
Abstract
Context
Dihydromyricetin (DMY) is extracted from vine tea, a traditional Chinese herbal medicine with anti-cancer, liver protection, and cholesterol-lowering effects.
Objective
This study investigated the mechanism of DMY against hepatocellular carcinoma (HCC).
Materials and methods
Potential DMY, HCC, and cholesterol targets were collected from relevant databases. PPI networks were created by STRING. Then, the hub genes of co-targets, screened using CytoHubba. GO and KEGG pathway enrichment, were performed by Metascape. Based on the above results, a series of in vitro experiments were conducted by using 40–160 μM DMY for 24 h, including transwell migration/invasion assay, western blotting, and Bodipy stain assay.
Results
Network pharmacology identified 98 common targets and 10 hub genes of DMY, HCC, and cholesterol, and revealed that the anti-HCC effect of DMY may be related to the positive regulation of lipid rafts. Further experiments confirmed that DMY inhibits the proliferation, migration, and invasion of HCC cells and reduces their cholesterol levels in vitro. The IC50 is 894.4, 814.4, 467.8, 1,878.8, 151.8, and 156.9 μM for 97H, Hep3B, Sk-Hep1, SMMC-7721, HepG2, and Huh7 cells, respectively. In addition, DMY downregulates the expression of lipid raft markers (CAV1, FLOT1), as well as EGFR, PI3K, Akt, STAT3, and Erk.
Discussion and conclusion
The present study reveals that DMY suppresses EGFR and its downstream pathways by reducing cholesterol to disrupt lipid rafts, thereby inhibiting HCC, which provides a promising candidate drug with low toxicity for the treatment of HCC.
Introduction
Hepatocellular carcinoma (HCC) is one of the global top 10 cancers, with high morbidity (4.7%) and mortality (8.3%) worldwide (Roth et al. Citation2018; Sung et al. Citation2021). HBV/HCV virus, aflatoxin B, alcoholic liver disease, and nonalcoholic steatohepatitis are the major risk factors (Perz et al. Citation2006; Yang and Roberts Citation2010). At present, HCC can be treated by hepatectomy, liver transplantation, locoregional treatments, and systemic therapies (Omata et al. Citation2017). However, since most patients with HCC are usually diagnosed at late stage, systematic therapy is the only option. Unfortunately, radiation therapy, sorafenib, and regorafenib are not satisfactory because they could cause serious side effects and drug resistance (Bentzen Citation2006; Randrup Hansen et al. Citation2017; Shoukat et al. Citation2019; Tang et al. Citation2020). Therefore, safe and effective drugs are urgently needed.
HCC is a type of cancer accompanied by dyshomeostasis of the internal environment, especially in lipid metabolism disorder (Luo et al. Citation2020; Li et al. Citation2022). There is a strong correlation between cholesterol and HCC. Generally, cholesterol is de novo synthesized, catabolized, and reverse-transported in the liver (Luo et al. Citation2020). Most of the cellular cholesterol is located on the plasma membrane, where it forms lipid rafts with sphingolipids and other proteins, serving as a platform for receptor trafficking and signal transduction that could promote the occurrence and development of cancers (Lingwood and Simons Citation2010; Greenlee et al. Citation2021). As early as 1990, studies have shown that cholesterol synthesis was enhanced in HCC (Kawata et al. Citation1990). Emerging evidence found that cholesterol affected HCC progression in various aspects. For example, increased cholesterol biosynthesis drives drug resistance of HCC, self-renewal of CSCs in HCC, and tumorigenesis (Wang et al. Citation2021; Mok et al. Citation2022). In addition, cholesterol accumulation reduces the antitumor immune response mediated by natural killer T cells in HCC (Tang et al. Citation2022). Moreover, cholesterol promotes the progression from non-alcoholic fatty liver disease to HCC by activating mTORC1 (Liu et al. Citation2022). Notably, cholesterol-lowering drugs are associated with a reduction in cancer-related mortality (Nielsen et al. Citation2012; Cardwell et al. Citation2014). Thus, lowering cholesterol might be an effective way to cancer prevention.
Dihydromyricetin (DMY), also known as ampelopsin, is a flavonoid compound derived from traditional Chinese herbal medicine vine tea. The DMY content in vine tea reaches as high as 30–40% (w/w) (Liu et al. Citation2019). In recent years, DMY has been gaining much attention because of its biological activities, including anti-inflammation (Dong et al. Citation2021), antioxidation (Zhang et al. Citation2019), liver protection (Silva et al. Citation2020), and anticancer (Ye et al. Citation2021). Furthermore, DMY has been found to promote cholesterol efflux and decreases serum total cholesterol levels in mice (Zeng et al. Citation2018; Song et al. Citation2022). Importantly, DMY is non-toxic, making it an ideal candidate for the treatment of HCC. Therefore, we propose that DMY exerts an anti-HCC effect by lowering cholesterol. To investigate this hypothesis, we evaluated the potential of DMY affecting cholesterol to anti-HCC through network pharmacology and conducted in vitro cell experiments to confirm the role and mechanism of DMY in HCC.
Materials and methods
Prediction of DMY-related targets
The Canonical SMILES number (CS) and the 3D chemical structure formula (SDF) of dihydromyricetin were provided by PubChem (https://pubchem.ncbi.nlm.nih.gov/) using the keyword ‘Dihydromyricetin’. DMY targets were collected from five data sources: PharmMapper (http://www.lilab-ecust.cn/pharmmapper/), SwissTarget (http://www.swisstargetprediction.ch/), TargetNet (http://targetnet.scbdd.com/), and TCMSP (https://old.tcmsp-e.com/tcmsp.php). After duplicates were deleted, the Uniprot database (https://www.uniprot.org/) transformed all targets into gene names.
