroenterol. 20(43):16079–16094.
- Garcia D, Shaw RJ. 2017. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. 66(6):789–800.
- He S, Guo Y, Zhao J, Xu X, Wang N, Liu Q. 2020. Ferulic acid ameliorates lipopolysaccharide-induced barrier dysfunction via microRNA-200c-3p-mediated activation of PI3K/AKT pathway in Caco-2 cells. Front Pharmacol. 11:376.
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. 2012. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 482(7384):179–185.
- Huang X, Chen W, Yan C, Yang R, Chen Q, Xu H, Huang Y. 2019. Gypenosides improve the intestinal microbiota of non-alcoholic fatty liver in mice and alleviate its progression. Biomed Pharmacother. 118:109258.
- Ji X, Shen Y, Guo X. 2018. Isolation, structures, and bioactivities of the polysaccharides from Gynostemma pentaphyllum (Thunb.) Makino: a review. Biomed Res Int. 2018:6285134.
- Jia L, Vianna CR, Fukuda M, Berglund ED, Liu C, Tao C, Sun K, Liu T, Harper MJ, Lee CE, et al. 2014. Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun. 5:3878.
- Jia N, Lin X, Ma S, Ge S, Mu S, Yang C, Shi S, Gao L, Xu J, Bo T, et al. 2018. Amelioration of hepatic steatosis is associated with modulation of gut microbiota and suppression of hepatic miR-34a in Gynostemma pentaphylla (Thunb.) Makino treated mice. Nutr Metab. 15:86.
- Kobayashi M, Fujii N, Narita T, Higami Y. 2018. SREBP-1c-Dependent metabolic remodeling of white adipose tissue by caloric restriction. Int J Mol Sci. 19(11):3335.
- Lambertz J, Weiskirchen S, Landert S, Weiskirchen R. 2017. Fructose: a dietary sugar in crosstalk with microbiota contributing to the development and progression of non-alcoholic liver disease. Front Immunol. 8:1159.
- Lee HS, Lim SM, Jung JI, Kim SM, Lee JK, Kim YH, Cha KM, Oh TK, Moon JM, Kim TY, et al. 2019. Gynostemma pentaphyllum extract ameliorates high-fat diet-induced obesity in C57BL/6N mice by upregulating SIRT1. Nutrients. 11(10):2475.
- Li S, Zheng X, Zhang X, Yu H, Han B, Lv Y, Liu Y, Wang X, Zhang Z. 2021. Exploring the liver fibrosis induced by deltamethrin exposure in quails and elucidating the protective mechanism of resveratrol. Ecotoxicol Environ Saf. 207:111501.
- Liu JP, Zou WL, Chen SJ, Wei HY, Yin YN, Zou YY, Lu FG. 2016. Effects of different diets on intestinal microbiota and nonalcoholic fatty liver disease development. World J Gastroenterol. 22(32):7353–7364.
- Maher JJ, Leon P, Ryan JC. 2008. Beyond insulin resistance: innate immunity in nonalcoholic steatohepatitis. Hepatology. 48(2):670–678.
- Mantovani A, Dalbeni A. 2021. Treatments for NAFLD: state of art. Int J Mol Sci. 22(5):2350.
- Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, et al. 2003. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 37(4):917–923.
- Milić S, Lulić D, Štimac D. 2014. Non-alcoholic fatty liver disease and obesity: biochemical, metabolic and clinical presentations. World J Gastroenterol. 20(28):9330–9337.
- Miura K, Ohnishi H. 2014. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J Gastroenterol. 20(23):7381–7391.
- Moon YA. 2017. The SCAP/SREBP pathway: a mediator of hepatic steatosis. Endocrinol Metab. 32(1):6–10.
- Neuschwander-Tetri BA. 2017. Non-alcoholic fatty liver disease. BMC Med. 15(1):45.
- O'Neill S, O'Driscoll L. 2015. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 16(1):1–12.
- Pagano G, Pacini G, Musso G, Gambino R, Mecca F, Depetris N, Cassader M, David E, Cavallo-Perin P, Rizzetto M. 2002. Nonalcoholic steatohepatitis, insulin resistance, and metabolic syndrome: further evidence for an etiologic association. Hepatology. 35(2):367–372.
- Pawlak M, Lefebvre P, Staels B. 2015. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 62(3):720–733.
- Peng L, Li ZR, Green RS, Holzman IR, Lin J. 2009. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr. 139(9):1619–1625.