Prediction of HCC and cholesterol-related targets
By searching the keywords ‘Hepatocellular Carcinoma’ and ‘cholesterol’ on OMIM (https://www.omim.org/), GeneCards (https://www.genecards.org/, ‘score’ >15), and NCBI (https://www.ncbi.nlm.nih.gov/), retrieve genes which met the criteria. Then, using the UniProt database, turn them into gene names.
PPI network and hub targets analyses
The Venn diagram from the collected targets of DMY, HCC, and cholesterol using Venny 2.1.0 (https://bioinfogp.cnb.csic.es/tools/venny/). Then, use of co-targets to establish PPI networks by uploading them to the STRING database (https://www. string-db. org/). The .tsv file was downloaded for importing into Cytoscape (https://cytoscape.org/) to create a PPI network based on degree value. The hub gene was screened by the CytoHubba plugin in Cytoscape.
Gene ontology and pathway enrichment analysis
The common collected targets were input into Gene ID by Uniprot (https://www.uniprot.org) and uploaded to Metascape (http://metascape.org/gp/index.html) for enrichment analysis. In the enrichment option, GO molecular function, GO biological processes, GO cellular components, and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways were selected for analysis.
In vitro experiments
Cell culture and reagents
HepG2, Hep3B, 97H, SMMC-7721, Sk-Hep1, Huh7, and LO2 cell lines were provided by the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cell lines were cultured in DMEM containing 10% fetal bovine serum and 1% streptomycin-penicillin. Using a humidified incubator maintained the cells at 37 °C with 5% CO2. DMY (N1849, APExBIO, USA; the purity is 99.04%) was dissolved as a 200 mM stock solution with dimethylsulfoxide (DMSO, Sigma-Aldrich, USA), and stored at −80 °C. The stock solution was diluted to a suitable concentration through the culture medium.
Cell counting Kit-8 assay (CCK-8)
Cell viability was assessed by the CCK-8 kit (k1018, APExBIO, USA) according to the manufacturer’s instructions as described previously (Zhang et al. Citation2021). The density of cells in 96-well plates was 6 × 103 cells per well.
Colony formation assay
4 × 103 cells per well were seeded in 6-well plates and incubated for about 7–10 days until the colonies were obviously visible. The cells were then fixed in 4% paraformaldehyde for 10 min and stained in crystal violet (Solarbio, China) for 10 min.
Micro total cholesterol (TC) content assay
Cholesterol content was examined by Micro TC Content Assay Kit (Solarbio, Beijing, China). Making a standard curve by a cholesterol standard solution. The cell extract supernatant was incubated with the detection reagent for 15 min at 37 °C, and optical density values were determined at 500 nm.
BODIPY stain
BODIPY 493/503 (No. GC42959, Glpbio, Montclair, CA, United States) was used to produce cellular cholesterol fluoresce. HepG2 cells were cultured in 6-well plates with indicated doses of DMY for 24 h. The was followed by incubation with 2 μM BODIPY staining solution in darkness at 37 °C for 15 min. Fluorescent intensity was captured with an OLYMPUS DP73 fluorescence microscope.
Wound healing assay
A wound healing assay was conducted as described previously (Zhang et al. Citation2021). The area covered by the migrated cells was quantified with Image J 1.53 T (National Institutes of Health, USA).
Transwell migration assay
The transwell migration assay followed previous descriptions (Zhang et al. Citation2021).
Transwell invasion assay
Invasion assays were similar to transwell migration assays, except the transwell inserts were coated with Matrigel (BD, Franklin Lakes, NJ, USA).
Western blotting
Western blots were performed in the same way as previously described (Zhang et al. Citation2021). GAPDH (1:10,000 dilution), β-Actin (1:3,000 dilution), EGFR (1:1,000 dilution), Caveolin-1 (CAV1, 1:1,000 dilution), Flotillin 1 (FLOT1, 1:2,000 dilution), Desmoplakin (DSP, 1:1,000 dilution), E-cadherin (E-CAD, 1:2,0000 dilution), N-cadherin (N-CAD, 1:4,000 dilution), Vimentin (Vim, 1:4,000 dilution), STAT3 (signal transducer and activator of transcription, 1:3,000 dilution), Erk (extracellular regulated protein kinases, 1:3,000 dilution), and p-Erk (1:3,000 dilution) were purchased from Proteintech. AKT (protein kinase B, 1:1,000 dilution) and p-AKT (1:1,000 dilution) were purchased from Abcam.
Statistical analysis
All experiments were independently repeated in triplicate. All data are represented as mean ± SD. Two group comparison was analyzed by Student’s t-test and multiple comparisons were analyzed by one-way ANOVA, using GraphPad Prism version 8.0 (La Jolla, CA, USA). p < 0.05 was taken as the statistical significance level.
Results
Targets of DMY, HCC, and cholesterol
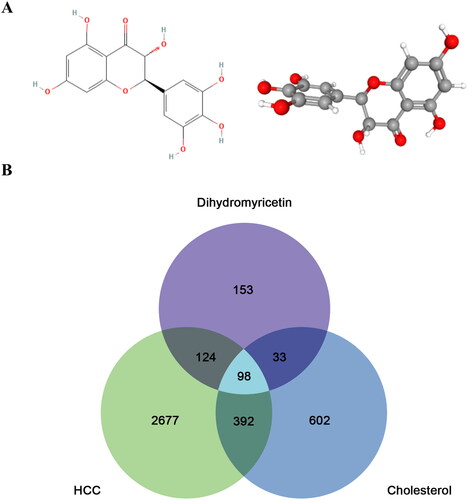
The chemical structure of DMY in 2D and 3D formats is presented in . Based on an extensive search of PharmMapper, SwissTarget, TargetNet, and TCMSP, a total of 408 DMY-related targets were identified. The number of HCC and cholesterol targets from GeneCards, OMIM, and NCBI are 1,125 and 3,291, respectively. The co-target of DMY, HCC, and cholesterol was 98, as illustrated in the Venn diagram (). These findings reveal that DMY may have a significant effect on HCC through its relationship with cholesterol, and further exploration into the mechanism of DMY on HCC is warranted.
Figure 1. (A) 2D (Left) and 3D (right) structures of DMY. (B) The co-targets Venn diagram.
PPI network
The PPI network, consisting of the 98 co-targets shared among DMY, HCC, and cholesterol, is comprised of 98 nodes and 961 edges (). The average node degree was 19.6, with a local clustering coefficient of 0.596, and p = 1.0 × 10−16, indicating a significant difference between this group of targets and random combinations. Outer circle degree values range from 0 to 10, intermediate circle degree values range from 11 to 50, and inner circle degree values range from 51 to 200. The thickness of the color in the PPI network indicates the degree of the target, the thicker the color, the higher the degree. The analysis of the PPI network revealed that ‘ALB’, ‘EGFR’, and ‘AKT1’ are the core targets of PPI.
Figure 2. (A) PPI network of 98 co-targets of DMY and cholesterol in HCC. They are colored according to ranking. The higher the ranking, the redder the color. (B) Ten hub genes from the PPI network. EGFR ranks first.
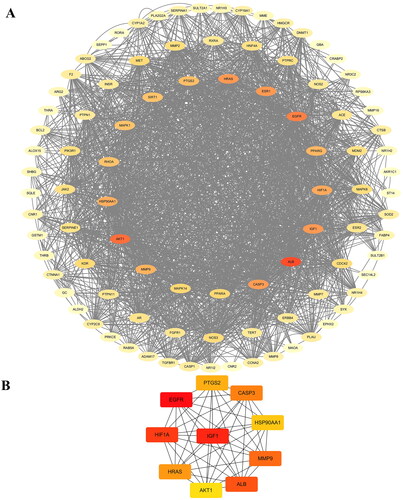
Prediction of core targets
The CytoHubba analysis showed the possible core targets of DMY include ‘EGFR’, ‘IGF1’, ‘HIF1A’, ‘ALB’, ‘MMP9’, ‘CASP3’, ‘HRAS’, ‘AKT1’, ‘PTGS2’ (). Notably, EGFR (epidermal growth factor receptor) ranks first in this list ().
Table 1. Top 10 core targets.
GO and KEGG analyses
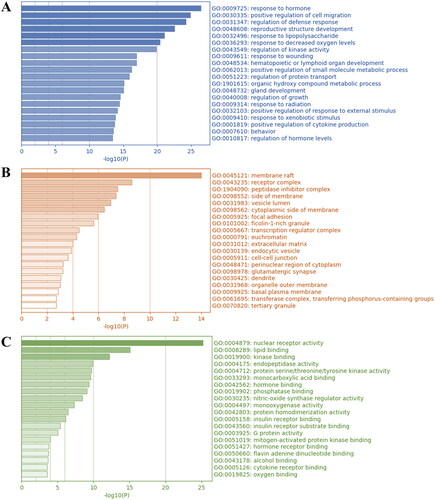
The 98 hub targets were subjected to GO and KEGG pathway enrichment analyses to further investigate the potential therapeutic mechanisms of DMY in HCC. The GO analysis result revealed that in the molecular function (MF) aspect DMY could be associated with response to hormone, positive regulation of cell migration, and regulation of defense response (). In terms of cellular component (CC), lipid raft might be involved (). Moreover, at biological processes (BP) level, DMY could have a primary impact on nuclear receptor activity, lipid binding, and kinase binding ().
Figure 3. GO analysis of DMY, HCC, and cholesterol co-targets. (A) GO molecular function analysis. (B) GO cellular component analysis. (C) GO biological processes analysis. On the Y-axis, the enriched GO items of target genes are displayed. The gene counts for the items are shown on the X-axis. The colors represent the p value. The darker the color shading, the higher the p value.
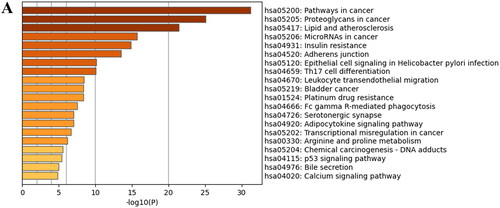
Furthermore, the top 3 significantly enriched pathways between DMY, cholesterol, and HCC were pathways in cancer, proteoglycans in cancer, and lipid and atherosclerosis. Specifically, ‘pathways in cancer’ include 12 pathways (). PI3K (phosphatidylinositol 3-Kinase)/Akt, MAPK (mitogen-activated protein kinase), and STAT pathway being one of those 12 pathways, and are also the downstream pathway for EGFR (Figure S1A) (Kuzu et al. Citation2016). In particular, the PI3K/Akt pathway activates epithelial-to-mesenchymal transition (EMT) progress (Greenlee et al. Citation2021), the MAPK pathway is involved in tumor proliferation, invasion, and metastasis (Guo et al. Citation2020), and the STAT pathway is implicated in cancer development and progression (Verhoeven et al. Citation2020). In addition, the activation of EGFR requires translocation to lipid rafts (Xu et al. Citation2012). Based on the above information, we hypothesize that DMY may exert its anti-HCC effect by decreasing cholesterol to disrupt lipid raft, affecting EGFR kinase interaction with its ligand, and suppressing the downstream pathway involved HCC cell proliferation, migration, and invasion.