- Porras D, Nistal E, Martínez-Flórez S, Pisonero-Vaquero S, Olcoz JL, Jover R, González-Gallego J, García-Mediavilla MV, Sánchez-Campos S. 2017. Protective effect of quercetin on high-fat diet-induced non-alcoholic fatty liver disease in mice is mediated by modulating intestinal microbiota imbalance and related gut-liver axis activation. Free Radic Biol Med. 102:188–202.
- Qiao J, Sun Z, Liang D, Li H. 2020. Lactobacillus salivarius alleviates inflammation via NF-κB signaling in ETEC K88-induced IPEC-J2 cells. J Anim Sci Biotechnol. 11:76.
- Radwan E, Bakr MH, Taha S, Sayed SA, Farrag AA, Ali M. 2020. Inhibition of endoplasmic reticulum stress ameliorates cardiovascular injury in a rat model of metabolic syndrome. J Mol Cell Cardiol. 143:15–25.
- Ryan MC, Wilson AM, Slavin J, Best JD, Jenkins AJ, Desmond PV. 2005. Associations between liver histology and severity of the metabolic syndrome in subjects with nonalcoholic fatty liver disease. Diabetes Care. 28(5):1222–1224.
- Sun X, Yang Q, Rogers CJ, Du M, Zhu MJ. 2017. AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ. 24(5):819–831.
- Tilg H, Zmora N, Adolph TE, Elinav E. 2020. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol. 20(1):40–54.
- Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, Knight R. 2018. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol. 15(7):397–411.
- Wang J, Yang JL, Zhou PP, Meng XH, Shi YP. 2017. Further new Gypenosides from Jiaogulan (Gynostemma pentaphyllum). J Agric Food Chem. 65(29):5926–5934.
- Wu W, Wang S, Liu Q, Shan T, Wang Y. 2018. Metformin protects against LPS-induced intestinal barrier dysfunction by activating AMPK pathway. Mol Pharm. 15(8):3272–3284.
- Xian YX, Weng JP, Xu F. 2020. MAFLD vs. NAFLD: shared features and potential changes in epidemiology, pathophysiology, diagnosis, and pharmacotherapy. Chin Med J. 134(1):8–19.
- Xie Z, Jiang H, Liu W, Zhang X, Chen D, Sun S, Zhou C, Liu J, Bao S, Wang X, et al. 2020. The triterpenoid sapogenin (2α-OH-protopanoxadiol) ameliorates metabolic syndrome via the intestinal FXR/GLP-1 axis through gut microbiota remodelling. Cell Death Dis. 11(9):770.
- Xu C, Wan X, Xu L, Weng H, Yan M, Miao M, Sun Y, Xu G, Dooley S, Li Y, et al. 2015. Xanthine oxidase in non-alcoholic fatty liver disease and hyperuricemia: one stone hits two birds. J Hepatol. 62(6):1412–1419.
- Xue L, He J, Gao N, Lu X, Li M, Wu X, Liu Z, Jin Y, Liu J, Xu J, et al. 2017. Probiotics may delay the progression of nonalcoholic fatty liver disease by restoring the gut microbiota structure and improving intestinal endotoxemia. Sci Rep. 7:45176.
- Yan Z, Yang F, Hong Z, Wang S, Jinjuan Z, Han B, Xie R, Leng F, Yang Q. 2019. Blueberry attenuates liver fibrosis, protects intestinal epithelial barrier, and maintains gut microbiota homeostasis. Can J Gastroenterol Hepatol. 2019:1–11.
- Yang Q, Han B, Xue J, Lv Y, Li S, Liu Y, Wu P, Wang X, Zhang Z. 2020. Hexavalent chromium induces mitochondrial dynamics disorder in rat liver by inhibiting AMPK/PGC-1α signaling pathway. Environ Pollut. 265(Pt A):114855.
- Yki-Järvinen H. 2005. Fat in the liver and insulin resistance. Ann Med. 37(5):347–356.
- Yki-Järvinen H. 2014. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2(11):901–910.
- Yoseph BP, Klingensmith NJ, Liang Z, Breed ER, Burd EM, Mittal R, Dominguez JA, Petrie B, Ford ML, Coopersmith CM. 2016. Mechanisms of intestinal barrier dysfunction in sepsis. Shock. 46(1):52–59.
- Zhang H-K, Ye Y, Li K-J, Zhao Z-n, He J-F. 2020. Gypenosides prevent H2O2-induced retinal ganglion cell apoptosis by concurrently suppressing the neuronal oxidative stress and inflammatory response. J Mol Neurosci. 70(4):618–630.