Figure 4. (A) KEGG Analyses of DMY, HCC, and cholesterol co-targets.
In vitro experiments
DMY suppresses the proliferation of HepG2 cells
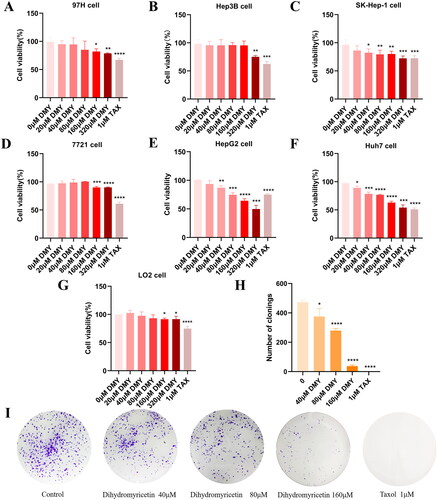
The capability of sustaining chronic proliferation is one of the most fundamental characteristics of cancer cells (Hanahan and Weinberg Citation2011). So, we first evaluated the cytotoxicity of DMY on normal hepatocyte and HCC cell lines. The CCK-8 assay was used to detect the viability of HCC cells and normal hepatocytes after treating them with DMY for 24 h at different concentrations. The results revealed that in contrast to 97H (IC50: 894.4 μM), Hep3B (IC50: 814.4 μM), Sk-Hep1 (IC50: 467.8 μM), and SMMC-7721 (IC50: 1878.8 μM) cell lines, HepG2 (IC50: 151.8 μM) and Huh7 (IC50: 156.9 μM) were more sensitive to DMY-mediated cytotoxicity (). We used HepG2 cells for subsequent validation experiments due to their lower IC50 value and the higher efficacy of DMY in HepG2 cells compared to the positive drug paclitaxel (Tax). Importantly, DMY has low toxicity to normal hepatocyte cell line LO2 (IC50: 2815 μM, ). Then, further analysis confirmed the role of DMY in inhibiting HepG2 proliferation through clonal formation assay. The result exhibited that DMY greatly reduced the colony formation of HepG2 cells (). These data suggest that DMY can effectively inhibit the proliferation of HepG2 cells.
Figure 5. DMY suppressed HepG2 cell proliferation. (A–G) The cytotoxicity of DMY (0, 20, 40, 80, 160, 320 μM) on 97H, Hep3B, Sk-Hep1, SMMC-7721, HepG2, Huh7, and LO2 cells were assessed by CCK-8 assay. (H) Numbers of colony statistics (I) Colony formation ability in HepG2 cells was detected by colony formation assay. n ≥ 3, *p < 0.05, **p < 0.01, ***p < 0.001 vs. the control.
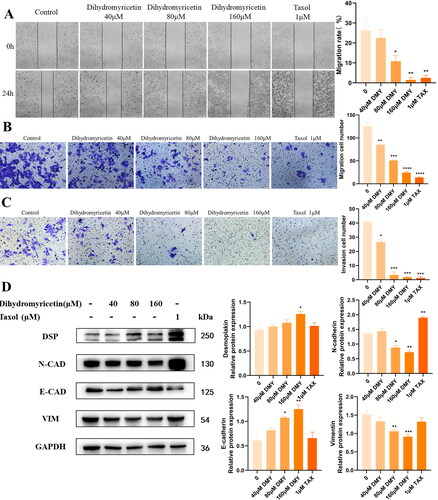
DMY inhibits the cell motility of HepG2 cells
Invasion and metastasis are critical hallmarks of cancers (Hanahan and Weinberg Citation2011), and also the mortality risk of HCC. Then we examined whether DMY affects HCC cell migration and invasion by conducting wound-healing, transwell migration, and transwell invasion assays. The wound-healing assay revealed that cells treated with DMY had larger wound closure areas, indicating that DMY had an inhibitory effect on cell migration (). To confirm this effect, transwell assays were used to detect the impact of DMY on HepG2 cell migration. As illustrated in , treatment with 160 μM DMY reduced cell migration by more than 80% and cell invasion by more than 50%. EMT is generally considered a key step in the migration and invasion of cancer cells, in which epithelial phenotype is lost and mesenchymal characteristics are acquired (Brabletz et al. Citation2018). When tumor cells undergo EMT, the most significant change is the decrease of epithelial cell markers, like E-CAD and DSP, while the increase of mesenchymal cell markers like N-CAD, Vim, and transcription factors (Snail, Zeb, Slug, Twist) (Brabletz et al. Citation2018). Then the expressions of EMT-related protein were further verified by western blotting. The result showed that DMY up-regulates E-CAD, DSP expression, and down-regulates N-CAD, Vim expression (), suggesting that DMY inhibited the cell motility of HepG2 cells.
Figure 6. DMY inhibited HepG2 cell invasion and migration. (A) Wound healing assay in DMY treated HepG2 cells. (B) the migration abilities of HepG2 cells with DMY treatment were evaluated by transwell migration assay. (C) the Invasive abilities of DMY treated HepG2 cells were detected by transwell invasion assays. (D) The expression of EMT marker proteins in DMY treated HepG2 cells was detected by Western blot. n = 3, *p < 0.05, **p < 0.01, ***p < 0.001 vs. the control.
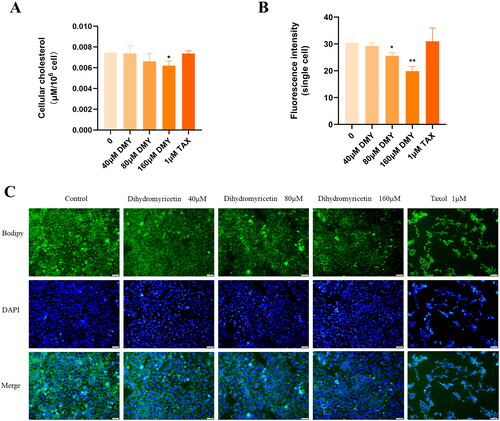
DMY reduces the cholesterol content of HepG2 cells
The effect of DMY on intracellular cholesterol content was examined by the method of micro TC content determination. The result showed that DMY reduced TC in HepG2 cells in a dose-dependent manner (). Furthermore, Bodipy staining confirmed this result. We applied single-cell fluorescence intensity to evaluate the cholesterol levels in HepG2 cells. Consistent with the result of the micro TC content assay, quantitative analysis showed that HepG2 cells treated with DMY had weaker single-cell fluorescence intensity (). Overall, these data provide compelling evidence that DMY reduces the cholesterol content of HepG2 cells.
Figure 7. DMY reduced HepG2 cell cholesterol content. (A) Quantification of intracellular TC levels. (B) Quantitative analysis of BODIPY staining single-cell fluorescence intensity. (C) BODIPY staining displayed intracellular cholesterol levels. n = 3, *p < 0.05, **p < 0.01, ***p < 0.001 vs. the control.
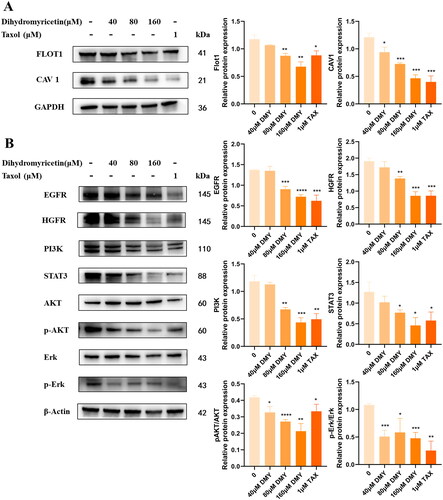
DMY disrupts lipid rafts and downregulates the expression of EGFR, PI3K, Akt, STAT3 and Erk in vitro
According to GO functional enrichment analysis, DMY mainly affects lipid rafts to exert anti-HCC effects in terms of cell composition. Intriguingly, EGFR, the top-ranked gene in the Hub gene results, has been reported to be located on the lipid raft. Therefore, the expression levels of EGFR, the key protein of EGFR downstream pathway (PI3K, Akt, p-Akt, STAT3, Erk, p-Erk) and the marker protein of lipid raft (CAV-1 and Flot1) were further analyzed by Western Blotting. The results demonstrated that DMY reduced the expression of EGFR, PI3K, Akt, STAT3, and Erkin HepG2 (). Meanwhile, the expression of lipid raft marker proteins CAV-1 and Flot1 were also decreased (). These data indicate that DMY might interfere with lipid rafts to inhibit EGFR and its downstream signaling pathways. In addition, HGFR is a member of the receptor tyrosine kinase superfamily. The combination of HGFR and its ligand HGF activates MAPK and PI3K/AKT signals, promoting tumor genesis (Recondo et al. Citation2020). We curiously detected the expression of HGFR; the result shows that DMY could inhibit HGFR as well.
Figure 8. (A) EGFR, HGFR, PI3K, Akt, STAT3, and Erk protein levels. (B) the expression of CAV-1 and FLOT-1. n = 3, *p < 0.05, **p < 0.01, ***p < 0.001 vs. The control.
Discussion and conclusions
DMY has been reported to exhibit anti-cancer activity in several types of cancers, including HCC, nasopharyngeal cancer, and ovarian cancer (Xu et al. Citation2017; Huang et al. Citation2022). Previous studies have shown the anti-HCC mechanisms of DMY involve the suppression of HCC cell proliferation by inducing G2/M arrest (Huang et al. Citation2013) and induction of HCC cell apoptosis by inhibiting the Notch1 pathway (Lu et al. Citation2017), up-regulating p53 expression (Liu et al. Citation2014), or reducing ROS accumulation (Liu et al. Citation2014). In addition, DMY also inhibits HCC cell migration and invasion through down-regulating MMP9 expression (Zhang et al. Citation2014) and improves Nedaplatin chemosensitivity by activating the p53/Bcl-2 pathway (Jiang et al. Citation2015). However, its effects on EMT, a key process during cancer metastasis, are still unclear. The present study provides evidence that DMY plays an inhibitory effect on the EMT progress of HCC cells.
Accumulating evidence linking cholesterol disorders to cancer occurrence and development. The level of cholesterol in HCC cells is abnormally high, as cholesterol biosynthesis and absorption are increased, while cholesterol export and excretion are reduced (Hryniewicz-Jankowska et al. Citation2019; Zhou and Sun Citation2021). On the one hand, abnormally elevated cholesterol can lead to increased risk factors for HCC such as lipotoxicity, inflammation, and fibrosis (Zhou and Sun Citation2021). Furthermore, the increase in cholesterol content facilitates the rapid growth of cancer cells and the formation of cancers (Snaebjornsson et al. Citation2020), as evidenced by the activation of the Hedgehog signal by cholesterol synthesis, leading to self-renewal and tumorigenesis (Wang et al. Citation2021). Conversely, a study has revealed that increased cholesterol efflux promotes human colon cancer cell death (Smith and Land Citation2012). In particular, cholesterol-lowering drugs are associated with a reduction in cancer-related mortality (Nielsen et al. Citation2012). As previously mentioned, DMY decreases cholesterol in macrophages by promoting cholesterol efflux, while the effect of DMY on cholesterol in cancer cells is uncertain. Our results reveal that DMY reduced the cholesterol content of HCC cells.
Notably, an elevated level of cholesterol has been shown to increase cholesterol-driven lipid raft content, thereby augmenting raft-mediated signals, including carcinogenic and metastatic signaling pathways, such as PI3K/Akt signaling for cancer cell EMT and migration, and VEGF/VEGFR2 (vascular endothelial cell growth factor receptor) for angiogenesis (Hryniewicz-Jankowska et al. Citation2019; Guo et al. Citation2020; Greenlee et al. Citation2021; Zhao et al. Citation2021). Here, our bioinformatic analysis and in vitro experiments results indicate that DMY might disrupt lipid rafts. Importantly, the disruption of lipid rafts is accompanied by the reduction of cholesterol, which makes the result more reliable. Moreover, lipid rafts are closely associated with EGFR, which is consistent with the Hub gene results.
The Hub gene analysis in our study identified EGFR as the top gene. EGFR is a transmembrane glycoprotein belonging to the tyrosine kinase receptor, and its aberrant activation in cancer can trigger MAPK signaling cascade, PI3K/Akt pathway, and STAT pathway, leading to tumor proliferation, metastasis, and invasion after ligand stimulation (Tomas et al. Citation2014). In particular, all of those three downstream pathways of EGFR are a part of ‘pathways in cancer’, the first result of KEGG enrichment. EGFR was reported enriched in lipid rafts (Xu et al. Citation2012), and the activation of its downstream PI3K/Akt pathway requires lipid rafts (Gao et al. Citation2011). As expected, our results suggest that DMY inhibits EGFR and downregulates PI3K/Akt, STAT3, and Erk, providing further support for our network pharmacological prediction.
In conclusion, this is the first time to integrate network pharmacology with experiments to elucidate the pharmacological mechanisms of DMY in the treatment of HCC. The anti-cancer effects of DMY may be to disrupt lipid rafts by decreasing cholesterol and then inhibit EGFR to suppress the downstream PI3K/Akt, MAPK, and STAT signaling, thus impairing EMT progression, eventually inhibiting HCC cell migration and invasion. More importantly, cholesterol-lowering strategies may offer novel perspectives for the prevention of cholesterol disorder diseases. Taken together, the present study provides a promising candidate adjuvant drug with low toxicity for the treatment of HCC. Nevertheless, it should be noted that the in vitro validation is only a preliminary investigation, and further experimental studies are necessary to corroborate our findings.
Consent form
All named authors have agreed to the publication of the work.
Supplemental Figure
Download MS Word (884.6 KB)Disclosure statement
The authors report there are no competing interests to declare.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Bentzen SM. 2006. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nat Rev Cancer. 6(9):702–713. doi: 10.1038/nrc1950.
- Brabletz T, Kalluri R, Nieto MA, Weinberg RA. 2018. EMT in cancer. Nat Rev Cancer. 18(2):128–134. doi: 10.1038/nrc.2017.118.
- Cardwell CR, Hicks BM, Hughes C, Murray LJ. 2014. Statin use after colorectal cancer diagnosis and survival: a population-based cohort study. J Clin Oncol. 32(28):3177–3183. doi: 10.1200/JCO.2013.54.4569.
- Dong S, Zhu M, Wang K, Zhao X, Hu L, Jing W, Lu H, Wang S. 2021. Dihydromyricetin improves DSS-induced colitis in mice via modulation of fecal-bacteria-related bile acid metabolism. Pharmacol Res. 171:105767. doi: 10.1016/j.phrs.2021.105767.
- Gao X, Lowry PR, Zhou X, Depry C, Wei Z, Wong GW, Zhang J. 2011. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc Natl Acad Sci U S A. 108(35):14509–14514. doi: 10.1073/pnas.1019386108.
- Greenlee JD, Subramanian T, Liu K, King MR. 2021. Rafting down the metastatic cascade: the role of lipid rafts in cancer metastasis, cell death, and clinical outcomes. Cancer Res. 81(1):5–17. doi: 10.1158/0008-5472.CAN-20-2199.
- Guo Y, Pan W, Liu S, Shen Z, Xu Y, Hu L. 2020. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 19(3):1997–2007. doi: 10.3892/etm.2020.8454.
- Guo D, Zhang D, Ren M, Lu G, Zhang X, He S, Li Y. 2020. THBS4 promotes HCC progression by regulating ITGB1 via FAK/PI3K/AKT pathway. Faseb J. 34(8):10668–10681. doi: 10.1096/fj.202000043R.
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. CELL. 144(5):646–674. doi: 10.1016/j.cell.2011.02.013.
- Hryniewicz-Jankowska A, Augoff K, Sikorski AF. 2019. The role of cholesterol and cholesterol-driven membrane raft domains in prostate cancer. Exp Biol Med (Maywood). 244(13):1053–1061. doi: 10.1177/1535370219870771.
- Huang H, Hu M, Zhao R, Li P, Li M. 2013. Dihydromyricetin suppresses the proliferation of hepatocellular carcinoma cells by inducing G2/M arrest through the Chk1/Chk2/Cdc25C pathway. Oncol Rep. 30(5):2467–2475. doi: 10.3892/or.2013.2705.
- Huang C, Su C, Wang P, Lu Y, Ho Y, Yang S, Hsin C, Lin C. 2022. Dihydromyricetin inhibits cancer cell migration and matrix metalloproteinases-2 expression in human nasopharyngeal carcinoma through extracellular signal-regulated kinase signaling pathway. Environ Toxicol. 37(5):1244–1253. doi: 10.1002/tox.23480.
- Jiang L, Zhang Q, Ren H, Ma S, Lu C, Liu B, Liu J, Liang J, Li M, Zhu R. 2015. Dihydromyricetin enhances the chemo-sensitivity of nedaplatin via regulation of the p53/Bcl-2 pathway in hepatocellular carcinoma cells. PLoS One. 10(4):e124994. doi: 10.1371/journal.pone.0124994.
- Kawata S, Takaishi K, Nagase T, Ito N, Matsuda Y, Tamura S, Matsuzawa Y, Tarui S. 1990. Increase in the active form of 3-hydroxy-3-methylglutaryl coenzyme A reductase in human hepatocellular carcinoma: possible mechanism for alteration of cholesterol biosynthesis. Cancer Res. 50(11):3270–3273.
- Kuzu OF, Noory MA, Robertson GP. 2016. The role of cholesterol in cancer. Cancer Res. 76(8):2063–2070. doi: 10.1158/0008-5472.CAN-15-2613.
- Li M, Tang Y, Wang D, Zhai X, Shen H, Zhong C, Yao M, Jin A, Zhou Z, Zhou S, et al. 2022. Sphingosine-1-phosphate transporter spinster homolog 2 is essential for iron-regulated metastasis of hepatocellular carcinoma. Mol Ther. 30(2):703–713. doi: 10.1016/j.ymthe.2021.09.012.
- Lingwood D, Simons K. 2010. Lipid rafts as a membrane-organizing principle. Science. 327(5961):46–50. doi: 10.1126/science.1174621.
- Liu D, Mao Y, Ding L, Zeng X. 2019. Dihydromyricetin: a review on identification and quantification methods, biological activities, chemical stability, metabolism and approaches to enhance its bioavailability. Trends Food Sci Technol. 91:586–597. doi: 10.1016/j.tifs.2019.07.038.
- Liu J, Shu Y, Zhang Q, Liu B, Xia J, Qiu M, Miao H, Li M, Zhu R. 2014. Dihydromyricetin induces apoptosis and inhibits proliferation in hepatocellular carcinoma cells. Oncol Lett. 8(4):1645–1651. doi: 10.3892/ol.2014.2330.
- Liu B, Tan X, Liang J, Wu S, Liu J, Zhang Q, Zhu R. 2014. A reduction in reactive oxygen species contributes to dihydromyricetin-induced apoptosis in human hepatocellular carcinoma cells. Sci Rep. 4:7041. doi: 10.1038/srep07041.
- Liu F, Tian T, Zhang Z, Xie S, Yang J, Zhu L, Wang W, Shi C, Sang L, Guo K, et al. 2022. Long non-coding RNA SNHG6 couples cholesterol sensing with mTORC1 activation in hepatocellular carcinoma. Nat Metab. 4(8):1022–1040. doi: 10.1038/s42255-022-00616-7.
- Lu C, He Y, Yuan W, Xiang L, Zhang J, Liang Y, Duan J, He Y, Li M. 2017. Dihydromyricetin-mediated inhibition of the Notch1 pathway induces apoptosis in QGY7701 and HepG2 hepatoma cells. World J Gastroenterol. 23(34):6242–6251. doi: 10.3748/wjg.v23.i34.6242.
- Luo J, Yang H, Song B. 2020. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. 21(4):225–245. doi: 10.1038/s41580-019-0190-7.
- Mok EHK, Leung CON, Zhou L, Lei MML, Leung HW, Tong M, Wong TL, Lau EYT, Ng IOL, Ding J, et al. 2022. Caspase-3-induced activation of SREBP2 drives drug resistance via promotion of cholesterol biosynthesis in hepatocellular carcinoma. Cancer Res. 82(17):3102–3115. doi: 10.1158/0008-5472.CAN-21-2934.
- Nielsen SF, Nordestgaard BG, Bojesen SE. 2012. Statin use and reduced cancer-related mortality. N Engl J Med. 367(19):1792–1802. doi: 10.1056/NEJMoa1201735.
- Omata M, Cheng A-L, Kokudo N, Kudo M, Lee JM, Jia J, Tateishi R, Han K-H, Chawla YK, Shiina S, et al. 2017. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol Int. 11(4):317–370. doi: 10.1007/s12072-017-9799-9.
- Perz JF, Armstrong GL, Farrington LA, Hutin YJF, Bell BP. 2006. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 45(4):529–538. doi: 10.1016/j.jhep.2006.05.013.
- Randrup Hansen C, Grimm D, Bauer J, Wehland M, Magnusson NE. 2017. Effects and side effects of using sorafenib and sunitinib in the treatment of metastatic renal cell carcinoma. IJMS. 18(2):461. doi: 10.3390/ijms18020461.
- Recondo G, Che J, Jänne PA, Awad MM. 2020. Targeting MET dysregulation in cancer. Cancer Discov. 10(7):922–934. doi: 10.1158/2159-8290.CD-19-1446.
- Roth GA, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, Abbastabar H, Abd-Allah F, Abdela J, Abdelalim A, et al. 2018. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 392(10159):1736–1788. doi: 10.1016/S0140-6736(18)32203-7.
- Shoukat S, Zheng D, Yusuf SW. 2019. Cardiotoxicity related to radiation therapy. Cardiol Clin. 37(4):449–458. doi: 10.1016/j.ccl.2019.07.010.
- Silva J, Yu X, Moradian R, Folk C, Spatz MH, Kim P, Bhatti AA, Davies DL, Liang J. 2020. Dihydromyricetin protects the liver via changes in lipid metabolism and enhanced ethanol metabolism. Alcohol Clin Exp Res. 44(5):1046–1060. doi: 10.1111/acer.14326.
- Smith B, Land H. 2012. Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Rep. 2(3):580–590. doi: 10.1016/j.celrep.2012.08.011.
- Snaebjornsson MT, Janaki-Raman S, Schulze A. 2020. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 31(1):62–76. doi: 10.1016/j.cmet.2019.11.010.
- Song Y, Sun L, Ma P, Xu L, Xiao P. 2022. Dihydromyricetin prevents obesity via regulating bile acid metabolism associated with the farnesoid X receptor in ob/ob mice. Food Funct. 13(5):2491–2503. doi: 10.1039/d1fo03971g.
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. 2021. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 71(3):209–249. doi: 10.3322/caac.21660.
- Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, Wang Q, Wang S, Rong D, Reiter FP, et al. 2020. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. 5:87.
- Tang W, Zhou J, Yang W, Feng Y, Wu H, Mok MTS, Zhang L, Liang Z, Liu X, Xiong Z, et al. 2022. Aberrant cholesterol metabolic signaling impairs antitumor immunosurveillance through natural killer T cell dysfunction in obese liver. Cell Mol Immunol. 19(7):834–847. doi: 10.1038/s41423-022-00872-3.
- Tomas A, Futter CE, Eden ER. 2014. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 24(1):26–34. doi: 10.1016/j.tcb.2013.11.002.
- Verhoeven Y, Tilborghs S, Jacobs J, De Waele J, Quatannens D, Deben C, Prenen H, Pauwels P, Trinh XB, Wouters A, et al. 2020. The potential and controversy of targeting STAT family members in cancer. Semin Cancer Biol. 60:41–56. doi: 10.1016/j.semcancer.2019.10.002.
- Wang Y, Wang J, Li X, Xion X, Wang J, Zhou Z, Zhu X, Gu Y, Dominissini D, He L, et al. 2021. N (1)-Methyladenosine methylation in tRNA drives liver tumorigenesis by regulating cholesterol metabolism. Nat Commun. 12(1):6314. doi: 10.1038/s41467-021-26718-6.
- Xu Y, Wang S, Chan HF, Lu H, Lin Z, He C, Chen M. 2017. Dihydromyricetin induces apoptosis and reverses drug resistance in ovarian cancer cells by p53-mediated downregulation of survivin. Sci Rep. 7:46060. doi: 10.1038/srep46060.
- Xu L, Zhang Y, Liu J, Qu J, Hu X, Zhang F, Zheng H, Qu X, Liu Y. 2012. TRAIL-activated EGFR by Cbl-b-regulated EGFR redistribution in lipid rafts antagonises TRAIL-induced apoptosis in gastric cancer cells. Eur J Cancer. 48(17):3288–3299. doi: 10.1016/j.ejca.2012.03.005.
- Yang JD, Roberts LR. 2010. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 7(8):448–458. doi: 10.1038/nrgastro.2010.100.
- Ye L, Yin G, Jiang M, Tu B, Li Z, Wang Y. 2021. Dihydromyricetin exhibits antitumor activity in nasopharyngeal cancer cell through antagonizing Wnt/β-catenin signaling. Integr Cancer Ther. 20:153473542199121. doi: 10.1177/1534735421991217.
- Zeng Y, Peng Y, Tang K, Wang YQ, Zhao ZY, Wei XY, Xu XL. 2018. Dihydromyricetin ameliorates foam cell formation via LXRα-ABCA1/ABCG1-dependent cholesterol efflux in macrophages. Biomed Pharmacother. 101:543–552. doi: 10.1016/j.biopha.2018.02.124.
- Zhang Q, Li R, Zeng G, Liu B, Liu J, Shu Y, Liu Z, Qiu Z, Wang D, Miao H, et al. 2014. Dihydromyricetin inhibits migration and invasion of hepatoma cells through regulation of MMP-9 expression. World J Gastroenterol. 20(29):10082–10093. doi: 10.3748/wjg.v20.i29.10082.
- Zhang X, Wang L, Peng L, Tian X, Qiu X, Cao H, Yang Q, Liao R, Yan F. 2019. Dihydromyricetin protects HUVECs of oxidative damage induced by sodium nitroprusside through activating PI3K/Akt/FoxO3a signalling pathway. J Cell Mol Med. 23(7):4829–4838. doi: 10.1111/jcmm.14406.
- Zhang C, Zhu N, Long J, Wu H, Wang Y, Liu B, Liao D, Qin L. 2021. Celastrol induces lipophagy via the LXRα/ABCA1 pathway in clear cell renal cell carcinoma. Acta Pharmacol Sin. 42(9):1472–1485. doi: 10.1038/s41401-020-00572-6.
- Zhao T, Guo B, Xiao C, Chen J, Lü C, Fang F, Li B. 2021. Aerobic exercise suppresses hepatocellular carcinoma by downregulating dynamin-related protein 1 through PI3K/AKT pathway. J Integr Med. 19(5):418–427. doi: 10.1016/j.joim.2021.08.003.
- Zhou F, Sun X. 2021. Cholesterol metabolism: a double-edged sword in hepatocellular carcinoma. Front Cell Dev Biol. 9:762828. doi: 10.3389/fcell.2021.762828.